1. Introduction
Enantioselective biocatalysis has long been used as an alternative to traditional methods for obtaining pure chemical isomers [
1]. It has been used in many industrial fields; in particular, the use of whole-cell biocatalysts and enzymes has become common in producing enantiomeric active drugs [
2,
3,
4,
5]. This is due to the associated highly regioselective and enantioselective reactions, carried out mainly in water under mild conditions [
6]. Biotransformations are applied to obtain enantiopure compounds by enantioselective reactions, such as deracemization, desymmetrization, or asymmetric synthesis [
7,
8]. Hydroxyphosphonates are among the countless different classes of compounds obtained by enantioselective biocatalysis [
9,
10]. These compounds have a wide range of biological properties, serving as antibacterial, antiviral, and anticancer agents, as well as enzyme inhibitors or pesticides; they can also be used as precursors of other biologically active compounds [
11,
12,
13,
14]. Their bioavailability depends on their three-dimensional structure; therefore, obtaining them as enantiopure isomers with a specific absolute configuration is crucial for therapeutic effectiveness and drug safety [
15].
Similar compounds, diethyl 1-carboxy-1-hydroxymethylphosphonate and its ester were previously synthesized, and enantioselective kinetic resolution of diethyl 1-carboxy-1-hydroxyphosphonate was investigated [
16]. Dimethyl, dibutyl and diisopropyl 1-carboxy-1-hydroxymethylphosphonates were also previously synthesized as intermediates of the synthesis to obtain dioxolanone-substituted dialkyl phosphonates, and their spectroscopic data were not determined [
17].
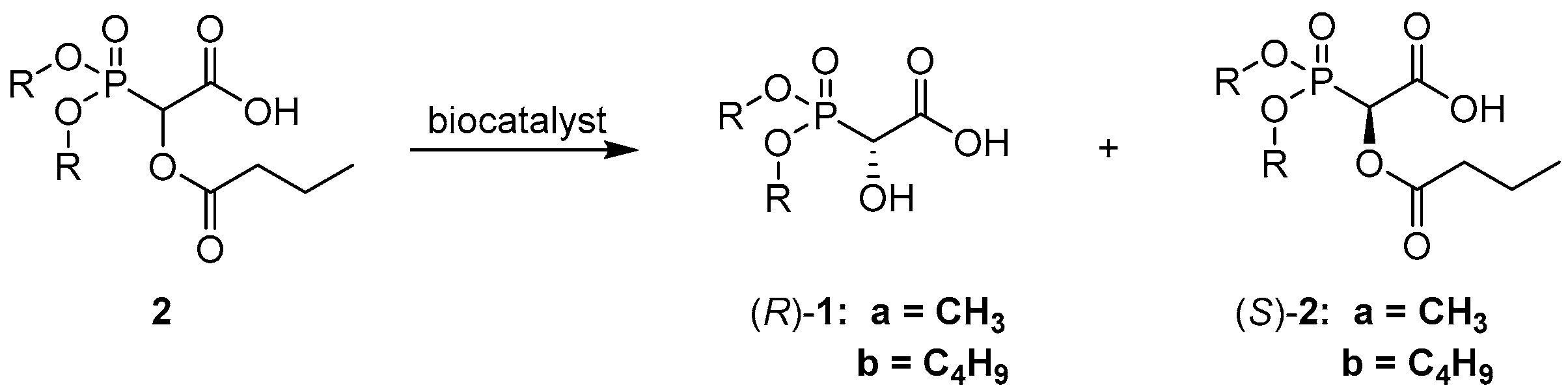
The aim of this study was to synthesize two—as yet unexploited—1-carboxy-1-hydroxymethylphosphonates, obtaining them with good enantiomeric excesses. For this purpose, biocatalytic hydrolysis by lipases was carried out. Despite the fact that the biological activity of carboxyhydroxyphosphonates is unknown, pure enantiomers of these compounds may be used as chiral auxiliaries, useful in
31P NMR spectroscopy to determine the enantiomeric purity and absolute configuration of different classes of compounds [
18]. Moreover, they can be used as precursors of aminophosphonates [
12], due to the fact that many of aminophosphonic acids are biologically active [
19]. Dimethyl 1-carboxy-1-hydroxymethylphosphonate can be transformed to its (4-nitrophenyl)methyl ester, which can be used in the synthesis of 2-substitued-3-carboxy carbapenem antibiotics [
20].
