
Enhanced Thermal Stability of Polyphosphate-Dependent Glucomannokinase by Directed Evolution

Abstract
:1. Introduction
2. Results
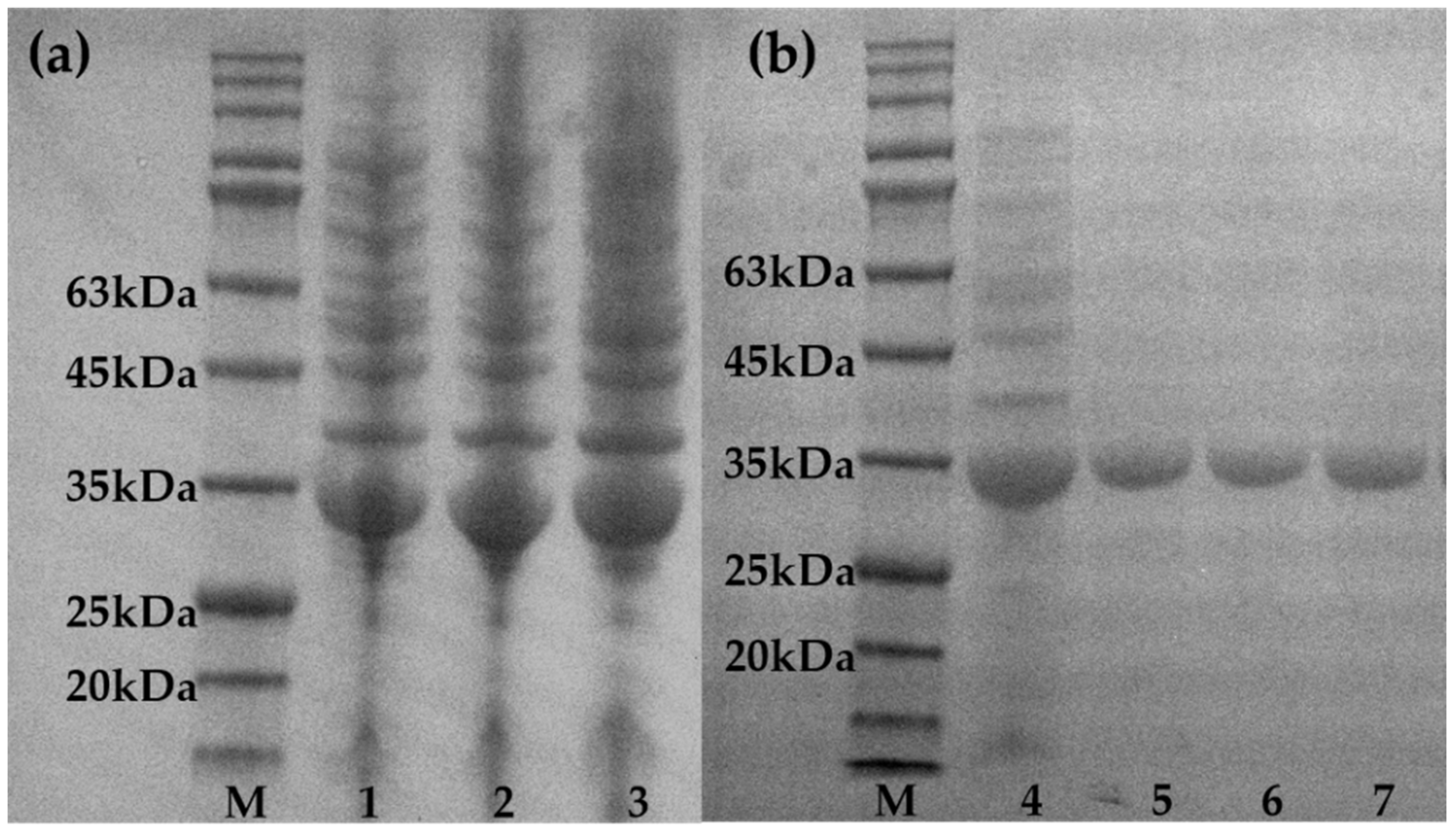
2.1. Construction and Screening of Random Mutation Libraries
2.2. Construction and Screening of Site-Directed Saturation Mutation Libraries
2.3. Combination and Screening of Mutation Sites
2.4. Characteristic Analysis of Mutants
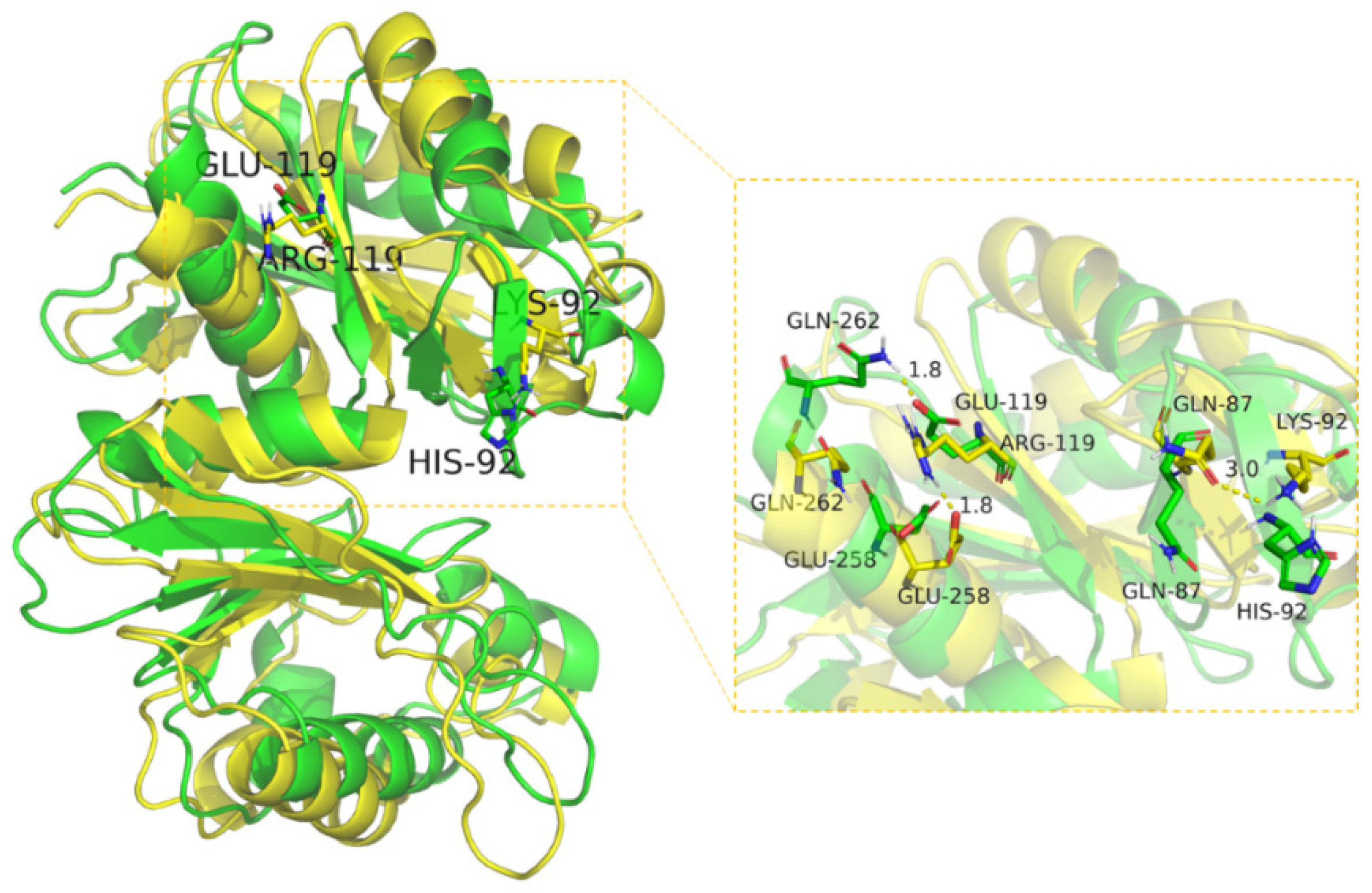
2.5. Structural Analysis of Mutants
3. Discussion
4. Materials and Methods
4.1. Strains, Plasmid and Reagents
4.2. Construction of Error-Prone PCR Mutant Library
4.3. Construction of Saturated Site-Directed Mutation Library and Combination of the Mutant Sites
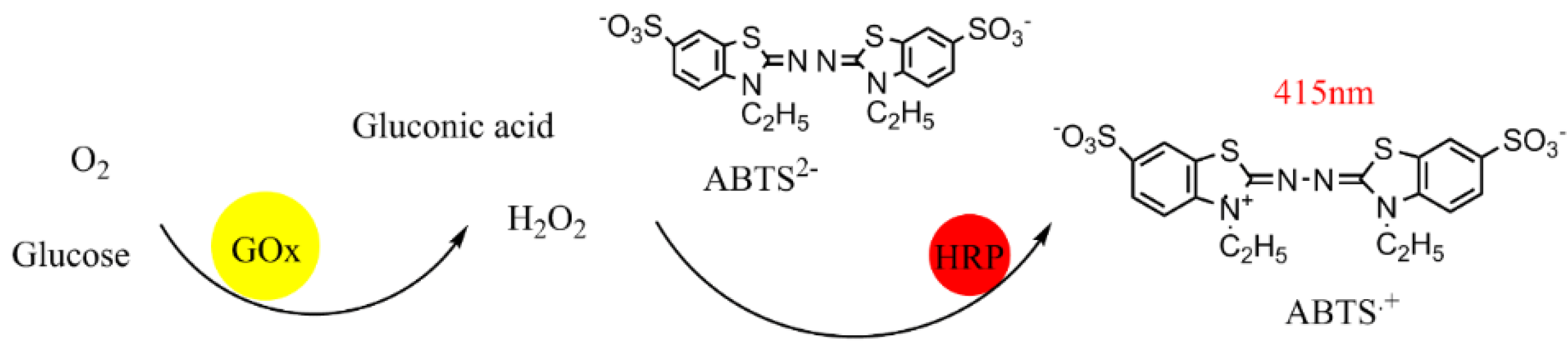
4.4. High-Throughput Screening
4.5. Enzyme Activity Determination
4.6. Enzymatic Properties
4.7. Structural Analysis
5. Conclusion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Mondal, B.; Dutta, T.; Sen Gupta, S. Dual enzyme responsive mannose-6-phosphate based vesicle for controlled lysosomal delivery. Chem. Commun. 2021, 57, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Kreiling, J.L.; Montgomery, M.A.; Wheeler, J.R.; Kopanic, J.L.; Connelly, C.M.; Zavorka, M.E.; Allison, J.L.; MacDonald, R.G. Dominant-negative effect of truncated mannose 6-phosphate/insulin-like growth factor II receptor species in cancer. FEBS J. 2012, 279, 2695–2713. [Google Scholar] [CrossRef] [PubMed]
- Girsch, J.H.; Jackson, W.; Carpenter, J.E.; Moninger, T.O.; Jarosinski, K.W.; Grose, C. Exocytosis of Progeny Infectious Varicella-Zoster Virus Particles via a Mannose-6-Phosphate Receptor Pathway without Xenophagy following Secondary Envelopment. J. Virol. 2020, 94, e00800-20. [Google Scholar] [CrossRef]
