
Single-Atom Co-Catalysts Employed in Titanium Dioxide Photocatalysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
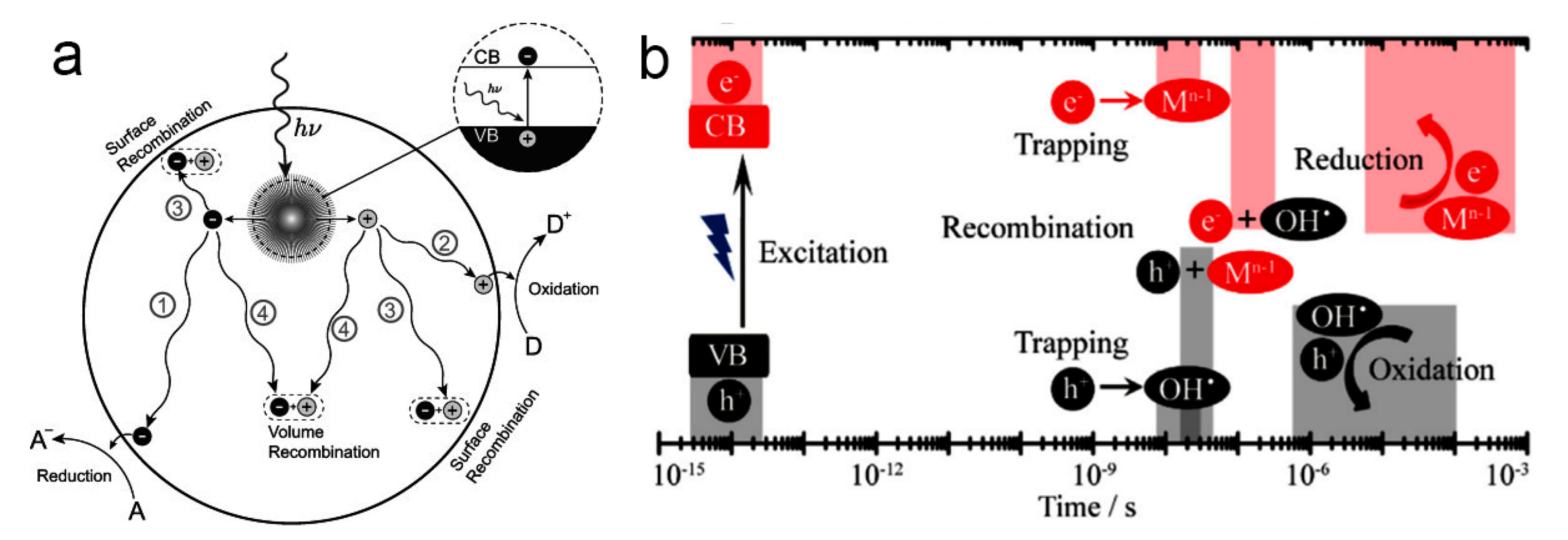
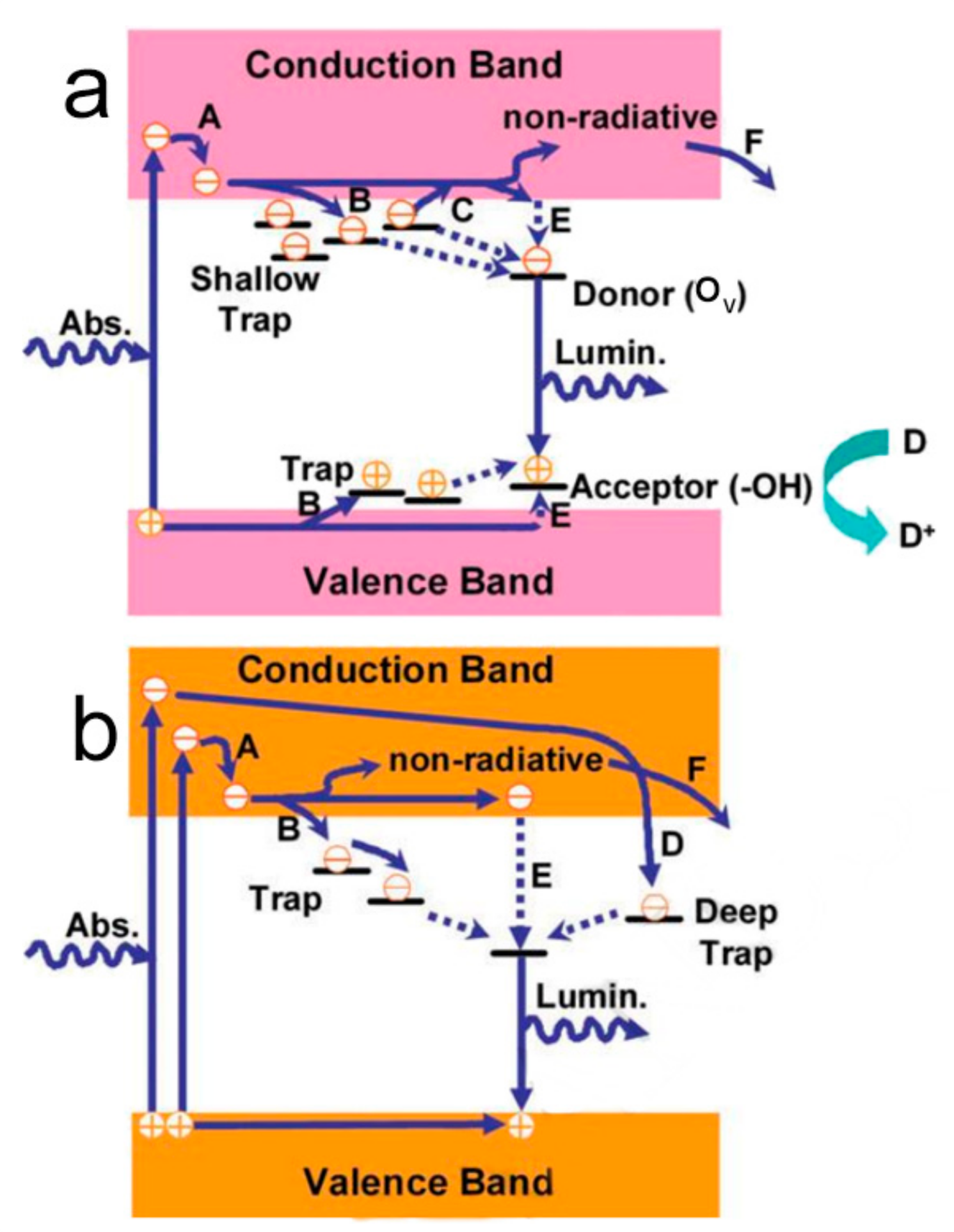
2. The Fundamental Principle of Photocatalysis

3. Accelerating Charge Transfer and Reaction Selectivity of Photocatalyst
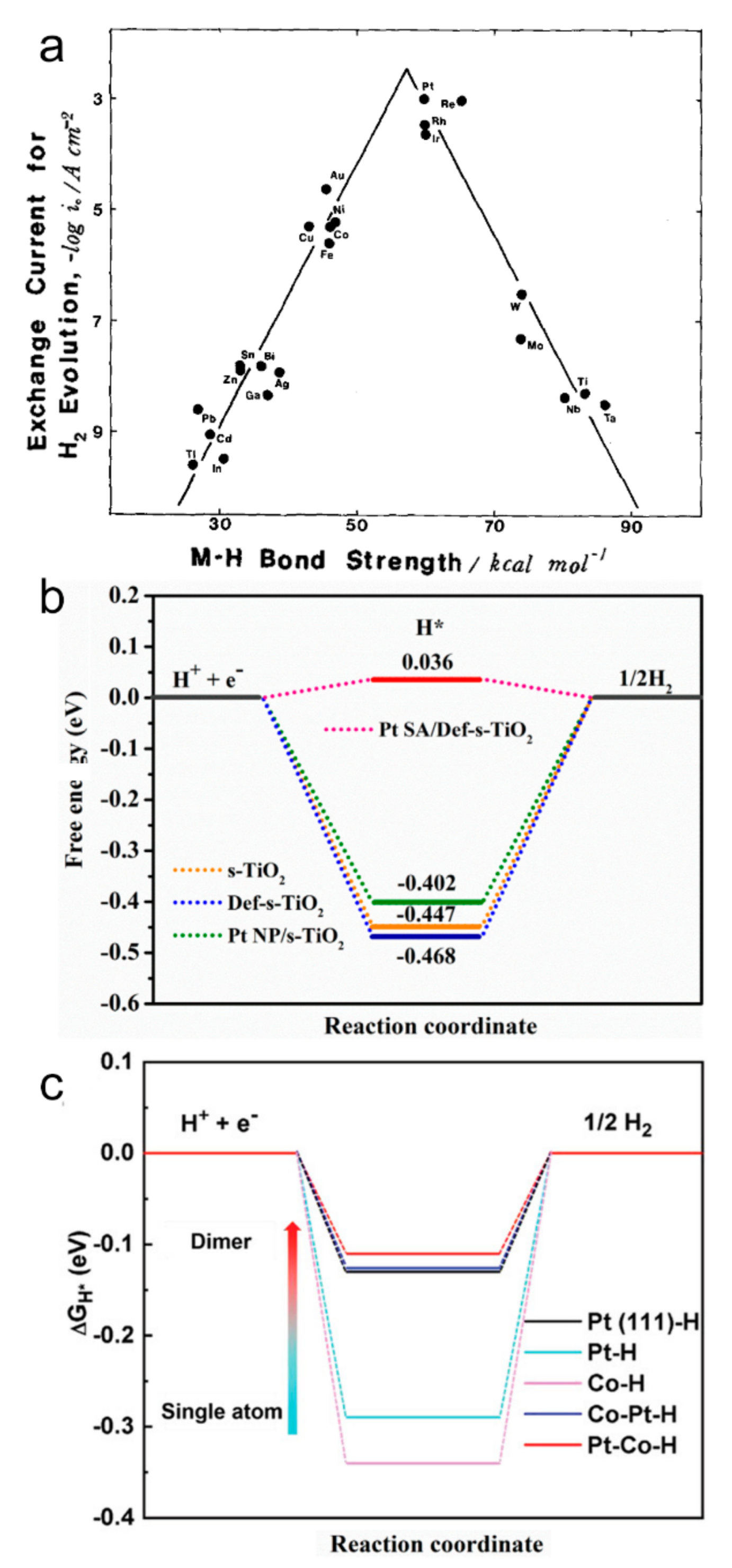
3.1. Co-Catalyst Approach in Photocatalysis
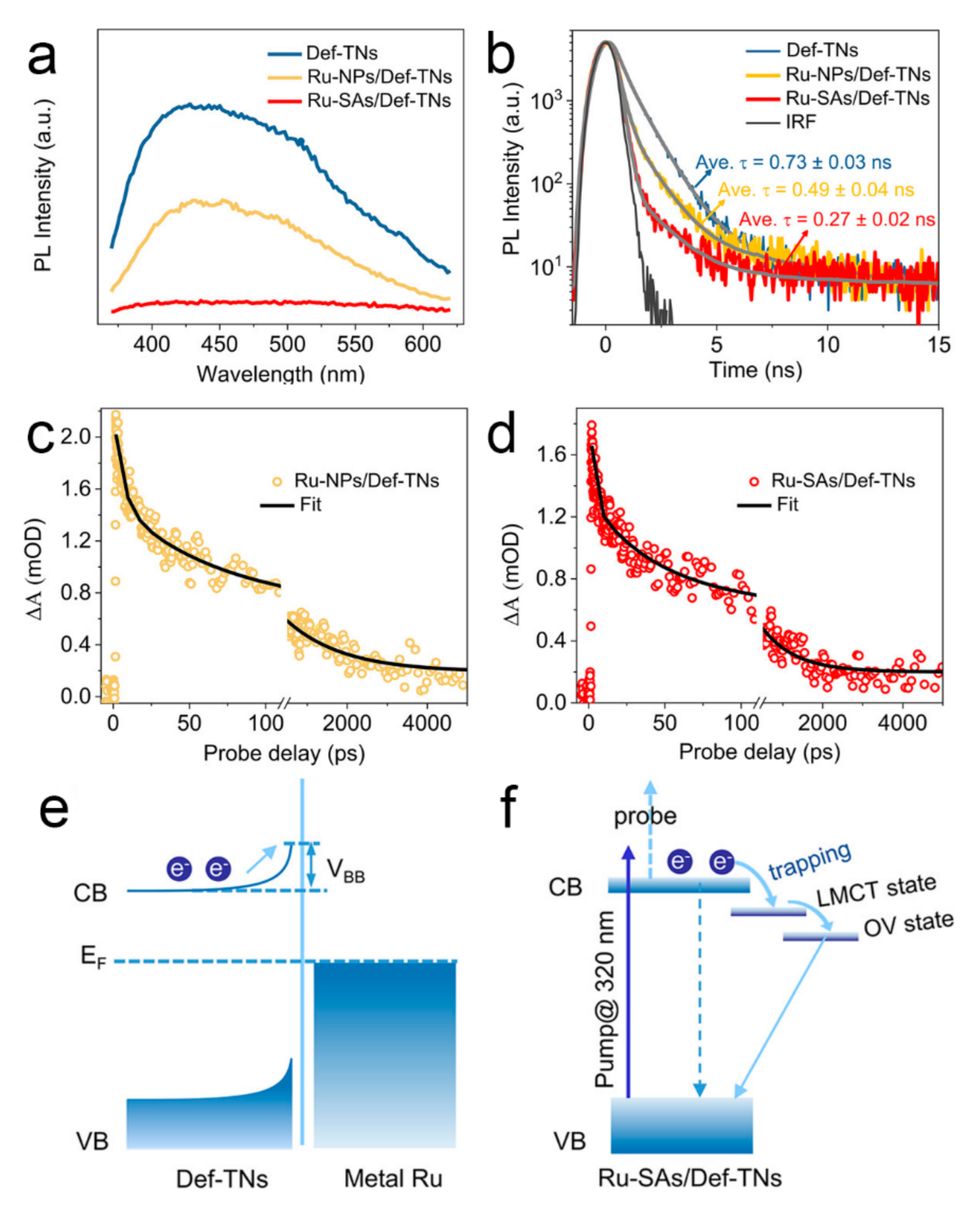
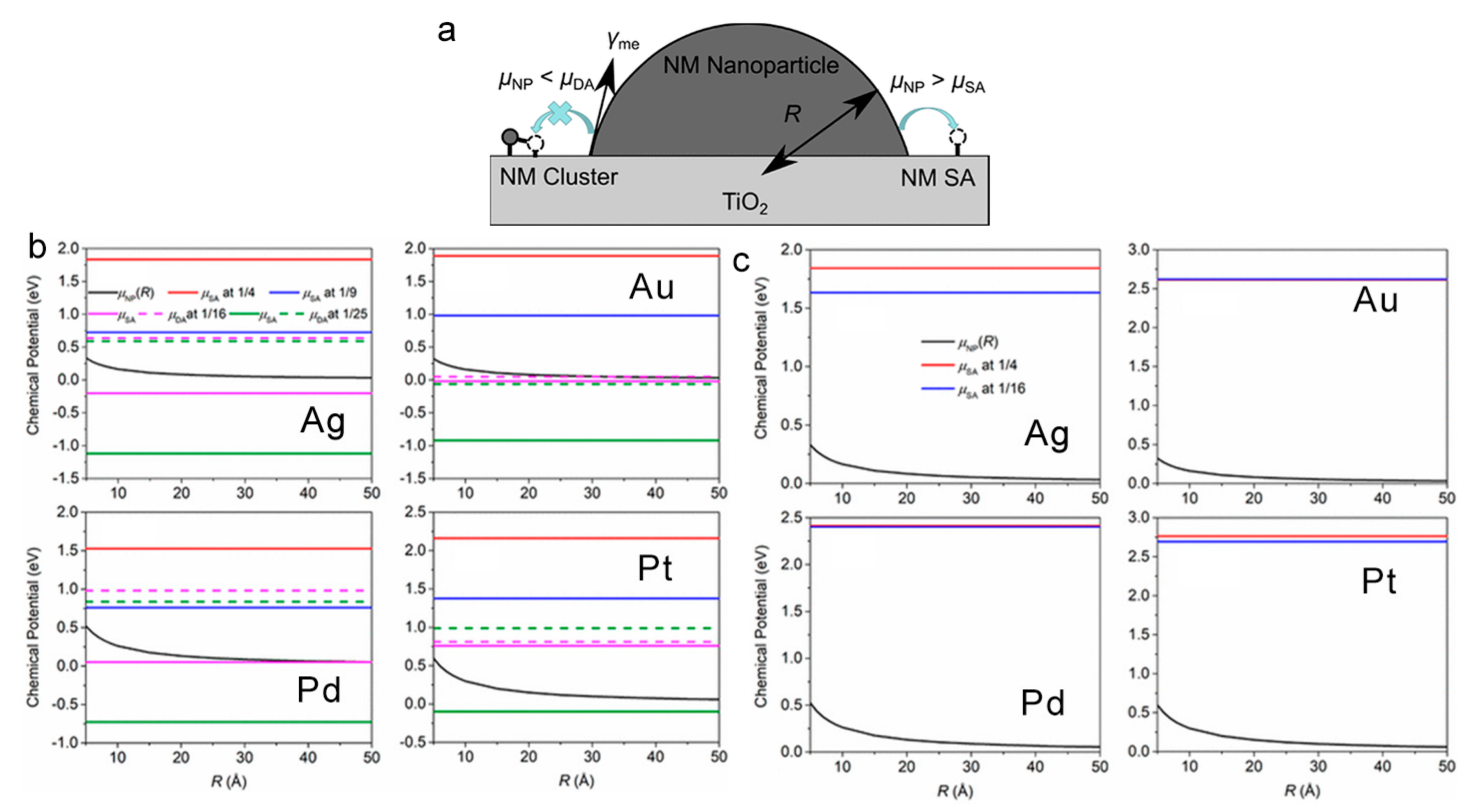
3.2. Absence of Schottky Junction in SA Co-Catalysts
3.3. SAs and Reaction Selectivity
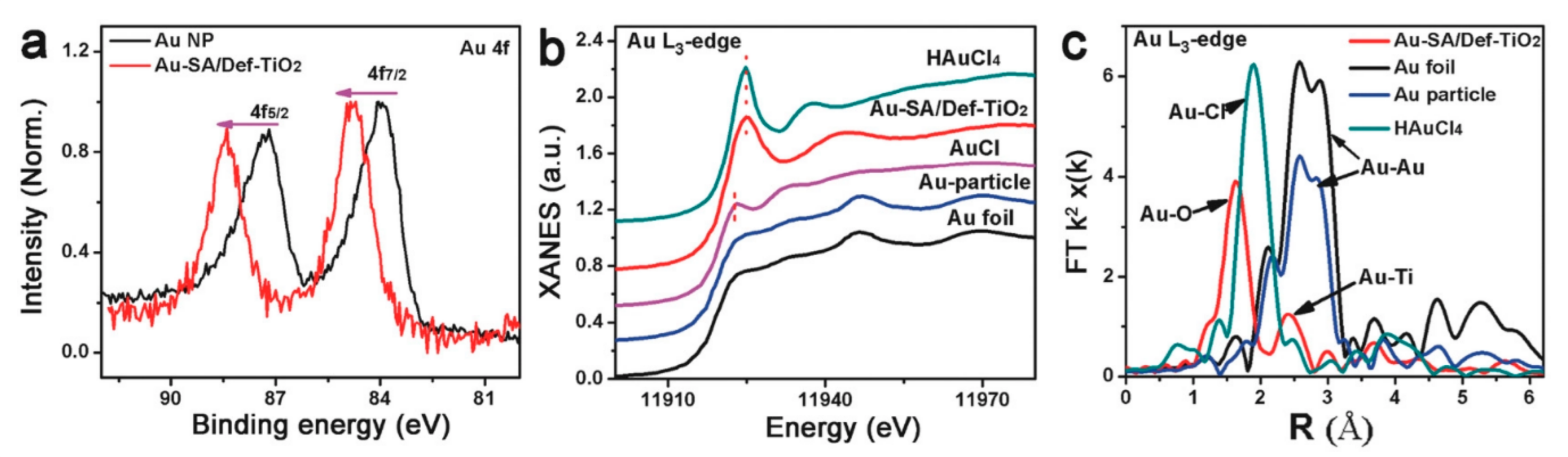
4. Synthesis and Anchoring of SAs
5. Other Aspects of Single Atom Co-Catalysts in Photocatalysis
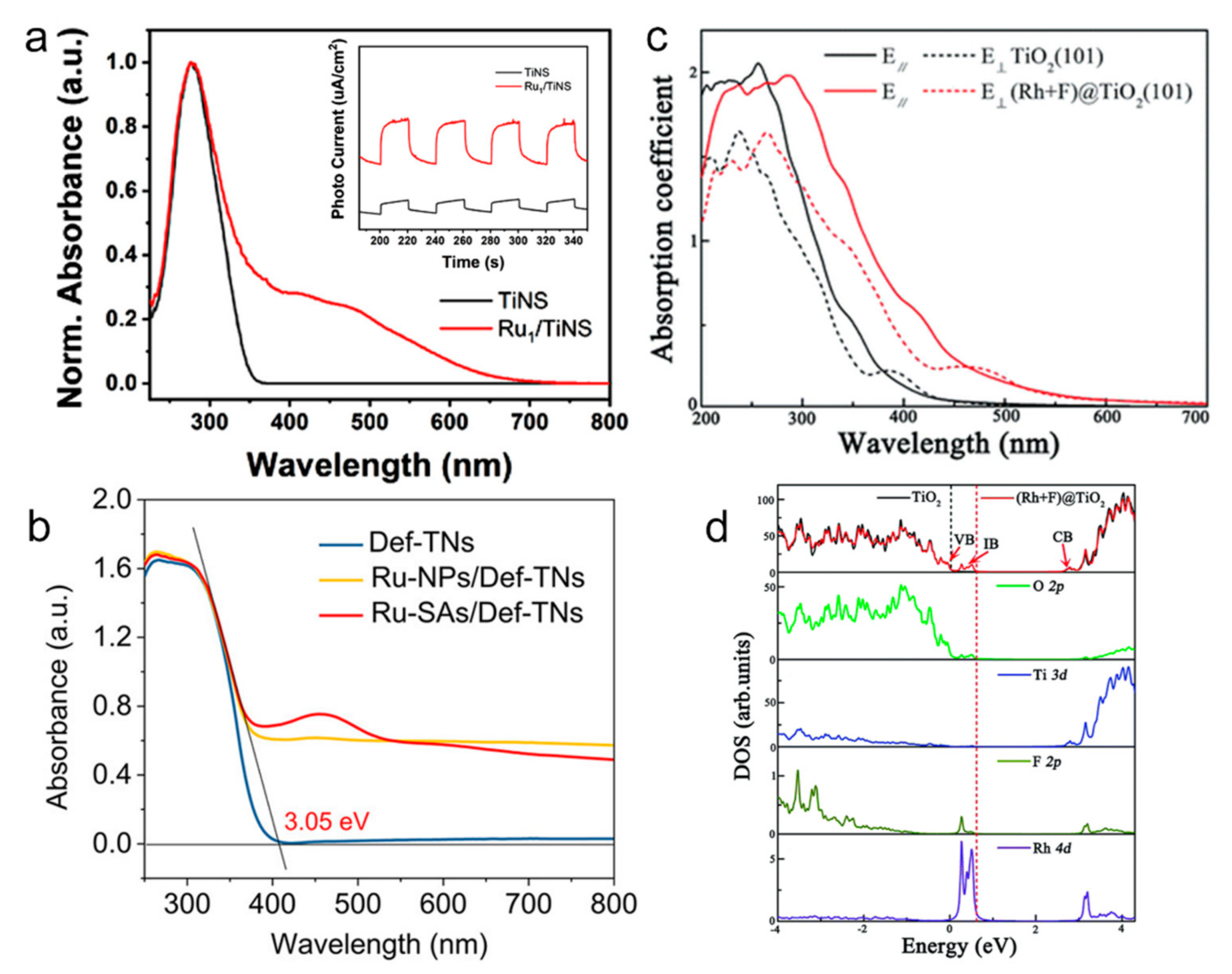
5.1. Light Absorption/Harvesting
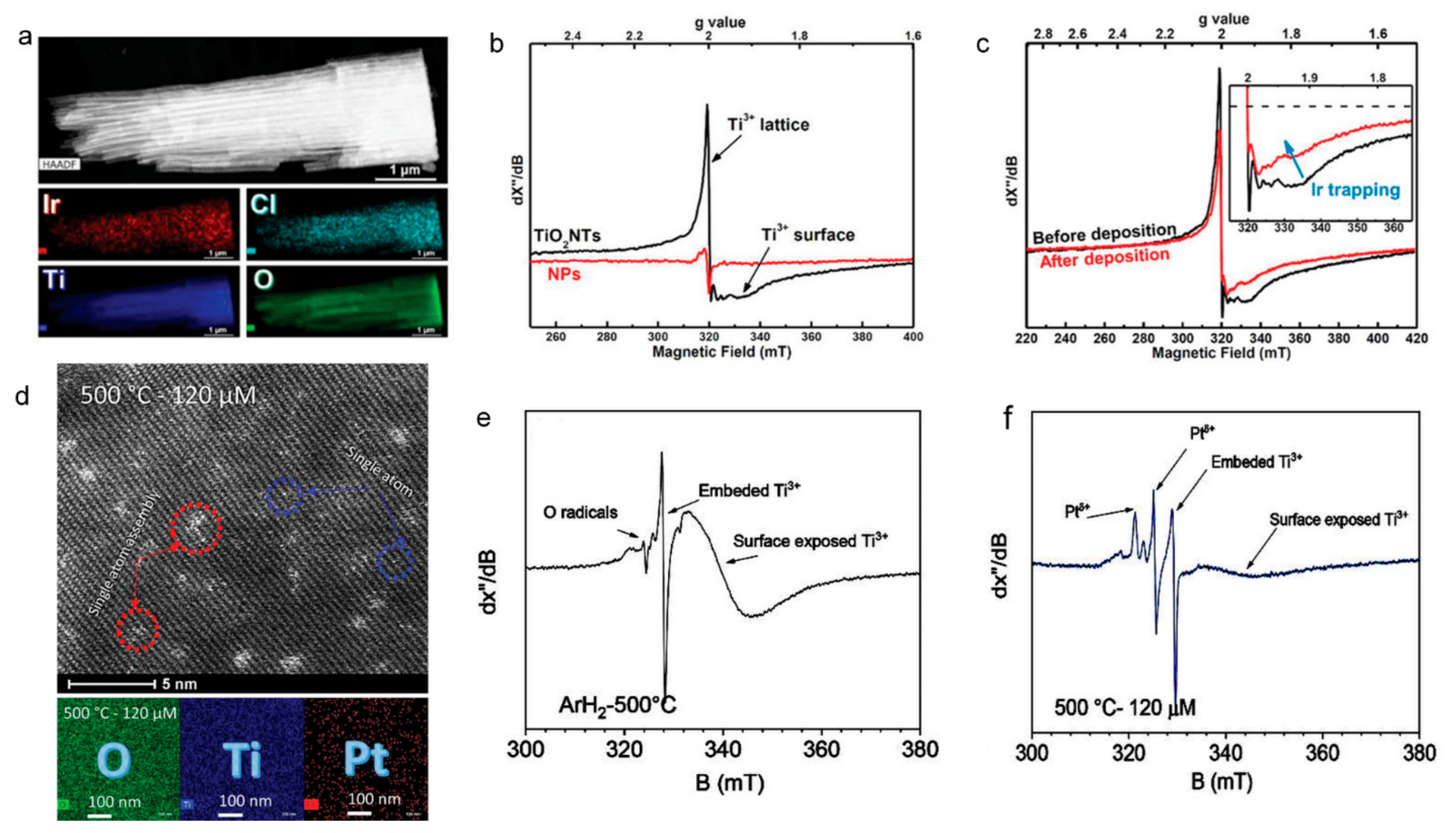
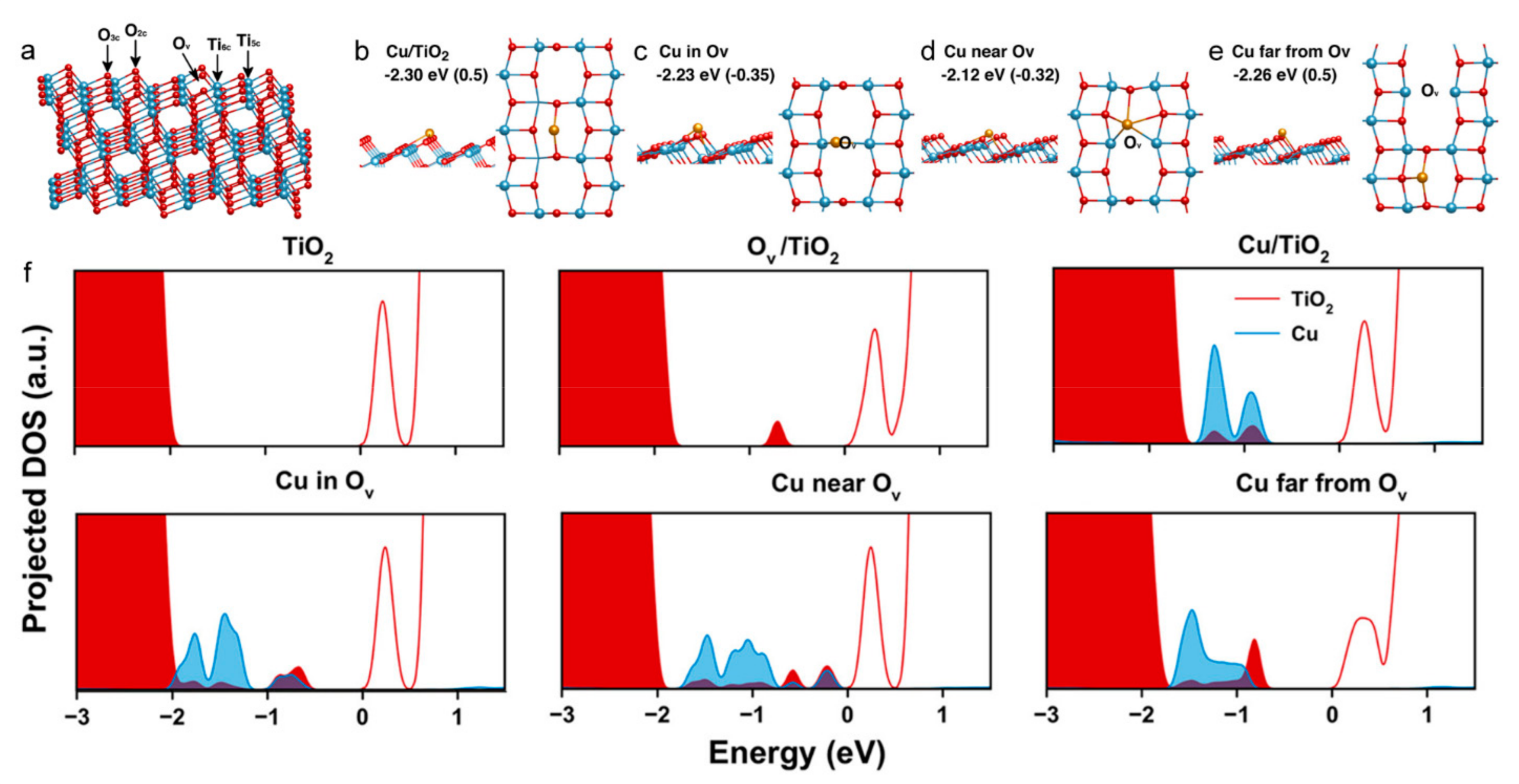
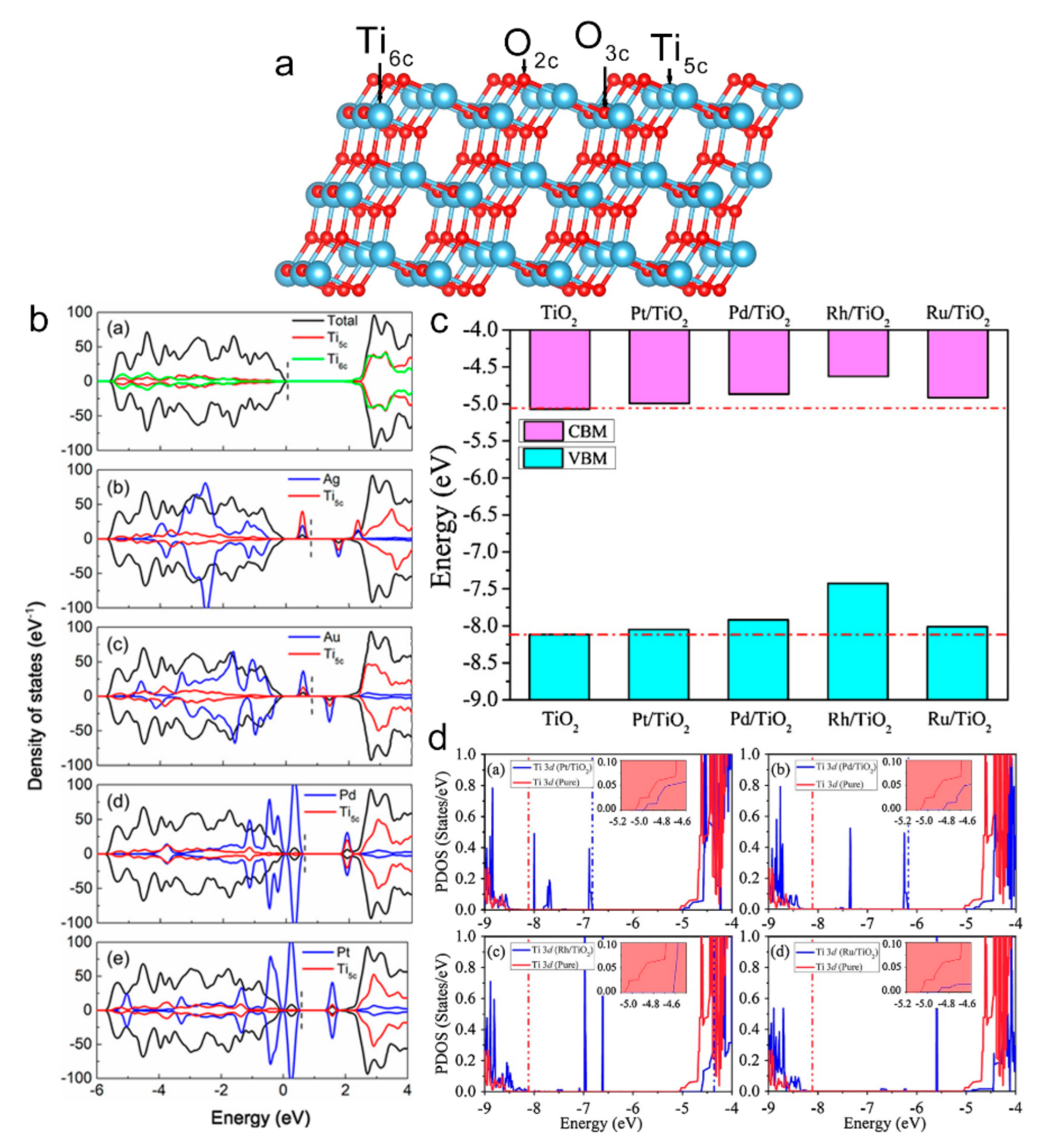
5.2. Single-Atoms and Local Electronic Structure
6. Applications of Single-Atom Co-Catalyst in Photocatalysis
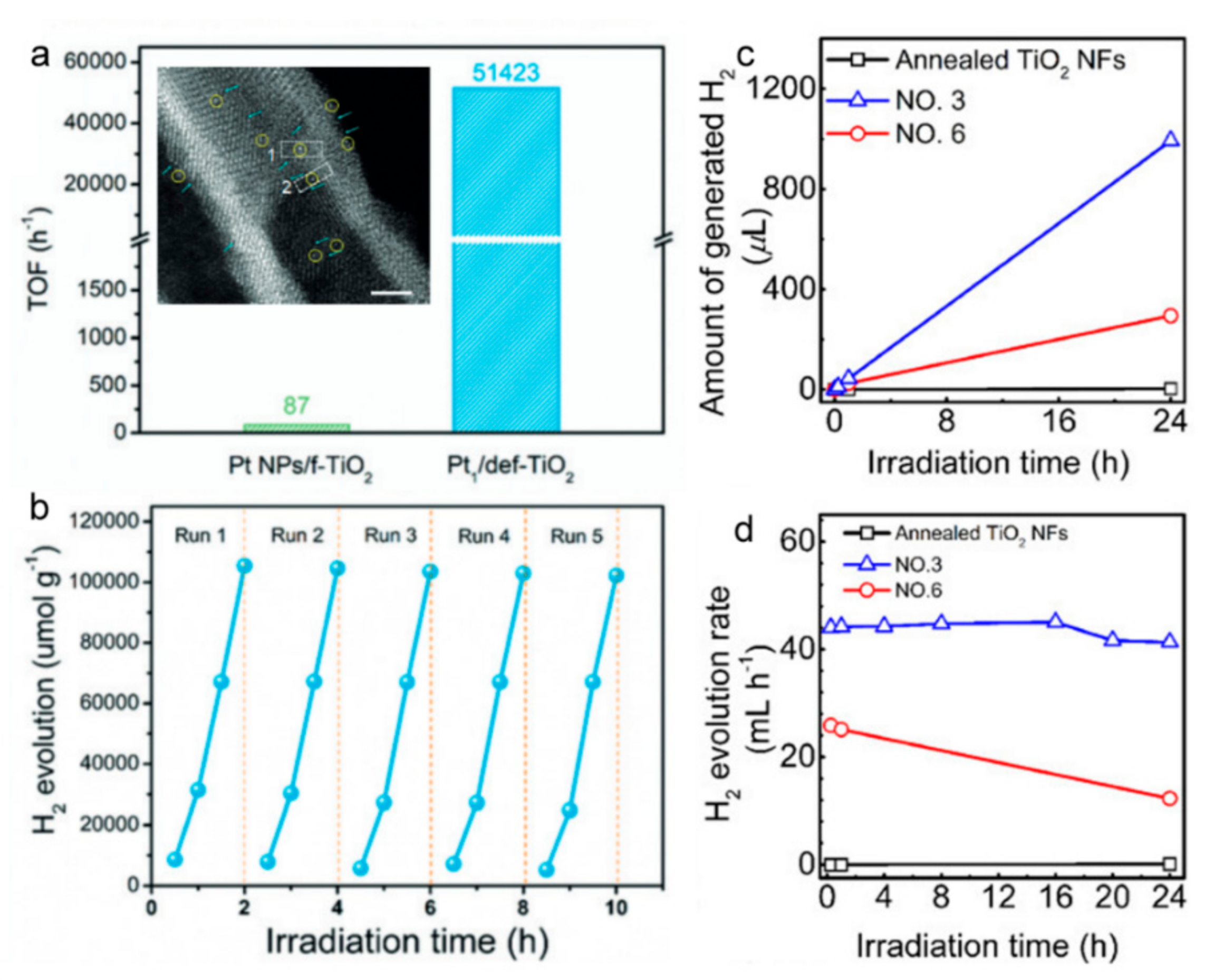
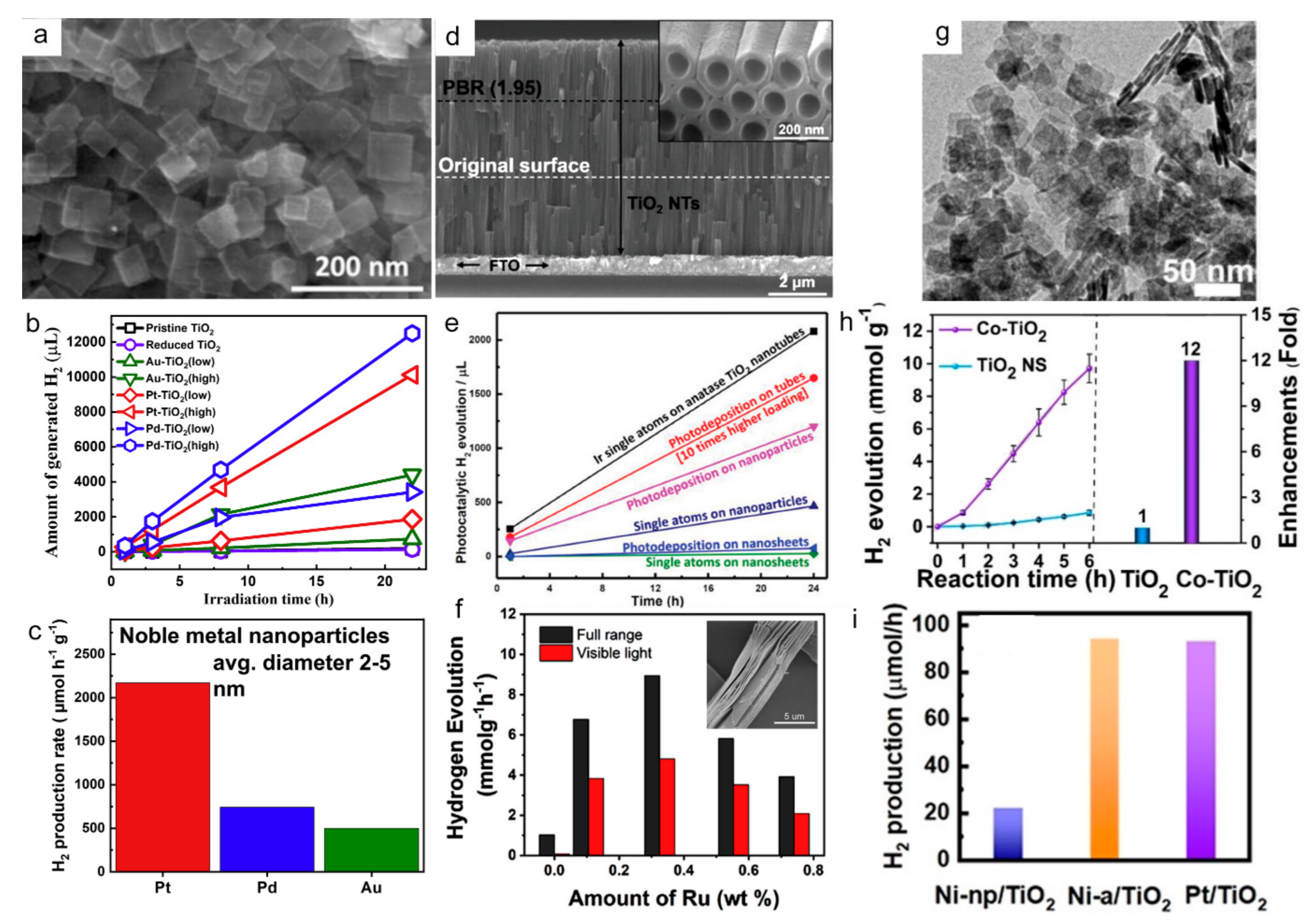
6.1. Hydrogen Production

6.2. Photocatalytic CO2 Reduction
6.3. Ammonia Production
6.4. Other Applications
6.4.1. Photoelectrochemical Water Splitting
6.4.2. Synthesis and Degradation of Organic Compounds
7. Summary and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Chorkendorff Niemantsverdriet, J.W.I. Concepts of Modern Catalysis and Kinetics; Chorkendorff, I., Niemantsverdriet, J.W., Eds.; John Wiley & Sons: Hoboken, NJ, USA; Incorporated: Newark, NJ, USA, 2017; pp. 1–21. [Google Scholar]
- Coronado, J.M. A Historical Introduction to Photocatalysis BT. In Design of Advanced Photocatalytic Materials for Energy and Environmental Applications; Coronado, J.M., Fresno, F., Hernández-Alonso, M.D., Portela, R., Eds.; Springer: London, UK, 2013; pp. 1–4. ISBN 978-1-4471-5061-9. [Google Scholar]
- Fujishima, A.; Honda, K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Dutta, S. A review on H2 production through photocatalytic reactions using TiO2/TiO2-assisted catalysts. Fuel 2018, 220, 607–620. [Google Scholar] [CrossRef]
- Zhang, F.; Zhao, P.; Niu, M.; Maddy, J. The survey of key technologies in hydrogen energy storage. Int. J. Hydrogen Energy 2016, 41, 14535–14552. [Google Scholar] [CrossRef]
- Yang, J.; Wang, D.; Han, H.; Li, C. Roles of Cocatalysts in Photocatalysis and Photoelectrocatalysis. Acc. Chem. Res. 2013, 46, 1900–1909. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, J.; Lan, R.; Tao, S. Development and Recent Progress on Ammonia Synthesis Catalysts for Haber–Bosch Process. Adv. Energy Sustain. Res. 2021, 2, 2000043. [Google Scholar] [CrossRef]
- Wang, B.; Cai, H.; Shen, S. Single Metal Atom Photocatalysis. Small Methods 2019, 3, 1–14. [Google Scholar] [CrossRef]
- Naldoni, A.; Guler, U.; Wang, Z.; Marelli, M.; Malara, F.; Meng, X.; Besteiro, L.V.; Govorov, A.O.; Kildishev, A.V.; Boltasseva, A.; et al. Broadband Hot-Electron Collection for Solar Water Splitting with Plasmonic Titanium Nitride. Adv. Opt. Mater. 2017, 5, 1601031. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Zhang, W.; Luo, N.; Xue, Z.; Hu, Q.; Zeng, W.; Xu, J. Bimetallic Nanocrystals: Structure, Controllable Synthesis and Applications in Catalysis, Energy and Sensing. Nanomaterials 2021, 11, 1926. [Google Scholar] [CrossRef]
- Bielan, Z.; Kowalska, E.; Dudziak, S.; Wang, K.; Ohtani, B.; Zielińska-Jurek, A. Mono- and bimetallic (Pt/Cu) titanium(IV) oxide core–shell photocatalysts with UV/Vis light activity and magnetic separability. Catal. Today 2021, 361, 198–209. [Google Scholar] [CrossRef]
- Yasuhara, A.; Sannomiya, T. Atomically Localized Ordered Phase and Segregation at Grain Boundaries in Au–Ag–Cu Ternary Alloy Nanoparticles. J. Phys. Chem. C 2022, 126, 1160–1167. [Google Scholar] [CrossRef]
- Yasuhara, A.; Kubo, K.; Yanagimoto, S.; Sannomiya, T. Thermodynamic Tuning of Au–Ag–Cu Nanoparticles with Phase Separation and Ordered Phase Formation. J. Phys. Chem. C 2020, 124, 15481–15488. [Google Scholar] [CrossRef]
- Cha, G.; Hwang, I.; Hejazi, S.; Dobrota, A.S.; Pašti, I.A.; Osuagwu, B.; Kim, H.; Will, J.; Yokosawa, T.; Badura, Z.; et al. As a single atom Pd outperforms Pt as the most active co-catalyst for photocatalytic H2 evolution. iScience 2021, 24, 102938. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, D.; Chen, Y.; Fu, W.F.; Lv, X.J. Single-Atom Catalysts for Photocatalytic Reactions. ACS Sustain. Chem. Eng. 2019, 7, 6430–6443. [Google Scholar] [CrossRef]
- Xia, B.; Zhang, Y.; Ran, J.; Jaroniec, M.; Qiao, S.Z. Single-Atom Photocatalysts for Emerging Reactions. ACS Cent. Sci. 2021, 7, 39–54. [Google Scholar] [CrossRef]
- Fu, Q.; Saltsburg, H.; Flytzani-Stephanopoulos, M. Active nonmetallic Au and Pt species on ceria-based water-gas shift catalysts. Science 2003, 301, 935–938. [Google Scholar] [CrossRef]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Chen, J.F.; Li, Y.H.; Yuan, W.T.; Zhou, Y.; Zheng, L.R.; Wang, H.F.; Hu, P.; Wang, Y.; Zhao, H.J.; et al. Stable isolated metal atoms as active sites for photocatalytic hydrogen evolution. Chem.—A Eur. J. 2014, 20, 2138–2144. [Google Scholar] [CrossRef]
- Jin, C.; Dai, Y.; Wei, W.; Ma, X.; Li, M.; Huang, B. Effects of single metal atom (Pt, Pd, Rh and Ru) adsorption on the photocatalytic properties of anatase TiO2. Appl. Surf. Sci. 2017, 426, 639–646. [Google Scholar] [CrossRef]
- Jiang, X.H.; Zhang, L.S.; Liu, H.Y.; Wu, D.S.; Wu, F.Y.; Tian, L.; Liu, L.L.; Zou, J.P.; Luo, S.L.; Chen, B.B. Silver Single Atom in Carbon Nitride Catalyst for Highly Efficient Photocatalytic Hydrogen Evolution. Angew. Chem.—Int. Ed. 2020, 59, 23112–23116. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Su, Y.; Quan, X.; Choi, W.; Zhang, G.; Liu, N.; Kim, B.; Chen, S.; Yu, H.; Zhang, S. Single-atom platinum confined by the interlayer nanospace of carbon nitride for efficient photocatalytic hydrogen evolution. Nano Energy 2020, 69, 104409. [Google Scholar] [CrossRef]