4. Materials and Methods
4.1. General Informations
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA), POCh (Gliwice, Poland), or BIOCORP (Warsaw, Poland), and were used without further purification.
NMR (nuclear magnetic resonance) spectra were measured using a Bruker Avance™ 600 (Bruker, Karlsruhe, Germany) at 600.58 MHz for 1H, 243.12 MHz for 31P, and 151.02 MHz for 13C; or on a Jeol ECZ 400S (Jeol Ltd., Tokyo, Japan) at 399.96 MHz for 1H, 161.92 MHz for 31P, and 100.6 MHz for 13C, in CDCl3 (99.8% of D, containing 0.03% v/v TMS) or in D2O (99.8% of D, containing 0.75% 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid). 1H NMR were referenced to the internal standards TMS (δ = 0.00) or 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid (δ = 0.00), 13C NMR spectra to the central line of CHCl3 (δ = 77.23), and 85% phosphoric acid in H2O for 31P NMR spectra of synthesized compounds was used as external reference (δ = 0.00). Chemical shifts (δ) are reported in ppm, in relation to standards. The biotransformation products were analyzed by 31P NMR, with quinine used as a chiral solvating a(CSA) and without the addition of an external standard for more readable spectra. The assignment of signals in 1H and 13C NMR was confirmed using 1H-1H COSY, 1H-13C HMQC, and 1H-13C HMBC spectra.
The optical rotation was measured in CHCl3, using a polAAr-31 polarimeter at 578 nm (Optical Activity Ltd., Cambridgeshire, UK).
MS spectra were obtained using a high-resolution mass spectrometer with time-of-flight analyzer (TOFMS) from LCT PremierTM XE (Waters, Milford, MA, USA).
The synthesized compounds were purified by a medium-pressure liquid chromatography system: Combi Flash® Rf 150 (Teledyne ISCO, Lincoln, NE, USA) on reversed phase column PuriFlash C18-HP, 15 μm, 120 g (Interchim, Montluçon, France) or by high-pressure liquid chromatography: ACCQ Prep HP125 UV–Vis (Teledyne ISCO, Lincoln, NE, USA) on reversed phase column Reprospher 100 C18, 5 μm, length: 250 mm, ID: 20 mm (Dr. Maish, Ammerbuch-Entringen, Germany).
The general procedure of purification using MPLC was as follows: 3 min of isocratic flow of pure water, 37 min from 0% to 100% of acetonitrile in water, 10 min of isocratic flow of pure acetonitrile; flow 10 mL/min, death time 3 min, Rf1a = 5 min, (compound 1a) Rf2a = 18 min (compound 2a), Rf1b = 26 min (compound 1b), and Rf2b = 30 min (compound 2b).
The general procedure of purification using HPLC was as follows: 12 min of isocratic flow of pure water, 8 min from 0% to 30% of acetonitrile in water, 12 min of isocratic flow of 30% acetonitrile in water, 4 min from 30% to 40% of acetonitrile in water, 15 min of isocratic flow of 40% acetonitrile in water, 10 min from 40% to 60% of acetonitrile in water, 10 min from 60% to 100% of acetonitrile in water, 10 min of isocratic flow of pure acetonitrile; flow 10 mL/min, death time 5 min, Rf3 = 60 min (compound 3).
4.2. General Procedure of Synthesis of Dimethyl and Dibutyl 1-Carboxy-1-Hydroxymethylphosphonates 1
The procedure of synthesis of compounds
1 was carried out according to the procedure for similar compounds described previously [
16,
23]. To diethyl or dibutyl phosphonate (20 mmol), glyoxylic acid monohydrate (1.84 g, 20 mmol) was added, followed by the addition of triethylamine (2.79 mL, 20 mmol). All compounds were stirred for 2 h at room temperature. The crude residue was dissolved in 10 mL distilled water and triethylamine was removed by ion exchange chromatography (Dowex
® 50WX8 50-100 mesh, Sigma Aldrich). The crude residue was purified by MPLC, giving the product as a colorless oily liquid.
Compound 1a:
Yield: 3.61 g, 98.1%
MS(TOF MS ES-) Calcd for C4H8O6P [M − H]− 183.0058; found: 183.0064.