- Janson, J.; Andersson, G.; Bergquist, L.; Eriksson, M.; Folgering, J.H.A. Impact of chemical modification of sulfamidase on distribution to brain interstitial fluid and to CSF after an intravenous administration in awake, freely-moving rats. Mol. Genet. Metab. Rep. 2020, 22, 100554. [Google Scholar] [CrossRef] [PubMed]
- Meunier, M.; Chapuis, E.; Lapierre, L.; Auriol, P.; Paulus, C.; Elbaum, B.; Simoni, E.D.; Sandre, J.; Auriol, D.; Scandolera, A.; et al. Mannose-6-phosphate complex and improvement in biomechanical properties of the skin. J. Cosmet. Dermatol. 2021, 20, 1598–1610. [Google Scholar] [CrossRef]
- Khanjin, N.A.; Montero, J.L. Synthesis of mannose-6-phosphate analogs: Large-scale preparation of isosteric mannose-6-phosphonate via cyclic sulfate precursor. Tetrahedron Lett. 2002, 43, 4017–4020. [Google Scholar] [CrossRef]
- Slein, M.W. [20] Synthesis of mannose-6-phosphate by hexokinase. Methods Enzymol. 1957, 3, 154–157. [Google Scholar]
- Zhu, W.L.; Gao, M.M.; Chen, B.Q.; Tan, T.W.; Cao, H.; Liu, L. The Synthesis of Mannose-6-Phosphate Using Polyphosphate-Dependent Mannose Kinase. Catalysts 2019, 9, 250. [Google Scholar] [CrossRef]
- Albi, T.; Serrano, A. Two strictly polyphosphate-dependent gluco (manno) kinases from diazotrophic Cyanobacteria with potential to phosphorylate hexoses from polyphosphates. Appl. Microbiol. Biotechnol. 2015, 99, 3887–3900. [Google Scholar] [CrossRef]
- Zhou, W.; Huang, R.; Zhu, Z.; Zhang, Y.-H.P.J. Coevolution of both thermostability and activity of polyphosphate glucokinase from Thermobifida fusca YX. Appl. Environ. Microbiol. 2018, 84, e01224-18. [Google Scholar] [CrossRef]