- Gao, G.; Jiao, Y.; Waclawik, E.R.; Du, A. Single Atom (Pd/Pt) Supported on Graphitic Carbon Nitride as an Efficient Photocatalyst for Visible-Light Reduction of Carbon Dioxide. J. Am. Chem. Soc. 2016, 138, 6292–6297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Dong, Y.; Zhao, S.; Wang, G.; Jiang, P.; Zhong, J.; Zhu, Y. Photochemical preparation of atomically dispersed nickel on cadmium sulfide for superior photocatalytic hydrogen evolution. Appl. Catal. B Environ. 2020, 261, 118233. [Google Scholar] [CrossRef]
- Zhao, G.; Liu, H.; Ye, J. Constructing and controlling of highly dispersed metallic sites for catalysis. Nano Today 2018, 19, 108–125. [Google Scholar] [CrossRef]
- Cao, H.; Xue, J.; Wang, Z.; Dong, J.; Li, W.; Wang, R.; Sun, S.; Gao, C.; Tan, Y.; Zhu, X.; et al. Construction of Atomically Dispersed Cu Sites and S Vacancies on CdS for Enhanced Photocatalytic CO2 Reduction. J. Mater. Chem. A 2021, 9, 16339–16344. [Google Scholar] [CrossRef]
- Pan, J.; Zhang, G.; Guan, Z.; Zhao, Q.; Li, G.; Yang, J.; Li, Q.; Zou, Z. Anchoring Ni single atoms on sulfur-vacancy-enriched ZnIn2S4 nanosheets for boosting photocatalytic hydrogen evolution. J. Energy Chem. 2021, 58, 408–414. [Google Scholar] [CrossRef]
- Kou, M.; Liu, W.; Wang, Y.; Huang, J.; Chen, Y.; Zhou, Y.; Chen, Y.; Ma, M.; Lei, K.; Xie, H.; et al. Photocatalytic CO2 conversion over single-atom MoN2 sites of covalent organic framework. Appl. Catal. B Environ. 2021, 291, 120146. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, G.; Shi, L.; Ye, J. Single-Atom Catalysts: Emerging Multifunctional Materials in Heterogeneous Catalysis. Adv. Energy Mater. 2018, 8, 1701343. [Google Scholar] [CrossRef]
- Liang, S.; Hao, C.; Shi, Y. The Power of Single-Atom Catalysis. ChemCatChem 2015, 7, 2559–2567. [Google Scholar] [CrossRef]
- Yang, M.; Flytzani-Stephanopoulos, M. Design of single-atom metal catalysts on various supports for the low-temperature water-gas shift reaction. Catal. Today 2017, 298, 216–225. [Google Scholar] [CrossRef]
- Kramar, B.V.; Phelan, B.T.; Sprague-Klein, E.A.; Diroll, B.T.; Lee, S.; Otake, K.; Palmer, R.; Mara, M.W.; Farha, O.K.; Hupp, J.T.; et al. Single-Atom Metal Oxide Sites as Traps for Charge Separation in the Zirconium-Based Metal–Organic Framework NDC–NU-1000. Energy Fuels 2021, 35, 19081–19095. [Google Scholar] [CrossRef]
- Peng, Y.; Lu, B.; Chen, S. Carbon-Supported Single Atom Catalysts for Electrochemical Energy Conversion and Storage. Adv. Mater. 2018, 30, 1801995. [Google Scholar] [CrossRef]
- Liu, P.; Zhao, Y.; Qin, R.; Mo, S.; Chen, G.; Gu, L.; Chevrier, D.M.; Zhang, P.; Guo, Q.; Zang, D.; et al. Catalysis: Photochemical route for synthesizing atomically dispersed palladium catalysts. Science 2016, 352, 797–801. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Bi, W.; Zhang, L.; Tao, S.; Chu, W.; Zhang, Q.; Luo, Y.; Wu, C.; Xie, Y. Single-Atom Pt as Co-Catalyst for Enhanced Photocatalytic H2 Evolution. Adv. Mater. 2016, 28, 2427–2431. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, H.; Yu, H.; Yi, K.; Zhang, W.; Yuan, X.; Huang, J.; Deng, Y.; Zeng, G. Single-Atom Catalysts for Hydrogen Generation: Rational Design, Recent Advances, and Perspectives. Adv. Energy Mater. 2022, 12, 2200875. [Google Scholar] [CrossRef]
- Di, J.; Chen, C.; Yang, S.Z.; Chen, S.; Duan, M.; Xiong, J.; Zhu, C.; Long, R.; Hao, W.; Chi, Z.; et al. Isolated single atom cobalt in Bi3O4Br atomic layers to trigger efficient CO2 photoreduction. Nat. Commun. 2019, 10, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Xu, Y.; Zhou, X.; Lv, C.; Huang, Q.; Chen, G.; Xie, H.; Ge, T.; Cao, J.; Zhan, J.; et al. Single-Atom Fe Triggers Superb CO2 Photoreduction on a Bismuth-Rich Catalyst. ACS Mater. Lett. 2021, 3, 364–371. [Google Scholar] [CrossRef]
- Gao, C.; Chen, S.; Wang, Y.; Wang, J.; Zheng, X.; Zhu, J.; Song, L.; Zhang, W.; Xiong, Y. Heterogeneous Single-Atom Catalyst for Visible-Light-Driven High-Turnover CO2 Reduction: The Role of Electron Transfer. Adv. Mater. 2018, 30, 1704624. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Qi, M.-Y.; Li, Y.-H.; Tang, Z.-R.; Wang, T.; Gong, J.; Xu, Y.-J. Activating two-dimensional Ti3C2Tx-MXene with single-atom cobalt for efficient CO2 photoreduction. Cell Rep. Phys. Sci. 2021, 2, 100371. [Google Scholar] [CrossRef]
- Wang, J.; Kim, E.; Kumar, D.P.; Rangappa, A.P.; Kim, Y.; Zhang, Y.; Kim, T.K. Highly durable and fully dispersed Co diatomic site catalysts for CO2 photoreduction to CH4. Angew. Chem. Int. Ed. 2021, 61, e202113044. [Google Scholar] [CrossRef]
- Niu, X.; Zhu, Q.; Jiang, S.; Zhang, Q. Photoexcited Electron Dynamics of Nitrogen Fixation Catalyzed by Ruthenium Single-Atom Catalysts. J. Phys. Chem. Lett. 2020, 11, 9579–9586. [Google Scholar] [CrossRef]
- Hou, T.; Peng, H.; Xin, Y.; Wang, S.; Zhu, W.; Chen, L.; Yao, Y.; Zhang, W.; Liang, S.; Wang, L. Fe Single-Atom Catalyst for Visible-Light-Driven Photofixation of Nitrogen Sensitized by Triphenylphosphine and Sodium Iodide. ACS Catal. 2020, 10, 5502–5510. [Google Scholar] [CrossRef]
- Liu, S.; Wang, Y.; Wang, S.; You, M.; Hong, S.; Wu, T.-S.S.; Soo, Y.-L.L.; Zhao, Z.; Jiang, G.; Qiu, J.; et al. Photocatalytic Fixation of Nitrogen to Ammonia by Single Ru Atom Decorated TiO2 Nanosheets. ACS Sustain. Chem. Eng. 2019, 7, 6813–6820. [Google Scholar] [CrossRef]
- Han, L.; Liu, X.; Chen, J.; Lin, R.; Liu, H.; Lü, F.; Bak, S.; Liang, Z.; Zhao, S.; Stavitski, E.; et al. Atomically Dispersed Molybdenum Catalysts for Efficient Ambient Nitrogen Fixation. Angew. Chem. Int. Ed. 2019, 58, 2321–2325. [Google Scholar] [CrossRef]
- Zhou, P.; Chao, Y.; Lv, F.; Wang, K.; Zhang, W.; Zhou, J.; Chen, H.; Wang, L.; Li, Y.; Zhang, Q.; et al. Metal single atom strategy greatly boosts photocatalytic methyl activation and C–C coupling for the coproduction of high-value-added multicarbon compounds and hydrogen. ACS Catal. 2020, 10, 9109–9114. [Google Scholar] [CrossRef]