31P NMR δ(ppm): 19.78; 1H NMR: δ(ppm) 3.91 (d, J = 11.0 Hz, 3H, OCH3), 3.92 (d, J = 10.7 Hz, 3H, OCH3), 4.62 (d, J = 16.3 Hz, 1H, PCH); 13C NMR δ(ppm): 54.71 (d, J = 6.9 Hz, 1C, OCH3), 55.28 (d, J = 6.9 Hz, 1C, OCH3), 68.48 (d, J = 157.7 Hz, 1C, PC), 170.36 (1C, COOH).
Compound 1b:
Yield: 4.41 g, 82.3%
MS(TOF MS ES+) Calcd for C10H22O6P [M + H]+ 269.1154; found: 269.1192, calcd for C10H21O6PNa [M + Na]+ 291.0974; found: 291.0971.
31P NMR δ(ppm): 17.27; 1H NMR: δ(ppm) 0.93 (t, J = 7.4 Hz, 6H, OCH2CH2CH2CH3), 1.38–1.45 (m, 4H, OCH2CH2CH2CH3), 1.65–1.71 (m, 4H, OCH2CH2CH2CH3), 4.15–4.23 (m, 4H, OCH2CH2CH2CH3), 4.61 (d, J = 16.8 Hz, 1H, PCH); 13C NMR δ(ppm): 13.67 (2C, OCH2CH2CH2CH3), 18.71 (2C, OCH2CH2CH2CH3), 32.53 (d, J = 5.0 Hz, 1C OCH2CH2CH2CH3), 32.56 (d, J = 5.0 Hz, 1C OCH2CH2CH2CH3), 68.12 (d, J = 7.2 Hz, 1C, OCH2CH2CH2CH3), 68.44 (d, J = 7.2 Hz, 1C, OCH2CH2CH2CH3), 68.55 (d, J = 156.2 Hz, 1C, PC), 170.64 (1C, COOH).
4.3. General Procedure of Synthesis of Dimethyl and Dibutyl 1-Butyryloxy-1-Carboxymethylphosphonates 2
The procedure of synthesis of compounds
2 was carried out according to the procedure for similar compounds described previously [
16,
24]. To diethyl or dibutyl phosphonate (20 mmol), glyoxylic acid monohydrate (1.84 g, 20 mmol) was added, followed by the addition of triethylamine (2.79 mL, 20 mmol). All compounds were stirred for 2 h at room temperature. The reaction mixture was placed in an ice bath, it was all dissolved in 100 mL of chloroform, and 2.07 mL (20 mmol) of butyryl chloride was slowly added dropwise. After completion of the reaction—which was monitored by TLC—the resulting solution was extracted with 100 mL of distilled water, and the organic phase was dried by anhydrous magnesium sulphate and evaporated. The crude residue was purified by MPLC, giving the product as a colorless oily liquid.
Compound 2a:
Yield: 1.34 g, 26.4%
MS(TOF MS ES-) Calcd for C8H16O7P [M − H]− 253.0477; found: 253.0478.
31P NMR δ(ppm): 17.25; 1H NMR: δ(ppm) 0.98 (t, J = 7.4 Hz, 3H, OCOCH2CH2CH3), 1.67–1.74 (m, 2H, OCOCH2CH2CH3), 2.41–2.51 (m, 2H, OCOCH2CH2CH3), 3.88 (d, J = 6.1 Hz, 3H, OCH3), 3.90 (d, J = 6.1 Hz, 3H, OCH3), 5.57 (d, J = 17.8 Hz, 1H, PCH); 13C NMR δ(ppm): 13.61 (1C, OCOCH2CH2CH3), 18.41 (1C, OCOCH2CH2CH3), 35.62 (1C, OCOCH2CH2CH3), 54.89 (t, J = 6.9 Hz, 2C, OCH3), 67.65 (d, J = 162.4 Hz, 1C, PC), 166.20 (1C, OCOCH2CH2CH3), 172.18 (d, J = 10.2 Hz, 1C, COOH).
Compound 2b:
Yield: 2.22 g, 32.8%
MS(TOF MS ES+) Calcd for C14H28O7P [M + H]+ 339.1573; found: 339.1572.