- Bashirova, A.; Pramanik, S.; Volkov, P.; Rozhkova, A.; Nemashkalov, V.; Zorov, I.; Gusakov, A.; Sinitsyn, A.; Schwaneberg, U.; Davari, M.D. Disulfide bond engineering of an endoglucanase from Penicillium verruculosum to improve its thermostability. Int. J. Mol. Sci. 2019, 20, 1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, E.L.; Smithson, R.; Kilbride, S.; Foster, J.; Hardy, F.J.; Ramachandran, S.; Tedstone, A.A.; Haigh, S.J.; Garforth, A.A.; Day, P.J. Directed evolution of an efficient and thermostable PET depolymerase. Nat. Catal. 2022, 5, 673–681. [Google Scholar] [CrossRef]
- Xu, Z.; Cen, Y.K.; Zou, S.P.; Xue, Y.P.; Zheng, Y.G. Recent advances in the improvement of enzyme thermostability by structure modification. Crit. Rev. Biotechnol. 2020, 40, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Chen, Q.M.; Zhang, W.L.; Mu, W.M. Overview of strategies for developing high thermostability industrial enzymes: Discovery, mechanism, modification and challenges. Crit. Rev. Food Sci. Nutr. 2021, 1970508, 1–18. [Google Scholar] [CrossRef]
- Tamaki, F.K. Directed evolution of enzymes. Emerg. Top. Life Sci. 2020, 4, 119–127. [Google Scholar] [CrossRef]
- Shivange, A.V.; Hoeffken, H.W.; Haefner, S.; Schwaneberg, U. Protein consensus-based surface engineering (ProCoS): A computer-assisted method for directed protein evolution. Biotechniques 2016, 61, 305–314. [Google Scholar] [CrossRef]
- Nirantar, S.R. Directed Evolution Methods for Enzyme Engineering. Molecules 2021, 26, 5599. [Google Scholar] [CrossRef]
- Schmidt, A.; Shvetsov, A.; Soboleva, E.; Kil, Y.; Sergeev, V.; Surzhik, M. Thermostability improvement of Aspergillus awamori glucoamylase via directed evolution of its gene located on episomal expression vector in Pichia pastoris cells. Protein Eng. Des. Sel. 2019, 32, 251–259. [Google Scholar] [CrossRef]
- Xing, Y.N.; Tan, J.; Wang, Y.H.; Wang, J.Q. Enhancing the thermostability of a mono- and diacylglycerol lipase from Malassizia globose by stabilizing a flexible loop in the catalytic pocket. Enzym. Microb. Technol. 2021, 149, 109849. [Google Scholar] [CrossRef]