- Vilé, G.; Sharma, P.; Nachtegaal, M.; Tollini, F.; Moscatelli, D.; Sroka-Bartnicka, A.; Tomanec, O.; Petr, M.; Filip, J.; Pieta, I.S.; et al. An Earth-Abundant Ni-Based Single-Atom Catalyst for Selective Photodegradation of Pollutants. Sol. RRL 2021, 5, 2100176. [Google Scholar] [CrossRef]
- Wang, X.; Tong, Y.; Fang, G. Advances of single-atom catalysts for applications in persulfate-based advanced oxidation technologies. Curr. Opin. Chem. Eng. 2021, 34, 100757. [Google Scholar] [CrossRef]
- Wang, T.; Zhu, Y.; Luo, Z.; Li, Y.; Niu, J.; Wang, C. Oxygen vacancy confining effect on photocatalytic efficiency of Pt1-black TiO2 single-atom photocatalysts for hydrogen generation and phenol decomposition. Environ. Chem. Lett. 2021, 19, 1815–1821. [Google Scholar] [CrossRef]
- Augugliaro, V.; Palmisano, G.; Palmisano, L.; Soria, J. Chapter 1—Heterogeneous Photocatalysis and Catalysis: An Overview of Their Distinctive Features. In Heterogeneous Photocatalysis—Relationships with Heterogeneous Catalysis and Perspectives; Marcì, G., Palmisano, L., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 1–24. ISBN 978-0-444-64015-4. [Google Scholar]
- Roduner, E. Understanding catalysis. Chem. Soc. Rev. 2014, 43, 8226–8239. [Google Scholar] [CrossRef] [Green Version]
- Takanabe, K. Photocatalytic Water Splitting: Quantitative Approaches toward Photocatalyst by Design. ACS Catal. 2017, 7, 8006–8022. [Google Scholar] [CrossRef]
- Van de Krol, R.; Grätzel, M. (Eds.) Photoelectrochemical Hydrogen Production; Electronic Materials: Science & Technology; Springer: Boston, MA, USA, 2012; Volume 102, ISBN 978-1-4614-1379-0. [Google Scholar]
- Sachs, M.; Pastor, E.; Kafizas, A.; Durrant, J.R. Evaluation of Surface State Mediated Charge Recombination in Anatase and Rutile TiO2. J. Phys. Chem. Lett. 2016, 7, 3742–3746. [Google Scholar] [CrossRef]
- Yalavarthi, R.; Naldoni, A.; Kment, Š.; Mascaretti, L.; Kmentová, H.; Tomanec, O.; Schmuki, P.; Zbořil, R. Radiative and Non-Radiative Recombination Pathways in Mixed-Phase TiO2 Nanotubes for PEC Water-Splitting. Catalysts 2019, 9, 204. [Google Scholar] [CrossRef] [Green Version]
- Heller, A. Conversion of sunlight into electrical power and photoassisted electrolysis of water in photoelectrochemical cells. Acc. Chem. Res. 1981, 14, 154–162. [Google Scholar] [CrossRef]
- Raghav, A.; Tri Hanindriyo, A.; Utimula, K.; Abbasnejad, M.; Maezono, R.; Panda, E. Intrinsic electronic defect states of anatase using density functional theory. Comput. Mater. Sci. 2020, 184, 109925. [Google Scholar] [CrossRef]
- DeRita, L.; Resasco, J.; Dai, S.; Boubnov, A.; Thang, H.V.; Hoffman, A.S.; Ro, I.; Graham, G.W.; Bare, S.R.; Pacchioni, G.; et al. Structural evolution of atomically dispersed Pt catalysts dictates reactivity. Nat. Mater. 2019, 18, 746–751. [Google Scholar] [CrossRef]
- Resasco, J.; DeRita, L.; Dai, S.; Chada, J.P.; Xu, M.; Yan, X.; Finzel, J.; Hanukovich, S.; Hoffman, A.S.; Graham, G.W.; et al. Uniformity Is Key in Defining Structure–Function Relationships for Atomically Dispersed Metal Catalysts: The Case of Pt/CeO2. J. Am. Chem. Soc. 2020, 142, 169–184. [Google Scholar] [CrossRef]
- Qin, S.; Shui, L.; Osuagwu, B.; Denisov, N.; Tesler, A.B.; Schmuki, P. Facet-Control versus Co-Catalyst-Control in Photocatalytic H2 Evolution from Anatase TiO2 Nanocrystals. ChemistryOpen 2022, 11, e202200010. [Google Scholar] [CrossRef]
- Habisreutinger, S.N.; Schmidt-Mende, L.; Stolarczyk, J.K. Photocatalytic Reduction of CO2 on TiO2 and Other Semiconductors. Angew. Chem. Int. Ed. 2013, 52, 7372–7408. [Google Scholar] [CrossRef]
- Yang, Q.; Dong, L.; Su, R.; Hu, B.; Wang, Z.; Jin, Y.; Wang, Y.; Besenbacher, F.; Dong, M. Nanostructured heterogeneous photo-catalysts for hydrogen production and water splitting: A comprehensive insight. Appl. Mater. Today 2019, 17, 159–182. [Google Scholar] [CrossRef]
- Nawaz, A.; Goudarzi, S.; Asghari, M.A.; Pichiah, S.; Selopal, G.S.; Rosei, F.; Wang, Z.M.; Zarrin, H. Review of Hybrid 1D/2D Photocatalysts for Light-Harvesting Applications. ACS Appl. Nano Mater. 2021, 4, 11323–11352. [Google Scholar] [CrossRef]
- Weng, B.; Liu, S.; Tang, Z.-R.; Xu, Y.-J. One-dimensional nanostructure based materials for versatile photocatalytic applications. RSC Adv. 2014, 4, 12685–12700. [Google Scholar] [CrossRef]
- He, Z.; Zhang, J.; Li, X.; Guan, S.; Dai, M.; Wang, S. 1D/2D Heterostructured Photocatalysts: From Design and Unique Properties to Their Environmental Applications. Small 2020, 16, 2005051. [Google Scholar] [CrossRef]
- Liu, Y.; Le Formal, F.; Boudoire, F.; Guijarro, N. Hematite Photoanodes for Solar Water Splitting: A Detailed Spectroelectrochemical Analysis on the pH-Dependent Performance. ACS Appl. Energy Mater. 2019, 2, 6825–6833. [Google Scholar] [CrossRef]
- Kang, X.; Liu, S.; Dai, Z.; He, Y.; Song, X.; Tan, Z. Titanium Dioxide: From Engineering to Applications. Catalysts 2019, 9, 191. [Google Scholar] [CrossRef] [Green Version]
- Altomare, M.; Pfoch, O.; Tighineanu, A.; Kirchgeorg, R.; Lee, K.; Selli, E.; Schmuki, P. Molten o-H3PO4: A New Electrolyte for the Anodic Synthesis of Self-Organized Oxide Structures—WO3 Nanochannel Layers and Others. J. Am. Chem. Soc. 2015, 137, 5646–5649. [Google Scholar] [CrossRef] [Green Version]
- Tayebi, M.; Lee, B.K. Recent advances in BiVO4 semiconductor materials for hydrogen production using photoelectrochemical water splitting. Renew. Sustain. Energy Rev. 2019, 111, 332–343. [Google Scholar] [CrossRef]
- Fatahi, P.; Roy, A.; Bahrami, M.; Hoseini, S.J. Visible-Light-Driven Efficient Hydrogen Production from CdS NanoRods Anchored with Co-catalysts Based on Transition Metal Alloy Nanosheets of NiPd, NiZn, and NiPdZn. ACS Appl. Energy Mater. 2018, 1, 5318–5327. [Google Scholar] [CrossRef]