31P NMR δ(ppm): 14.45; 1H NMR: δ(ppm) 0.93 (t, J = 7.4 Hz, 6H, OCH2CH2CH2CH3), 0.98 (t, J = 7.4 Hz, 3H, OCOCH2CH2CH3), 1.37–1.45 (m, 4H, OCH2CH2CH2CH3), 1.62–1.74 (m, 6H, OCOCH2CH2CH3, OCH2CH2CH2CH3), 2.37–2.51 (m, 2H, OCOCH2CH2CH3), 4.14–4.24 (m, 4H, OCH2CH2CH2CH3), 5.54 (d, J = 17.8 Hz, 1H, PCH); 13C NMR δ(ppm): 13.69 (3C, OCOCH2CH2CH3, OCH2CH2CH2CH3), 18.44 (1C, OCOCH2CH2CH3), 18.73 (1C, OCH2CH2CH2CH3), 18.74 (1C, OCH2CH2CH2CH3), 32.50 (d, J = 2.9 Hz, 1C, OCH2CH2CH2CH3), 32.54 (d, J = 2.9 Hz, 1C, OCH2CH2CH2CH3), 35.70 (1C, OCOCH2CH2CH3), 68.40 (d, J = 6.7 Hz, 1C, OCH2CH2CH2CH3), 68.49 (d, J = 6.4 Hz, 1C, OCH2CH2CH2CH3), 67.94 (d, J = 148.1 Hz, 1C, PC), 166.55 (1C, OCOCH2CH2CH3), 172.17 (d, J = 10.2 Hz, 1C, COOH).
4.4. Enzyme Source
ANL, Amano lipase A from Aspergillus niger (Sigma-Aldrich); CAL, Lipase A from Candida antarctica (Fluka, Buchs, Switzerland); CRL, Candida rugosa lipase (Sigma-Aldrich); PFL, Amano lipase AK, from Pseudomonas fluorescens (Sigma-Aldrich); MCL, Mucor circinelloides lipase (gift from Lodz University of Technology, Lodz, Poland); PCL, Amano Lipase G, from Penicillium camemberti (Sigma-Aldrich); PPL, Lipase Type II, Crude from Porcine Pancreas (Sigma-Aldrich); TLL, Lipase from Termomyces lanuginosus (Fluka); BCL, Amano Lipase PS from Burkholderia cepacia (Sigma-Aldrich); MRL, Mucor Racemosus lipase (gift from Lodz University of Technology); RNL, Lipase from Rhizopus niveus (Sigma-Aldrich); MJL, Amano Lipase M from Mucor javanicus (Sigma-Aldrich); RSL, Lipase from Rhizopus sp. (SERVA, Heidelberg, Germany); RML, Lipase from Rhizomucoer miehei produced in Aspergillus oryzae (Sigma-Aldrich); N435, Novozym® 435, immobilised lipase from Candida antarctica B (Novozymes, Bagsværd, Denmark); ROL, Lipase from Rhizopus oryzae (Sigma-Aldrich).
4.5. Enzymatic Hydrolysis General Procedure
Lipase-catalyzed reactions were prepared according to a procedure described previously [
22]. Reactions were carried out in a biphasic system (3.8 mL) consisting of 0.05 M phosphate buffer (pH 7.0, 3.0 mL) and a mixture of diisopropyl ether (0.2 mL) with hexane (0.6 mL). After addition of 0.2 mmol of substrate (51 mg or 68 mg respectively) and 100 mg of a suitable lipase, reactions were carried out at room temperature with shaking (150 rpm) and stopped after certain periods of time, when the conversion degree reached up to 50%, by the addition of 2 mL of acetone and the filtration of precipitated protein. The solvent was evaporated, and the residues were dissolved in 5 mL of distilled water. The obtained solutions were purified by ion exchange chromatography on a column filled with Dowex (200–400 mesh), with the water as eluent. Then, the organic solvent was evaporated, and the products were analyzed by means of
31P NMR spectroscopy. In the case where there was no clear separation of signals derived from enantiomers, the analysis was repeated with quinine as a chiral solvating agent.
4.6. Enantioselectivity Assignment
The mixtures of biotransformation products (alcohol and unreacted ester) were analyzed by
31P NMR spectroscopy using quinine as a chiral solvating agent. The degree of enantiomeric excess was expressed as a percentage (%), and is defined as:
where
P1 and
P2 are the values of the area under the signals coming from the major and minor enantiomers of the product or substrate, respectively.
The enantiomeric ratio (E) was computed from the following formula [
25]:
where ee
p is the enantiomeric excess of product and ee
s is the enantiomeric excess of substrate.