- Xiang, L.; Lu, Y.H.; Wang, H.; Wang, M.X.; Zhang, G.M. Improving the specific activity and pH stability of xylanase XynHBN188A by directed evolution. Bioresour. Bioprocess. 2019, 6, 25. [Google Scholar] [CrossRef]
- Basit, A.; Tajwar, R.; Sadaf, S.; Zhang, Y.; Akhtar, M.W. Improvement in activity of cellulase Cel12A of Thermotoga neapolitana by error prone PCR. J. Biotechnol. 2019, 306, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Menghiu, G.; Ostafe, V.; Prodanovic, R.; Fischer, R.; Ostafe, R. A High-Throughput Screening System Based on Fluorescence-Activated Cell Sorting for the Directed Evolution of Chitinase A. Int. J. Mol. Sci. 2021, 22, 3041. [Google Scholar] [CrossRef]
- Gao, H.F.; Zhu, R.T.; Li, Z.L.; Wang, W.Y.; Liu, Z.D.; Hu, N. Improving the catalytic efficiency and substrate affinity of a novel esterase from marine Klebsiella aerogenes by random and site-directed mutation. World J. Microbiol. Biotechnol. 2021, 37, 106. [Google Scholar] [CrossRef]
- Balaz, A.M.; Stevanovic, J.; Ostafe, R.; Blazic, M.; Durdic, K.I.; Fischer, R.; Prodanovic, R. Semi-rational design of cellobiose dehydrogenase for increased stability in the presence of peroxide. Mol. Divers. 2020, 24, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Bao, Z.H.; Zhao, H.M. High Throughput Screening and Selection Methods for Directed Enzyme Evolution. Ind. Eng. Chem. Res. 2015, 54, 4011–4020. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.D.; Yang, C.C.; Yu, H.W. From molecular engineering to process engineering: Development of high-throughput screening methods in enzyme directed evolution. Appl. Microbiol. Biotechnol. 2018, 102, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.H.; Petrounia, I.P.; Yagasaki, M.; Bandara, G.; Arnold, F.H. Expression and stabilization of galactose oxidase in Escherichia coli by directed evolution. Protein Eng. 2001, 14, 699–704. [Google Scholar] [CrossRef]