- Tuo, Y.; Lu, Q.; Chen, C.; Liu, T.; Pan, Y.; Zhou, Y.; Zhang, J. The facile synthesis of core-shell PtCu nanoparticles with superior electrocatalytic activity and stability in the hydrogen evolution reaction. RSC Adv. 2021, 11, 26326–26335. [Google Scholar] [CrossRef]
- Lee, K.; Mazare, A.; Schmuki, P. One-Dimensional Titanium Dioxide Nanomaterials: Nanotubes. Chem. Rev. 2014, 114, 9385–9454. [Google Scholar] [CrossRef] [Green Version]
- Lettieri, S.; Pavone, M.; Fioravanti, A.; Santamaria Amato, L.; Maddalena, P. Charge Carrier Processes and Optical Properties in TiO2 and TiO2-Based Heterojunction Photocatalysts: A Review. Materials 2021, 14, 1645. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, K.; Emori, M.; Yamamoto, S.; Yukawa, R.; Yamamoto, S.; Hobara, R.; Fujikawa, K.; Sakama, H.; Matsuda, I. Electron–Hole Recombination Time at TiO2 Single-Crystal Surfaces: Influence of Surface Band Bending. J. Phys. Chem. Lett. 2014, 5, 1953–1957. [Google Scholar] [CrossRef]
- Yamada, Y.; Kanemitsu, Y. Determination of electron and hole lifetimes of rutile and anatase TiO2 single crystals. Appl. Phys. Lett. 2012, 101, 133907. [Google Scholar] [CrossRef] [Green Version]
- Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.; Horiuchi, Y.; Anpo, M.; Bahnemann, D.W. Understanding TiO2 Photocatalysis: Mechanisms and Materials. Chem. Rev. 2014, 114, 9919–9986. [Google Scholar] [CrossRef]
- Hoffmann, M.R.; Martin, S.T.; Choi, W.; Bahnemann, D.W. Environmental Applications of Semiconductor Photocatalysis. Chem. Rev. 1995, 95, 69–96. [Google Scholar] [CrossRef]
- Qian, R.; Zong, H.; Schneider, J.; Zhou, G.; Zhao, T.; Li, Y.; Yang, J.; Bahnemann, D.W.; Pan, J.H. Charge carrier trapping, recombination and transfer during TiO2 photocatalysis: An overview. Catal. Today 2019, 335, 78–90. [Google Scholar] [CrossRef]
- Augustynski, J. The role of the surface intermediates in the photoelectrochemical behaviour of anatase and rutile TiO2. Electrochim. Acta 1993, 38, 43–46. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, P.; Liu, J.; Yu, J. New understanding of the difference of photocatalytic activity among anatase, rutile and brookite TiO2. Phys. Chem. Chem. Phys. 2014, 16, 20382–20386. [Google Scholar] [CrossRef]
- Wang, X.; Feng, Z.; Shi, J.; Jia, G.; Shen, S.; Zhou, J.; Li, C. Trap states and carrier dynamics of TiO2 studied by photoluminescence spectroscopy under weak excitation condition. Phys. Chem. Chem. Phys. 2010, 12, 7083–7090. [Google Scholar] [CrossRef]
- Hakki, A.; AlSalka, Y.; Mendive, C.B.; Ubogui, J.; dos Santos Claro, P.C.; Bahnemann, D. Hydrogen Production by Heterogeneous Photocatalysis. In Encyclopedia of Interfacial Chemistry: Surface Science and Electrochemistry; Wandelt, K., Ed.; Elsevier: Oxford, UK, 2018; pp. 413–419. ISBN 978-0-12-809894-3. [Google Scholar]
- Bai, S.; Yin, W.; Wang, L.; Li, Z.; Xiong, Y. Surface and interface design in cocatalysts for photocatalytic water splitting and CO2 reduction. RSC Adv. 2016, 6, 57446–57463. [Google Scholar] [CrossRef]
- Trasatti, S. Work function, electronegativity, and electrochemical behaviour of metals: III. Electrolytic hydrogen evolution in acid solutions. J. Electroanal. Chem. Interfacial Electrochem. 1972, 39, 163–184. [Google Scholar] [CrossRef]
- Ooka, H.; Huang, J.; Exner, K.S. The Sabatier Principle in Electrocatalysis: Basics, Limitations, and Extensions. Front. Energy Res. 2021, 9, 654460. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A.; Kitchin, J.R.; Chen, J.G.; Pandelov, S.; Stimming, U. Trends in the Exchange Current for Hydrogen Evolution. J. Electrochem. Soc. 2005, 152, J23. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Bi, H.; Wang, Z.; Zhou, G. Insight into enhanced hydrogen evolution of single-atom Cu1/TiO2 catalysts from first principles. Int. J. Hydrogen Energy 2022, 47, 4653–4661. [Google Scholar] [CrossRef]
- Kasarevic-Popovic, Z.; Behar, D.; Rabani, J. Role of Excess Electrons in TiO2 Nanoparticles Coated with Pt in Reduction Reactions Studied in Radiolysis of Aqueous Solutions. J. Phys. Chem. B 2004, 108, 20291–20295. [Google Scholar] [CrossRef]
- Hu, X.; Song, J.; Luo, J.; Zhang, H.; Sun, Z.; Li, C.; Zheng, S.; Liu, Q. Single-atomic Pt sites anchored on defective TiO2 nanosheets as a superior photocatalyst for hydrogen evolution. J. Energy Chem. 2021, 62, 1–10. [Google Scholar] [CrossRef]
- Wang, C.; Wang, K.; Feng, Y.Y.; Li, C.; Zhou, X.; Gan, L.; Feng, Y.Y.; Zhou, H.; Zhang, B.; Qu, X.; et al. Co and Pt Dual-Single-Atoms with Oxygen-Coordinated Co–O–Pt Dimer Sites for Ultrahigh Photocatalytic Hydrogen Evolution Efficiency. Adv. Mater. 2021, 33, 2003327. [Google Scholar] [CrossRef]
- Kasap, O. Principles of Electronic Materials and Devices; McGraw-Hill Education (India) Pvt Limited: New Delhi, India, 2007; ISBN 9780070648203. [Google Scholar]
- Kraya, R.A.; Kraya, L.Y. The role of contact size on the formation of Schottky barriers and ohmic contacts at nanoscale metal-semiconductor interfaces. J. Appl. Phys. 2012, 111, 64302. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, L.; Chen, Z.; Hu, J.; Li, S.; Wang, Z.; Liu, J.; Wang, X. Semiconductor heterojunction photocatalysts: Design, construction, and photocatalytic performances. Chem. Soc. Rev. 2014, 43, 5234–5244. [Google Scholar] [CrossRef]
- Yang, X.-F.; Wang, A.; Qiao, B.; Li, J.; Liu, J.; Zhang, T. Single-atom catalysts: A new frontier in heterogeneous catalysis. Acc. Chem. Res. 2013, 46, 1740–1748. [Google Scholar]
- Lopez, N.; Janssens, T.V.W.; Clausen, B.S.; Xu, Y.; Mavrikakis, M.; Bligaard, T.; Nørskov, J.K. On the origin of the catalytic activity of gold nanoparticles for low-temperature CO oxidation. J. Catal. 2004, 223, 232–235. [Google Scholar] [CrossRef]
- Valden, M.; Lai, X.; Goodman, D.W. Onset of Catalytic Activity of Gold Clusters on Titania with the Appearance of Nonmetallic Properties. Science 1998, 281, 1647–1650. [Google Scholar] [CrossRef] [Green Version]
