2.1. Experimental Results on the Hirao Reaction of Iodobenzene and Diarylphosphine Oxides
It is well-known from earlier studies that the Cu
+-catalysis is less efficient than the Pd- or Ni-promoted cases. To compensate the lower reactivity of the Cu
+-catalyst, the more reactive iodobenzene was selected as the reactant with diphenylphosphine oxide under MW-assisted conditions. The first set of P–C couplings was carried out using CuI as the catalyst and triethylamine as the base in ethanol as the medium at a temperature of 165 °C (
Table 1). In the absence of catalyst, there was practically no reaction (
Table 1/Entry 1). Using 10% of CuI and 1 equivalent of each of Ph
2P(O)H and NEt
3, the conversion was 44% after a 3 h of irradiation (
Table 1/Entry 2). Offering a 20% excess of Ph
2P(O)H to act as a P-ligand, the conversion decreased to 35% (
Table 1/Entry 3). At the same time, measuring in 2 equivalents of TEA, the conversion increased to 51% (
Table 1/Entry 4) meaning that Cu
+ may prefer TEA to Ph
2P(O)H as the ligand. The P–C couplings became somewhat more efficient, when 20% of the catalyst was applied. The tendencies were the same: the use of 40% excess of Ph
2P(O)H was not advantageous (
Table 1/Entry 7). At the same time, applying NEt
3 in a 2 equivalents’ quantity, the conversion increased to 63% (
Table 1/Entry 8). Extension of the reaction time to 4 h was useful, as conversions of 61 and 75% could be attained as compared to the 3 h expositions (
Table 1/Entries 6 and 9 vs. entries 5 and 8). It is noteworthy that the formation of Ph
2(EtO)P(O) as a side-product (δ
P(CDCl
3) 31.5, [M + H]
+found = 247.0882, C
14H
16O
2P requires 247.0888) was inevitable during the coupling reactions by the participation of the solvent. Its quantity fell in the range of 1–7%.
In the next stage, different Cu
+ precursors were tested in the model reaction selected. Applying 20% of CuBr as the catalyst at 165 °C for 3 h at a 1:1 ratio of Ph
2P(O)H and NEt
3, the conversion was 75% (
Table 2/Entry 2). Using 1.4 equivalents of the P-reagent, the conversion dropped to 50% (
Table 2/Entry 3). However, when the quantity of NEt
3 was increased to two equivalents, a higher conversion of 90% was attained (
Table 2/Entry 4). Triphenylphosphine oxide (
1) was obtained in 65 and 85% yields from the better experiments (
Table 2/Entries 2 and 4). One can see that in the P–C coupling reaction under discussion, CuBr is a more efficient catalyst than CuI.
Then, CuCl was applied as the catalyst precursor. The tendencies observed were similar experienced with CuBr, but the conversions were somewhat lower. Applying Ph
2P(O)H and the base in a one equivalent quantity, the conversion was 71% as compared to 75% (
Table 2/Entry 5 vs. entry 2). Increasing the quantity of the P-reagent to 1.4 equivalents, the conversion was 47% against the value of 50% (
Table 2/Entry 6 vs. entry 3). When NEt
3 was used in an excess (two equivalents), a conversion of 84% was observed that was somewhat lower than the value of 90% obtained with CuBr (
Table 2/Entry 7 vs. entry 4). A longer reaction time of 4 h led to a conversion of 88% (
Table 2/Entry 8).