- Mukai, T.; Kawai, S.; Matsukawa, H.; Matuo, Y.; Murata, K. Characterization and molecular cloning of a novel enzyme, inorganic polyphosphate/ATP-glucomannokinase, of Arthrobacter sp strain KM. Appl. Environ. Microbiol. 2003, 69, 3849–3857. [Google Scholar] [CrossRef]
- Paul, R.K.; Nath, V.; Kumar, V. Structure based virtual screening of natural compounds and molecular dynamics simulation: Butirosin as Dipeptidyl peptidase (DPP-IV) inhibitor. Biocatal. Agric. Biotechnol. 2021, 35, 102042. [Google Scholar] [CrossRef]
- Parra-Cruz, R.; Jager, C.M.; Li Lau, P.; Gomes, R.L.; Pordea, A. Rational Design of Thermostable Carbonic Anhydrase Mutants Using Molecular Dynamics Simulations. J. Phys. Chem. B 2018, 122, 8526–8536. [Google Scholar] [CrossRef]
- Purmonen, M.; Valjakka, J.; Takkinen, K.; Laitinen, T.; Rouvinen, J. Molecular dynamics studies on the thermostability of family 11 xylanases. Protein Eng. Des. Sel. 2007, 20, 551–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, R.; Bennion, B.J.; Ham, S.; Daggett, V. Increasing temperature accelerates protein unfolding without changing the pathway of unfolding. J. Mol. Biol. 2002, 322, 189–203. [Google Scholar] [CrossRef]
- Mukai, T.; Kawai, S.; Mori, S.; Mikami, B.; Murata, K. Crystal structure of bacterial inorganic polyphosphate/ATP-glucomannokinase—Insights into kinase evolution. J. Biol. Chem. 2004, 279, 50591–50600. [Google Scholar] [CrossRef] [PubMed]
- Borgo, B.; Havranek, J.J. Automated selection of stabilizing mutations in designed and natural proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 1494–1499. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutants | Km/mM | Vmax/μmol∙min−1∙mg−1 | Kcat/Km/s−1∙mM−1 |