- Campbell, C.T. Electronic perturbations. Nat. Chem. 2012, 4, 597–598. [Google Scholar] [CrossRef]
- Jin, H.; Li, P.; Cui, P.; Shi, J.; Zhou, W.; Yu, X.; Song, W.; Cao, C. Unprecedentedly high activity and selectivity for hydrogenation of nitroarenes with single atomic Co1-N3P1 sites. Nat. Commun. 2022, 13, 723. [Google Scholar] [CrossRef]
- Ji, S.; Chen, Y.; Wang, X.; Zhang, Z.; Wang, D.; Li, Y. Chemical Synthesis of Single Atomic Site Catalysts. Chem. Rev. 2020, 120, 11900–11955. [Google Scholar] [CrossRef]
- Guo, J.; Liu, H.; Li, D.; Wang, J.; Djitcheu, X.; He, D.; Zhang, Q. A minireview on the synthesis of single atom catalysts. RSC Adv. 2022, 12, 9373–9394. [Google Scholar] [CrossRef]
- Wu, Z.-Y.; Zhu, P.; Cullen, D.A.; Hu, Y.; Yan, Q.-Q.; Shen, S.-C.; Chen, F.-Y.; Yu, H.; Shakouri, M.; Arregui-Mena, J.D.; et al. A general synthesis of single atom catalysts with controllable atomic and mesoporous structures. Nat. Synth. 2022, 1, 658–667. [Google Scholar] [CrossRef]
- Li, J.; Yang, Z.; Li, Y.; Zhang, G. Advances in single-atom catalysts: Design, synthesis and environmental applications. J. Hazard. Mater. 2022, 429, 128285. [Google Scholar] [CrossRef]
- Chen, Y.; Ji, S.; Chen, C.; Peng, Q.; Wang, D.; Li, Y. Single-Atom Catalysts: Synthetic Strategies and Electrochemical Applications. Joule 2018, 2, 1242–1264. [Google Scholar] [CrossRef] [Green Version]
- Asikin-Mijan, N.; Mohd Sidek, H.; AlSultan, A.G.; Azman, N.A.; Adzahar, N.A.; Ong, H.C. Single-Atom Catalysts: A Review of Synthesis Strategies and Their Potential for Biofuel Production. Catalysts 2021, 11, 1470. [Google Scholar] [CrossRef]
- Zhang, A.; Zhou, M.; Liu, S.; Chai, M.; Jiang, S. Synthesis of Single-Atom Catalysts Through Top-Down Atomization Approaches. Front. Catal. 2021, 1, 11. [Google Scholar] [CrossRef]
- Xi, J.; Jung, H.S.; Xu, Y.; Xiao, F.; Bae, J.W.; Wang, S. Synthesis Strategies, Catalytic Applications, and Performance Regulation of Single-Atom Catalysts. Adv. Funct. Mater. 2021, 31, 2008318. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, P.; Zhao, J.; Liu, Y.; Huang, Y.; Huang, H.; Yang, C.; Zhao, Y.; Wu, K.; Fu, X.; et al. The Hole-Tunneling Heterojunction of Hematite-Based Photoanodes Accelerates Photosynthetic Reaction. Angew. Chem. Int. Ed. 2021, 60, 16009–16018. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Z.; Wu, Y.; Li, Y. Fabrication of Single-Atom Catalysts with Precise Structure and High Metal Loading. Adv. Mater. 2018, 30, 1801649. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zuo, S.; Qiu, M.; Wang, S.; Zhang, Y.; Zhang, J.; Lou, X.W.D. Direct probing of atomically dispersed Ru species over multi-edged TiO2 for highly efficient photocatalytic hydrogen evolution. Sci. Adv. 2020, 6, 2–10. [Google Scholar] [CrossRef]
- Qin, S.; Denisov, N.; Will, J.; Kolařík, J.; Spiecker, E.; Schmuki, P. A Few Pt Single Atoms Are Responsible for the Overall Co-Catalytic Activity in Pt/TiO2 Photocatalytic H2 Generation. Sol. RRL 2022, 6, 2101026. [Google Scholar] [CrossRef]
- Vajda, S.; White, M.G. Catalysis Applications of Size-Selected Cluster Deposition. ACS Catal. 2015, 5, 7152–7176. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, G.; Gauquelin, N.; Chen, N.; Zhou, J.; Yang, S.; Chen, W.; Meng, X.; Geng, D.; Banis, M.N.; et al. Single-atom Catalysis Using Pt/Graphene Achieved through Atomic Layer Deposition. Sci. Rep. 2013, 3, 1775. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Cheng, H.; Yi, H.; Lin, Y.; Yao, T.; Wang, C.; Li, J.; Wei, S.; Lu, J. Single-Atom Pd1/Graphene Catalyst Achieved by Atomic Layer Deposition: Remarkable Performance in Selective Hydrogenation of 1,3-Butadiene. J. Am. Chem. Soc. 2015, 137, 10484–10487. [Google Scholar] [CrossRef] [PubMed]
- Hejazi, S.; Killian, M.S.; Mazare, A.; Mohajernia, S. Single-Atom-Based Catalysts for Photocatalytic Water Splitting on TiO2 Nanostructures. Catalysts 2022, 12, 905. [Google Scholar] [CrossRef]
- Fonseca, J.; Lu, J. Single-Atom Catalysts Designed and Prepared by the Atomic Layer Deposition Technique. ACS Catal. 2021, 11, 7018–7059. [Google Scholar] [CrossRef]
- Ouyang, R.; Liu, J.-X.; Li, W.-X. Atomistic Theory of Ostwald Ripening and Disintegration of Supported Metal Particles under Reaction Conditions. J. Am. Chem. Soc. 2013, 135, 1760–1771. [Google Scholar] [CrossRef]
- Cai, J.; Cao, A.; Wang, Z.; Lu, S.; Jiang, Z.; Dong, X.Y.; Li, X.; Zang, S.Q. Surface oxygen vacancies promoted Pt redispersion to single-atoms for enhanced photocatalytic hydrogen evolution. J. Mater. Chem. A 2021, 9, 13890–13897. [Google Scholar] [CrossRef]
- Wei, S.; Li, A.; Liu, J.-C.; Li, Z.; Chen, W.; Gong, Y.; Zhang, Q.; Cheong, W.-C.; Wang, Y.; Zheng, L.; et al. Direct observation of noble metal nanoparticles transforming to thermally stable single atoms. Nat. Nanotechnol. 2018, 13, 856–861. [Google Scholar] [CrossRef]
- Huang, Z.; Gu, X.; Cao, Q.; Hu, P.; Hao, J.; Li, J.; Tang, X. Catalytically Active Single-Atom Sites Fabricated from Silver Particles. Angew. Chem. Int. Ed. 2012, 51, 4198–4203. [Google Scholar] [CrossRef]
- Liu, J.-C.; Wang, Y.-G.; Li, J. Toward Rational Design of Oxide-Supported Single-Atom Catalysts: Atomic Dispersion of Gold on Ceria. J. Am. Chem. Soc. 2017, 139, 6190–6199. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Zhou, Z.; Zhang, X.; Huang, Z.; Li, M.; Guo, L. First-Principles Study on Stability and HER Activity of Noble Metal Single Atoms on TiO2: The Effect of Loading Density. J. Phys. Chem. C 2018, 122, 2546–2553. [Google Scholar] [CrossRef]
- Zhao, Y.; Jiang, W.-J.; Zhang, J.; Lovell, E.C.; Amal, R.; Han, Z.; Lu, X. Anchoring Sites Engineering in Single-Atom Catalysts for Highly Efficient Electrochemical Energy Conversion Reactions. Adv. Mater. 2021, 33, 2102801. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Low, J.; Long, R.; Kong, T.; Zhu, J.; Xiong, Y. Heterogeneous Single-Atom Photocatalysts: Fundamentals and Applications. Chem. Rev. 2020, 120, 12175–12216. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Sharma, V.; Gaikwad, R.P.; Fornasiero, P.; Zbořil, R.; Gawande, M.B. Single-Atom Catalysts: A Sustainable Pathway for the Advanced Catalytic Applications. Small 2021, 17, 2006473. [Google Scholar] [CrossRef]