4.7. Procedure of Determination of Optical Rotation and Determination of the Absolute Configuration
4.7.1. Preparation of Non-Equimolar Mixture of Enantiomers of Compounds 1 and 2 for Determination of Optical Rotation and of the Absolute Configuration
Butyryloxyphosphonates 2 were partially hydrolyzed by Amano Lipase PS from Burkholderia cepacia (compound 2b) or by Aspergillus niger lipase (compound 2a), according to the general procedure of enzymatic hydrolysis. Unreacted butyryloxyphosphonates 2 were separated from hydroxyphosphonates 1 by MPLC. Separated compounds were analyzed by NMR with quinine as a chiral solvating compound and optical rotation was measured.
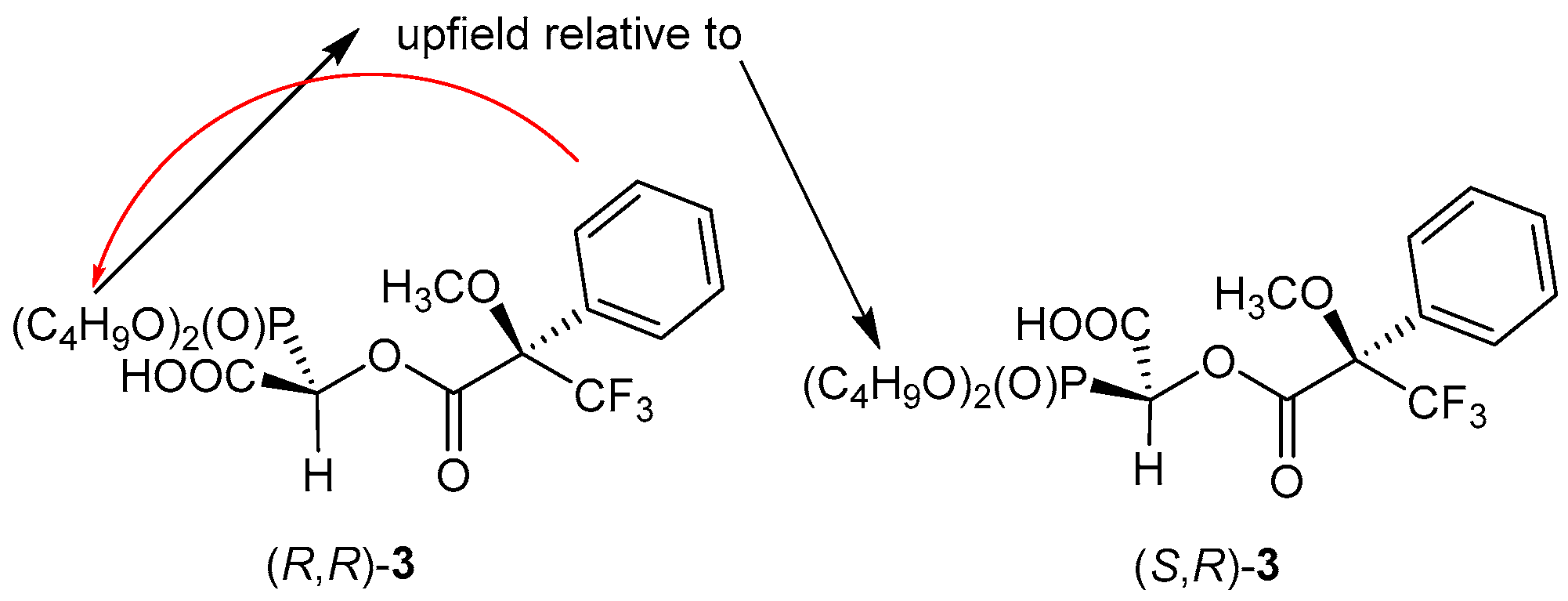
4.7.2. Synthesis of Dibutyl 1-Carboxy-1-(3,3,3-Trifluoro-2-Methoxy-2-Phenylpropanoxy)Methylphosphonate (compound 3)
The non-equimolar mixture of enantiomers of compound
1b obtained according to the procedure described in
Section 4.7.1, was acylated by (
S)-(+)MTPA-Cl, according to the literature [
17]. Dry pyridine (300 μL) was added to a dry bottle with septum using a syringe with a needle. Then, also using a syringe, (
S)-(+)MTPA-Cl (50 μL, 0.27 mmol) was added. After that, compound
1b (36 mg, 0.15 mmol) dissolved in dry chloroform (300 μL) was added. The mixture was left for 24 h at room temperature. An excess of 3-dimethylamino-1-propylamine (50 μL, 0.40 mmol) was then added and, after 5 min at room temperature, the mixture was diluted with chloroform (10 mL), washed with cold dilute HCl (10 mL) and water (10 mL), and dried over anhydrous magnesium sulphate. After filtration of the drying agent, the ether was evaporated and compound
3 was purified by HPLC.