|---|---|---|---|
| WT | 152.5 | 5.11 × 103 | 8.2 |
| H92K | 140.3 | 4.55 × 103 | 7.9 |
| E119R | 24.5 | 2.72 × 103 | 27.0 |
| H92K/E119R | 13.6 | 2.86 × 103 | 51.5 |
| Primer | Oligonucleotide Sequence |
|---|---|
| T7 | 5′-TAATACGACTCACTATAGGG-3′ |
| T7-Term | 5′-GCTAGTTATTGCTCAGCGG-3′ |
| H92-F | 5′-CAGCATGGTGTAGTTNNKTCTGCAGCTAACGTTGACAAGAGCTGGC-3′ |
| H92-R | 5′-CGTTAGCTGCAGAMNNAACTACACCATGCTGGATGATGCCTGGGAA-3′ |
| E119-F | 5′-TCGTCCAGTTNNKGTCATCAACGACGCTGATGCAGCTGGTCT-3′ |
| E119-R | 5′-TCGTTGATGACMNNAACTGGACGACCCAGACGTGCAGTCAGC-3′ |
| A138-F | 5′-ATGGTGCAGGTNNKGGTGTCAAAGGTACTGTACTGGTCATCAC-3′ |
| A138-R | 5′-CCTTTGACACCMNNACCTGCACCATAACGAGCTTCAGCCAGACC-3′ |
| E92K-F | 5′-GTAGTTAAGTCTGCAGCTAACGTTGACA-3′ |
| E92K-R | 5′-TGCAGACTTAACTACACCATGCTGGATG-3′ |
| E119R-F | 5′-CCAGTTCGTGTCATCAACGACGCTGATG-3′ |
| E119R-R | 5′-GATGACACGAACTGGACGACCCAGACGT-3′ |
| A138R-F | 5′-GCAGGTCGGGGTGTCAAAGGTACTGTAC-3′ |
| A138R-R | 5′-GACACCCCGACCTGCACCATAACGAGC-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, H.; Zhu, W.; Zhang, Q.; Zheng, R.; Liu, L.; Cao, H. Enhanced Thermal Stability of Polyphosphate-Dependent Glucomannokinase by Directed Evolution. Catalysts 2022, 12, 1112. https://doi.org/10.3390/catal12101112
Sun H, Zhu W, Zhang Q, Zheng R, Liu L, Cao H. Enhanced Thermal Stability of Polyphosphate-Dependent Glucomannokinase by Directed Evolution. Catalysts. 2022; 12(10):1112. https://doi.org/10.3390/catal12101112
Chicago/Turabian StyleSun, Heming, Wenlong Zhu, Qinfei Zhang, Ruonan Zheng, Luo Liu, and Hui Cao. 2022. "Enhanced Thermal Stability of Polyphosphate-Dependent Glucomannokinase by Directed Evolution" Catalysts 12, no. 10: 1112. https://doi.org/10.3390/catal12101112