- Jiao, L.; Jiang, H.-L. Metal-Organic-Framework-Based Single-Atom Catalysts for Energy Applications. Chem 2019, 5, 786–804. [Google Scholar] [CrossRef] [Green Version]
- Hejazi, S.; Mohajernia, S.; Osuagwu, B.; Zoppellaro, G.; Andryskova, P.; Tomanec, O.; Kment, S.; Zbořil, R.; Schmuki, P. On the Controlled Loading of Single Platinum Atoms as a Co-Catalyst on TiO2 Anatase for Optimized Photocatalytic H2 Generation. Adv. Mater. 2020, 32, 1908505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Hwang, I.; Cha, G.; Qin, S.; Tomanec, O.; Badura, Z.; Kment, S.; Zboril, R.; Schmuki, P. Optimized Pt Single Atom Harvesting on TiO2 Nanotubes—Towards a Most Efficient Photocatalyst. Small 2022, 18, 2104892. [Google Scholar] [CrossRef]
- Elibol, K.; Mangler, C.; O’Regan, D.D.; Mustonen, K.; Eder, D.; Meyer, J.C.; Kotakoski, J.; Hobbs, R.G.; Susi, T.; Bayer, B.C. Single Indium Atoms and Few-Atom Indium Clusters Anchored onto Graphene via Silicon Heteroatoms. ACS Nano 2021, 15, 14373–14383. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Huang, B.; Yi, Y.; Guo, Y.; Zuo, Z.; Li, Y.; Jia, Z.; Liu, H.; Li, Y. Anchoring zero valence single atoms of nickel and iron on graphdiyne for hydrogen evolution. Nat. Commun. 2018, 9, 1460. [Google Scholar] [CrossRef]
- Song, H.; Du, R.; Wang, Y.; Zu, D.; Zhou, R.; Cai, Y.; Wang, F.; Li, Z.; Shen, Y.; Li, C. Anchoring single atom cobalt on two-dimensional MXene for activation of peroxymonosulfate. Appl. Catal. B Environ. 2021, 286, 119898. [Google Scholar] [CrossRef]
- Wang, A.; Li, J.; Zhang, T. Heterogeneous single-atom catalysis. Nat. Rev. Chem. 2018, 2, 65–81. [Google Scholar] [CrossRef]
- Qi, K.; Chhowalla, M.; Voiry, D. Single atom is not alone: Metal–support interactions in single-atom catalysis. Mater. Today 2020, 40, 173–192. [Google Scholar] [CrossRef]
- Lewis, N.S. Toward Cost-Effective Solar Energy Use. Science 2007, 315, 798–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Hwang, I.; Tomanec, O.; Fehn, D.; Mazare, A.; Zboril, R.; Meyer, K.; Schmuki, P. Advanced Photocatalysts: Pinning Single Atom Co-Catalysts on Titania Nanotubes. Adv. Funct. Mater. 2021, 31, 2102843. [Google Scholar] [CrossRef]
- Wan, J.; Chen, W.; Jia, C.; Zheng, L.; Dong, J.; Zheng, X.; Wang, Y.; Yan, W.; Chen, C.; Peng, Q.; et al. Defect Effects on TiO2 Nanosheets: Stabilizing Single Atomic Site Au and Promoting Catalytic Properties. Adv. Mater. 2018, 30, 1705369. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Long, J.; Gu, Q.; Huang, H.; Lin, H.; Wang, X. Single-site nickel-grafted anatase TiO2 for hydrogen production: Toward understanding the nature of visible-light photocatalysis. J. Catal. 2014, 320, 147–159. [Google Scholar] [CrossRef]
- Gu, Q.; Long, J.; Zhou, Y.; Yuan, R.; Lin, H.; Wang, X. Single-site tin-grafted anatase TiO2 for photocatalytic hydrogen production: Toward understanding the nature of interfacial molecular junctions formed in semiconducting composite photocatalysts. J. Catal. 2012, 289, 88–99. [Google Scholar] [CrossRef]
- Chen, J.; Iyemperumal, S.K.; Fenton, T.; Carl, A.; Grimm, R.; Li, G.; Deskins, N.A. Synergy between Defects, Photoexcited Electrons, and Supported Single Atom Catalysts for CO2 Reduction. ACS Catal. 2018, 8, 10464–10478. [Google Scholar] [CrossRef]
- Saravanan, R.; Gracia, F.; Stephen, A. Basic Principles, Mechanism, and Challenges of Photocatalysis. In Nanocomposites for Visible Light-induced Photocatalysis; Khan, M.M., Pradhan, D., Sohn, Y., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 19–40. ISBN 978-3-319-62446-4. [Google Scholar]
- Li, J.; Yi, D.; Zhan, F.; Zhou, B.; Gao, D.; Guo, D.; Liu, S.; Wang, X.; Yao, J. Monolayered Ru1/TiO2 nanosheet enables efficient visible-light-driven hydrogen evolution. Appl. Catal. B Environ. 2020, 271, 118925. [Google Scholar] [CrossRef]
- Krogh-Jespersen, K.; Schugar, H.J. Detailed correlations between the ligand-to-metal charge-transfer (LMCT) spectra of copper(II) and ruthenium(III) imidazoles and imidazolates. Electronic structures of carbon-bound ruthenium(III) imidazoles and imidazolates. Inorg. Chem. 1984, 23, 4390–4393. [Google Scholar] [CrossRef]
- Nguyen-Phan, T.-D.; Luo, S.; Vovchok, D.; Llorca, J.; Sallis, S.; Kattel, S.; Xu, W.; Piper, L.F.J.; Polyansky, D.E.; Senanayake, S.D.; et al. Three-dimensional ruthenium-doped TiO2 sea urchins for enhanced visible-light-responsive H2 production. Phys. Chem. Chem. Phys. 2016, 18, 15972–15979. [Google Scholar] [CrossRef]
- Fu, Y.; Sun, D.; Chen, Y.; Huang, R.; Ding, Z.; Fu, X.; Li, Z. An Amine-Functionalized Titanium Metal–Organic Framework Photocatalyst with Visible-Light-Induced Activity for CO2 Reduction. Angew. Chem. Int. Ed. 2012, 51, 3364–3367. [Google Scholar] [CrossRef]
- Wang, J.; Huang, J.; Meng, J.; Li, Q.; Yang, J. Enhanced photoelectrochemical performance of anatase TiO2 for water splitting via surface codoping. RSC Adv. 2017, 7, 39877–39884. [Google Scholar] [CrossRef] [Green Version]
- Cha, G.; Mazare, A.; Hwang, I.; Denisov, N.; Will, J.; Yokosawa, T.; Badura, Z.; Zoppellaro, G.; Tesler, A.B.; Spiecker, E.; et al. A facile “dark”-deposition approach for Pt single-atom trapping on facetted anatase TiO2 nanoflakes and use in photocatalytic H2 generation. Electrochim. Acta 2022, 412, 140129. [Google Scholar] [CrossRef]