MS (TOF MS ES-) Calcd for C20H27O8PF3 [M − H]− 483.1396; found: 483.1396.
Isomer (R,R)
31P NMR δ(ppm): 12.84; 1H NMR: δ(ppm) 0.82 (t, J = 7.4 Hz, 3H, OCH2CH2CH2CH3), 0.87 (t, J = 7.4 Hz, 3H, OCH2CH2CH2CH3), 1.19–1.67 (m, 8H, OCH2CH2CH2CH3), 3.68 (s, 3H, OCH3), 3.78-4.20 (m, 4H, OCH2CH2CH2CH3), 5.60 (d, J = 16.5 Hz, 1H, PCH), 7.37–7.43 (m, 3H, PC6H5), 7.67–7.71 (m, 2H, PC6H5); 13C NMR δ(ppm): 13.66 (1C, OCH2CH2CH2CH3), 13.69 (1C, OCH2CH2CH2CH3), 18.55 (1C, OCH2CH2CH2CH3), 18.67 (1C, OCH2CH2CH2CH3), 32.35 (d, J = 6.1 Hz, 1C OCH2CH2CH2CH3), 32.42 (d, J = 6.6 Hz, 1C OCH2CH2CH2CH3), 56.22 (1C, OCH3), 68.71 (d, J = 6.9 Hz, 1C, OCH2CH2CH2CH3), 69.02 (d, J = 7.1 Hz, 1C, OCH2CH2CH2CH3), 69.56 (d, J = 145.6 Hz, 1C, PC), 84.59-85.35 (C(CF3)), 123.22 (q, J = 288.9 Hz, CF3), 127.58-132.06 (C(C6H5)), 163.88 (COO), 165.78 (d, J = 11.3 Hz, COOH).
Isomer (S,R)
31P NMR δ(ppm): 13.10; 1H NMR: δ(ppm) 0.86 (t, J = 7.4 Hz, 3H, OCH2CH2CH2CH3), 0.90 (t, J = 7.4 Hz, 3H, OCH2CH2CH2CH3), 1.19–1.67 (m, 8H, OCH2CH2CH2CH3), 3.55 (s, 3H, OCH3), 3.78–4.20 (m, 4H, OCH2CH2CH2CH3), 5.62 (d, J = 16.5 Hz, 1H, PCH), 7.37–7.43 (m, 3H, PC6H5), 7.61–7.65 (m, 2H, PC6H5); 13C NMR δ(ppm): 13.68 (1C, OCH2CH2CH2CH3), 13.70 (1C, OCH2CH2CH2CH3), 18.65 (1C, OCH2CH2CH2CH3), 18.72 (1C, OCH2CH2CH2CH3), 32.49 (d, J = 6.1 Hz, 1C OCH2CH2CH2CH3), 32.44 (d, J = 6.0 Hz, 1C OCH2CH2CH2CH3), 55.75 (1C, OCH3), 68.66 (d, J = 7.0 Hz, 1C, OCH2CH2CH2CH3), 68.97 (d, J = 6.9 Hz, 1C, OCH2CH2CH2CH3), 69.49 (d, J = 145.1 Hz, 1C, PC), 84.59–85.35 (C(CF3)), 123.19 (q, J = 288.9 Hz, CF3), 127.58–132.06 (C(C6H5)), 164.74 (COO), 165.70 (d, J = 11.3 Hz, COOH).