- He, H.; Wang, H.H.; Liu, J.; Liu, X.; Li, W.; Wang, Y. Research Progress and Application of Single-Atom Catalysts: A Review. Molecules 2021, 26, 6501. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Li, B.; Cui, X.; Tan, S.; Wang, B. Photoresponses of Supported Au Single Atoms on TiO2(110) through the Metal-Induced Gap States. J. Phys. Chem. Lett. 2019, 10, 4683–4691. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yates, J.T. Band Bending in Semiconductors: Chemical and Physical Consequences at Surfaces and Interfaces. Chem. Rev. 2012, 112, 5520–5551. [Google Scholar] [CrossRef]
- Louie, S.G.; Cohen, M.L. Electronic structure of a metal-semiconductor interface. Phys. Rev. B 1976, 13, 2461–2469. [Google Scholar] [CrossRef]
- Tersoff, J. Schottky Barrier Heights and the Continuum of Gap States. Phys. Rev. Lett. 1984, 52, 465–468. [Google Scholar] [CrossRef]
- Wendt, S.; Sprunger, P.T.; Lira, E.; Madsen, G.K.; Li, Z.; Hansen, J.Ø.; Matthiesen, J.; Blekinge-Rasmussen, A.; Lægsgaard, E.; Hammer, B.; et al. The Role of Interstitial Sites in the Ti3d Defect State in the Band Gap of Titania. Science 2008, 320, 1755–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finegold, L.; Cude, J.L. Biological sciences: One and two-dimensional structure of alpha-helix and beta-sheet forms of poly(L-Alanine) shown by specific heat measurements at low temperatures (1.5–20 K). Nature 1972, 238, 38–40. [Google Scholar] [CrossRef]
- Chen, Y.; Ji, S.; Sun, W.; Lei, Y.; Wang, Q.; Li, A.; Chen, W.; Zhou, G.; Zhang, Z.; Wang, Y.; et al. Engineering the Atomic Interface with Single Platinum Atoms for Enhanced Photocatalytic Hydrogen Production. Angew. Chem. Int. Ed. 2020, 59, 1295–1301. [Google Scholar] [CrossRef]
- Qin, S.; Denisov, N.; Sarma, B.B.; Hwang, I.; Doronkin, D.E.; Tomanec, O.; Kment, S.; Schmuki, P. Pt Single Atoms on TiO2 Polymorphs—Minimum Loading with a Maximized Photocatalytic Efficiency. Adv. Mater. Interfaces 2022, 9, 2200808. [Google Scholar] [CrossRef]
- Wang, Y.; Hwang, I.; Wu, Z.; Schmuki, P. Self-assembled monolayers enhance the efficiency of Pt single atom co-catalysts in photocatalytic H2 generation. Electrochem. Commun. 2021, 133, 107166. [Google Scholar] [CrossRef]
- Wu, X.; Zuo, S.; Qiu, M.; Li, Y.; Zhang, Y.; An, P.; Zhang, J.; Zhang, H.; Zhang, J. Atomically defined Co on two-dimensional TiO2 nanosheet for photocatalytic hydrogen evolution. Chem. Eng. J. 2021, 420, 127681. [Google Scholar] [CrossRef]
- Xiao, M.; Zhang, L.; Luo, B.; Lyu, M.; Wang, Z.; Huang, H.; Wang, S.; Du, A.; Wang, L. Molten-Salt-Mediated Synthesis of an Atomic Nickel Co-catalyst on TiO2 for Improved Photocatalytic H2 Evolution. Angew. Chem. Int. Ed. 2020, 59, 7230–7234. [Google Scholar] [CrossRef]
- Hwang, I.; Mazare, A.; Will, J.; Yokosawa, T.; Spiecker, E.; Schmuki, P. Inhibition of H2 and O2 Recombination: The Key to a Most Efficient Single-Atom Co-Catalyst for Photocatalytic H2 Evolution from Plain Water. Adv. Funct. Mater. 2022, 2207849. [Google Scholar] [CrossRef]
- Lee, B.H.; Park, S.; Kim, M.; Sinha, A.K.; Lee, S.C.; Jung, E.; Chang, W.J.; Lee, K.S.; Kim, J.H.; Cho, S.P.; et al. Reversible and cooperative photoactivation of single-atom Cu/TiO2 photocatalysts. Nat. Mater. 2019, 18, 620–626. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, J.; Wang, H.; Xiao, B.; Zhang, W.; Zhao, X.; Lv, T.; Thangamuthu, M.; Zhang, J.; Guo, Y.; et al. Single-atom Cu anchored catalysts for photocatalytic renewable H2 production with a quantum efficiency of 56%. Nat. Commun. 2022, 13, 58. [Google Scholar] [CrossRef] [PubMed]
- Umeda, M.; Niitsuma, Y.; Horikawa, T.; Matsuda, S.; Osawa, M. Electrochemical Reduction of CO2 to Methane on Platinum Catalysts without Overpotentials: Strategies for Improving Conversion Efficiency. ACS Appl. Energy Mater. 2020, 3, 1119–1127. [Google Scholar] [CrossRef] [Green Version]
- Cai, S.; Wang, L.; Heng, S.; Li, H.; Bai, Y.; Dang, D.; Wang, Q.; Zhang, P.; He, C. Interaction of Single-Atom Platinum-Oxygen Vacancy Defects for the Boosted Photosplitting Water H2 Evolution and CO2 Photoreduction: Experimental and Theoretical Study. J. Phys. Chem. C 2020, 124, 24566–24579. [Google Scholar] [CrossRef]
- Kwak, B.S.; Kang, M. Photocatalytic reduction of CO2 with H2O using perovskite CaxTiyO3. Appl. Surf. Sci. 2015, 337, 138–144. [Google Scholar] [CrossRef]
- Lee, B.-H.; Gong, E.; Kim, M.; Park, S.; Kim, H.H.R.; Lee, J.; Jung, E.; Lee, C.W.; Bok, J.; Jung, Y.; et al. Electronic interaction between transition metal single-atoms and anatase TiO2 boosts CO2 photoreduction with H2O. Energy Environ. Sci. 2022, 15, 601–609. [Google Scholar] [CrossRef]
- Lan, Y.; Xie, Y.; Chen, J.; Hu, Z.; Cui, D. Selective photocatalytic CO2 reduction on copper-titanium dioxide: A study of the relationship between CO production and H2 suppression. Chem. Commun. 2019, 55, 8068–8071. [Google Scholar] [CrossRef]
- Zhu, S.; Liang, S.; Tong, Y.; An, X.; Long, J.; Fu, X.; Wang, X. Photocatalytic reduction of CO2 with H2O to CH4 on Cu(i) supported TiO2 nanosheets with defective {001} facets. Phys. Chem. Chem. Phys. 2015, 17, 9761–9770. [Google Scholar] [CrossRef]
- Jiang, Z.; Sun, W.; Miao, W.; Yuan, Z.; Yang, G.; Kong, F.; Yan, T.; Chen, J.; Huang, B.; An, C.; et al. Living Atomically Dispersed Cu Ultrathin TiO2 Nanosheet CO2 Reduction Photocatalyst. Adv. Sci. 2019, 6, 3–7. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Hung, S.F.; Tang, Z.R.; Chen, H.M.; Xiong, Y.; Xu, Y.J. Dynamic Evolution of Atomically Dispersed Cu Species for CO2 Photoreduction to Solar Fuels. ACS Catal. 2019, 9, 4824–4833. [Google Scholar] [CrossRef]
- Andrews, E.; Fang, Y.; Flake, J. Electrochemical reduction of CO2 at CuAu nanoparticles: Size and alloy effects. J. Appl. Electrochem. 2018, 48, 435–441. [Google Scholar] [CrossRef]
- Dickinson, H.L.A.; Symes, M.D. Recent progress in CO2 reduction using bimetallic electrodes containing copper. Electrochem. Commun. 2022, 135, 107212. [Google Scholar] [CrossRef]
- Yu, Y.; Dong, X.; Chen, P.; Geng, Q.; Wang, H.; Li, J.; Zhou, Y.; Dong, F. Synergistic Effect of Cu Single Atoms and Au-Cu Alloy Nanoparticles on TiO2 for Efficient CO2 Photoreduction. ACS Nano 2021, 15, 14453–14464. [Google Scholar] [CrossRef]