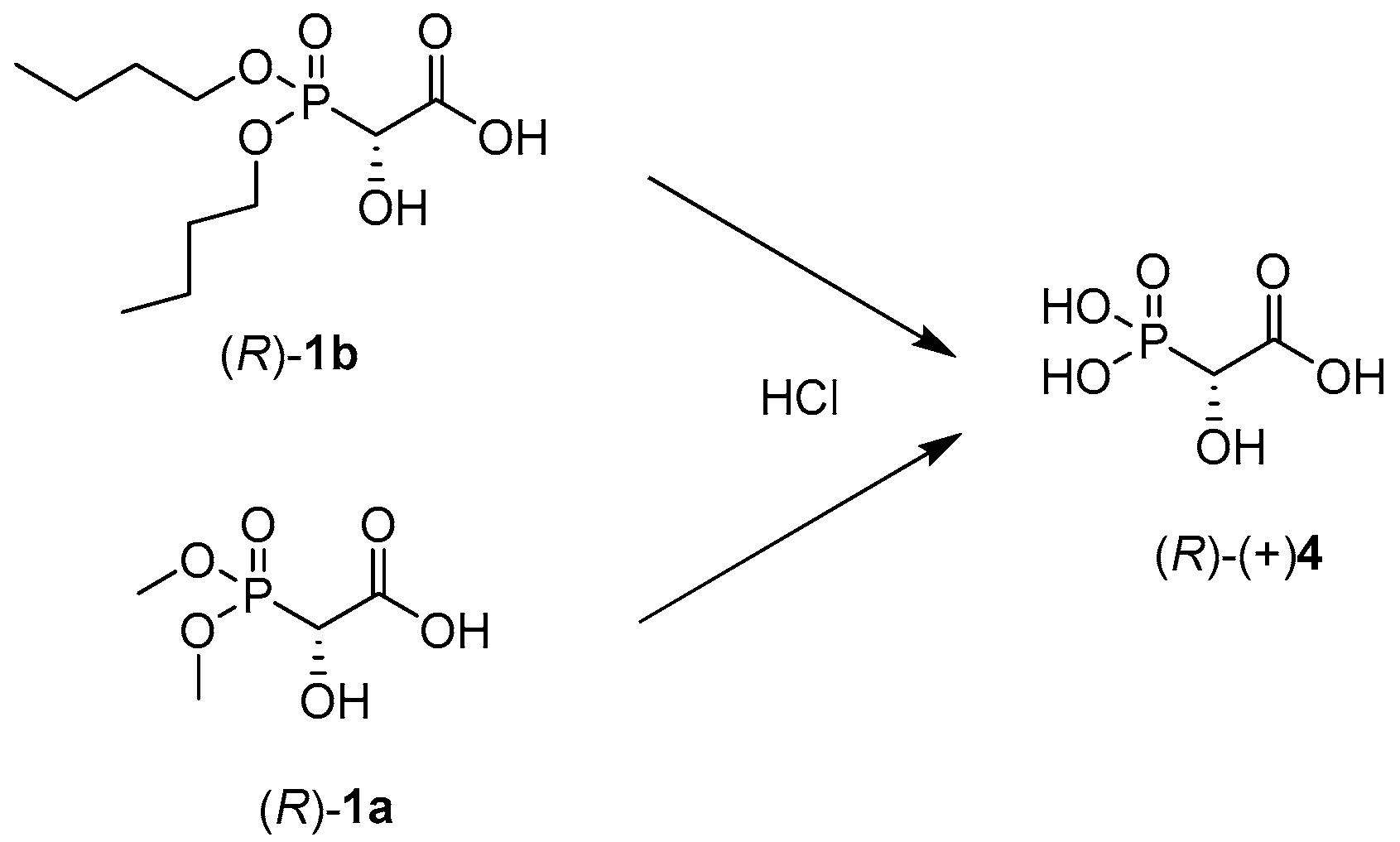
4.7.3. Synthesis of 1-Carboxy-1-Methylphosphonic Acid 4
The non-equimolar mixture of enantiomers of compounds 1a and 1b was hydrolyzed by HCl. To compound 1 (1a, 46 mg, 0.25 mmol, ee = 18%; 1b, 54 mg, 0.20 mmol, ee = 32%), distilled water (10 mL) and 36% HCl (10 mL) were added. The hydrolysis reaction was carried out under reflux for 2 h. After cooling the mixture, the solvents were evaporated and compound 4 was purified by MPLC.
MS(TOF MS ES-) Calcd for C2H5O6P [M − H]− 154.9745; found: 154.9750.
31P NMR δ(ppm): 14.25; 1H NMR: δ(ppm) 4.63 (d, J = 18.2 Hz, 1H, PCH); 13C NMR δ(ppm): 71.89 (d, J = 147.0 Hz, 1C, PC), 175.99 (d, J = 1.8 Hz, 1C, COOH).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}