- Pan, H.; Wang, X.; Xiong, Z.; Sun, M.; Murugananthan, M.; Zhang, Y. Enhanced photocatalytic CO2 reduction with defective TiO2 nanotubes modified by single-atom binary metal components. Environ. Res. 2021, 198, 111176. [Google Scholar] [CrossRef] [PubMed]
- Galloway, J.N.; Dentener, F.J.; Capone, D.G.; Boyer, E.W.; Howarth, R.W.; Seitzinger, S.P.; Asner, G.P.; Cleveland, C.C.; Green, P.A.; Holland, E.A.; et al. Nitrogen Cycles: Past, Present, and Future. Biogeochemistry 2004, 70, 153–226. [Google Scholar] [CrossRef]
- Erisman, J.W.; Sutton, M.A.; Galloway, J.; Klimont, Z.; Winiwarter, W. How a century of ammonia synthesis changed the world. Nat. Geosci. 2008, 1, 636–639. [Google Scholar] [CrossRef]
- Valera-Medina, A.; Amer-Hatem, F.; Azad, A.K.; Dedoussi, I.C.; de Joannon, M.; Fernandes, R.X.; Glarborg, P.; Hashemi, H.; He, X.; Mashruk, S.; et al. Review on Ammonia as a Potential Fuel: From Synthesis to Economics. Energy Fuels 2021, 35, 6964–7029. [Google Scholar] [CrossRef]
- Wang, L.; Xia, M.; Wang, H.; Huang, K.; Qian, C.; Maravelias, C.T.; Ozin, G.A. Greening Ammonia toward the Solar Ammonia Refinery. Joule 2018, 2, 1055–1074. [Google Scholar] [CrossRef] [Green Version]
- Van der Ham, C.J.M.; Koper, M.T.M.; Hetterscheid, D.G.H. Challenges in reduction of dinitrogen by proton and electron transfer. Chem. Soc. Rev. 2014, 43, 5183–5191. [Google Scholar] [CrossRef]
- Smith, C.; Hill, A.K.; Torrente-Murciano, L. Current and future role of Haber–Bosch ammonia in a carbon-free energy landscape. Energy Environ. Sci. 2020, 13, 331–344. [Google Scholar] [CrossRef]
- Ranjit, K.T.; Viswanathan, B. Photocatalytic reduction of dinitrogen to ammonia. Indian J. Chem.—Sect. A Inorg. Phys. Theor. Anal. Chem. 1996, 35, 443–453. [Google Scholar]
- Huang, Y.; Wang, C.; Yu, Y.; Yu, Y.; Wang, W.; Zhang, B. Atomically Dispersed Ru-Decorated TiO2 Nanosheets for Thermally Assisted Solar-Driven Nitrogen Oxidation into Nitric Oxide. CCS Chem. 2021, 4, 1468–1476. [Google Scholar] [CrossRef]
- Peter, L.M. CHAPTER 1 Photoelectrochemistry: From Basic Principles to Photocatalysis. In Photocatalysis: Fundamentals and Perspectives; The Royal Society of Chemistry: London, UK, 2016; pp. 1–28. ISBN 978-1-78262-041-9. [Google Scholar]
- McMichael, S.; Fernández-Ibáñez, P.; Byrne, J.A. A Review of Photoelectrocatalytic Reactors for Water and Wastewater Treatment. Water 2021, 13, 1198. [Google Scholar] [CrossRef]
- Marwat, M.A.; Humayun, M.; Afridi, M.W.; Zhang, H.; Abdul Karim, M.R.; Ashtar, M.; Usman, M.; Waqar, S.; Ullah, H.; Wang, C.; et al. Advanced Catalysts for Photoelectrochemical Water Splitting. ACS Appl. Energy Mater. 2021, 4, 12007–12031. [Google Scholar] [CrossRef]
- Tesler, A.B.; Sannomiya, T.; Hejazi, S.; Mohammadi, R.; Vogel, N.; Altomare, M.; Schmuki, P. Metallic nanoparticle-on-mirror: Multiple-band light harvesting and efficient photocurrent generation under visible light irradiation. Nano Energy 2021, 90, 106609. [Google Scholar] [CrossRef]
- Cheng, C.; Fang, W.-H.; Long, R.; Prezhdo, O.V. Water Splitting with a Single-Atom Cu/TiO2 Photocatalyst: Atomistic Origin of High Efficiency and Proposed Enhancement by Spin Selection. JACS Au 2021, 1, 550–559. [Google Scholar] [CrossRef]
- Pang, Y.; Zang, W.; Kou, Z.; Zhang, L.; Xu, G.; Lv, J.; Gao, X.; Pan, Z.; Wang, J.; Wu, Y. Assembling of Bi atoms on TiO2 nanorods boosts photoelectrochemical water splitting of semiconductors. Nanoscale 2020, 12, 4302–4308. [Google Scholar] [CrossRef]
- Hu, Y.; Cao, Y.; Wang, P.; Li, D.; Chen, W.; He, Y.; Fu, X.; Shao, Y.; Zheng, Y. A new perspective for effect of Bi on the photocatalytic activity of Bi-doped TiO2. Appl. Catal. B Environ. 2012, 125, 294–303. [Google Scholar] [CrossRef]
- Wang, T.; Tao, X.; Li, X.; Zhang, K.; Liu, S.; Li, B. Synergistic Pd Single Atoms, Clusters, and Oxygen Vacancies on TiO2 for Photocatalytic Hydrogen Evolution Coupled with Selective Organic Oxidation. Small 2021, 17, 1–10. [Google Scholar] [CrossRef]
- Weon, S.; Suh, M.-J.; Chu, C.; Huang, D.; Stavitski, E.; Kim, J.-H. Site-Selective Loading of Single-Atom Pt on TiO2 for Photocatalytic Oxidation and Reductive Hydrodefluorination. ACS ES&T Eng. 2021, 1, 512–522. [Google Scholar] [CrossRef]
- Hu, Z.; Li, X.; Zhang, S.; Li, Q.; Fan, J.; Qu, X.; Lv, K. Fe1/TiO2 Hollow Microspheres: Fe and Ti Dual Active Sites Boosting the Photocatalytic Oxidation of NO. Small 2020, 16, 1–6. [Google Scholar] [CrossRef]
- Selcuk, S.; Selloni, A. Facet-dependent trapping and dynamics of excess electrons at anatase TiO2 surfaces and aqueous interfaces. Nat. Mater. 2016, 15, 1107–1112. [Google Scholar] [CrossRef]
- Yu, J.; Low, J.; Xiao, W.; Zhou, P.; Jaroniec, M. Enhanced photocatalytic CO2-Reduction activity of anatase TiO2 by Coexposed {001} and {101} facets. J. Am. Chem. Soc. 2014, 136, 8839–8842. [Google Scholar] [CrossRef]
- Pharmacology, A. Toxicity and Toxicokinetics of Perfluorooctanoic. J. Toxicol. Sci. 2003, 28, 49–57. [Google Scholar]
- Trofimovaite, R.; Parlett, C.M.A.; Kumar, S.; Frattini, L.; Isaacs, M.A.; Wilson, K.; Olivi, L.; Coulson, B.; Debgupta, J.; Douthwaite, R.E.; et al. Single atom Cu(I) promoted mesoporous titanias for photocatalytic Methyl Orange depollution and H2 production. Appl. Catal. B Environ. 2018, 232, 501–511. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kerketta, U.; Tesler, A.B.; Schmuki, P. Single-Atom Co-Catalysts Employed in Titanium Dioxide Photocatalysis. Catalysts 2022, 12, 1223. https://doi.org/10.3390/catal12101223
Kerketta U, Tesler AB, Schmuki P. Single-Atom Co-Catalysts Employed in Titanium Dioxide Photocatalysis. Catalysts. 2022; 12(10):1223. https://doi.org/10.3390/catal12101223
Chicago/Turabian StyleKerketta, Ujjaval, Alexander B. Tesler, and Patrik Schmuki. 2022. "Single-Atom Co-Catalysts Employed in Titanium Dioxide Photocatalysis" Catalysts 12, no. 10: 1223. https://doi.org/10.3390/catal12101223