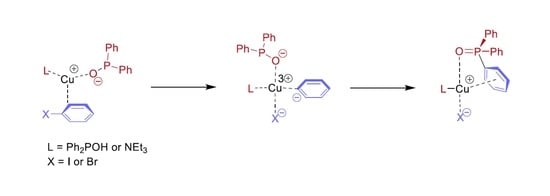
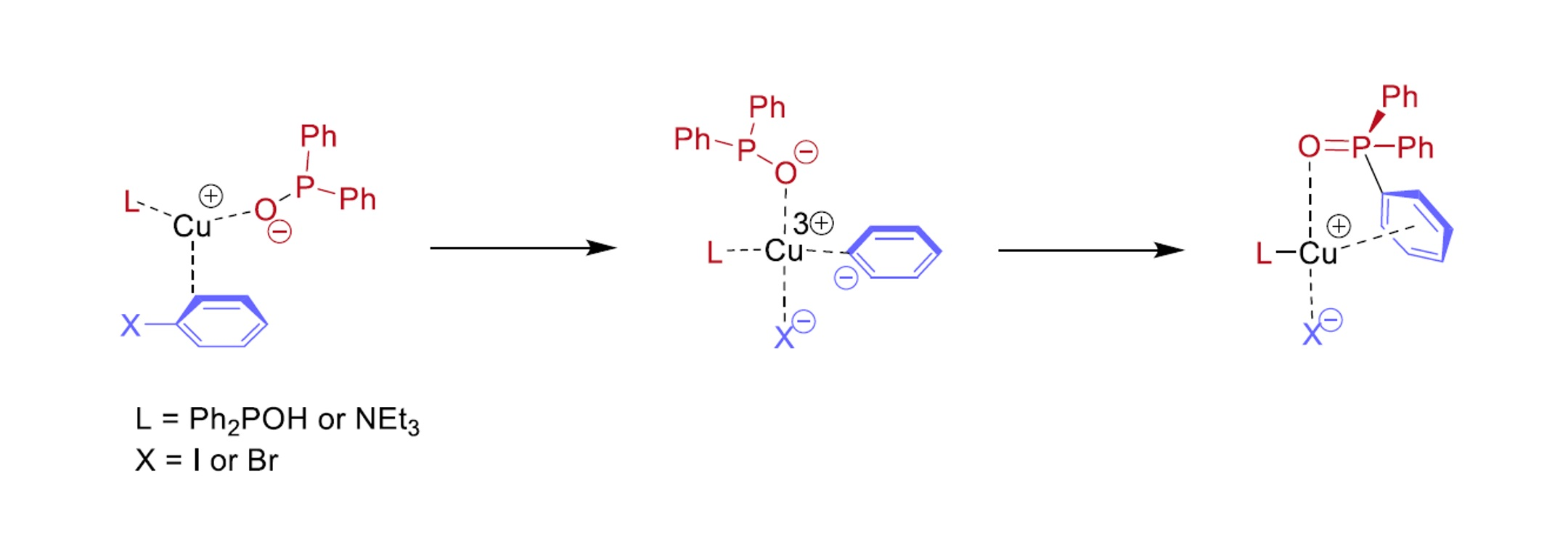
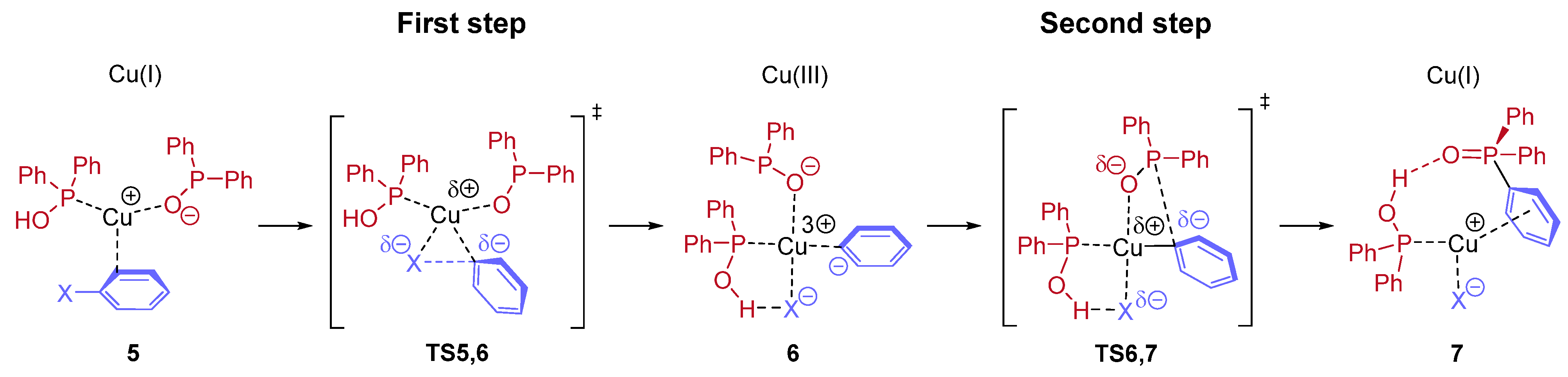
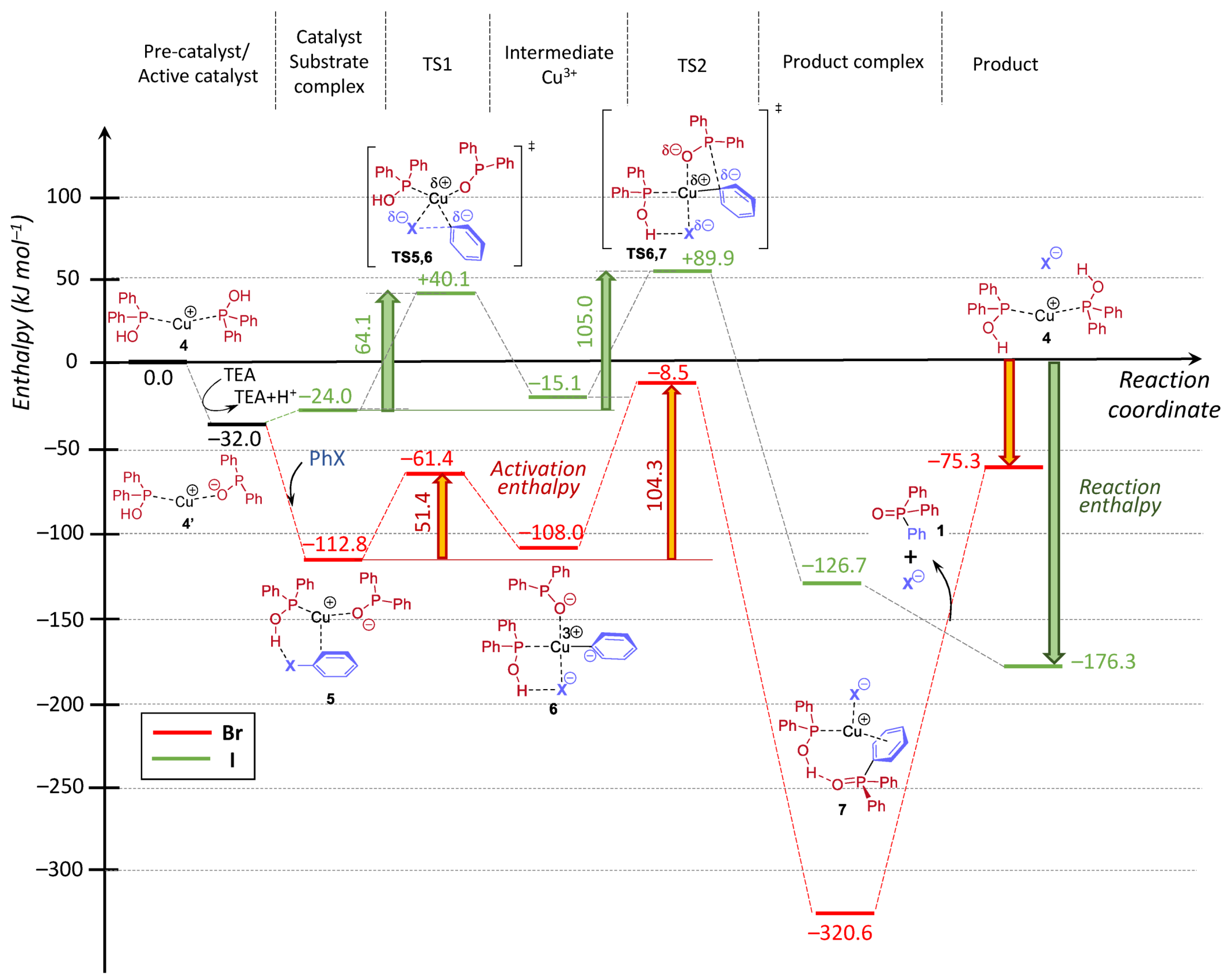
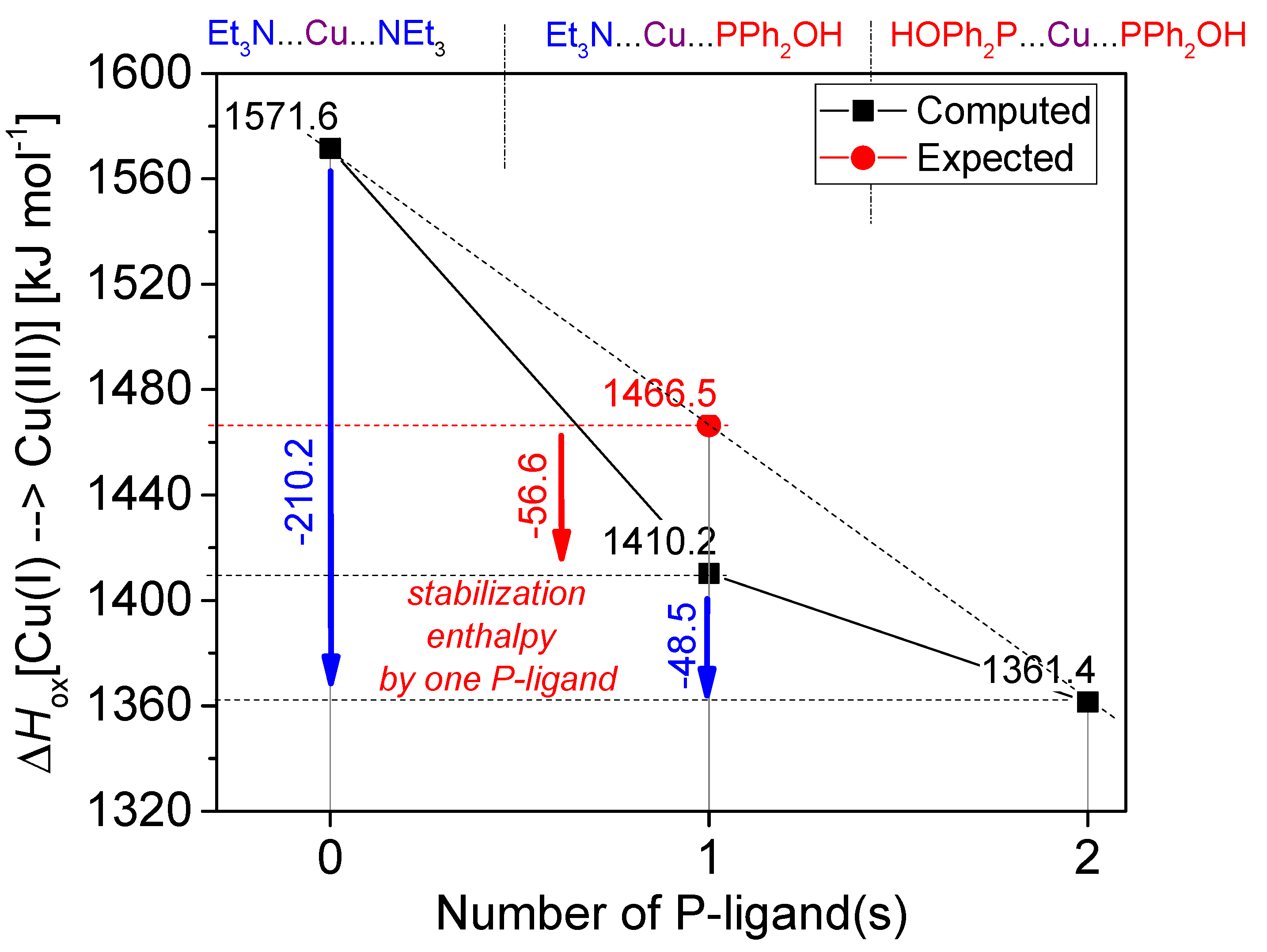
In conclusion, the higher conversions around 89% belonging to CuBr and CuCl catalyst precursors were obtained using two equivalents of NEt
3. One may see that CuBr was found the most efficient catalyst precursor in the series investigated. On the other hand, it may be observed from the experimental data that probably not the P---Cu(I)---P (
A) complex is the active species, but rather the P---Cu(I)---N (
B) or the N---Cu(I)---N (
C) complex. The mixed ligation may be more realistic, as in this case, the reacting P moiety is also included. This problem will be discussed in the theoretical part, see
Section 2.4.2.)
Bis(4-methylphenyl)phosphine oxide and bis(3,5-dimethylphenyl)phosphine oxide were also tested in the P–C coupling reactions applying Cu salt catalyst precursors (20%) at 165 °C in EtOH as the solvent. Applying CuCl in reaction with the 4-MePh derivative, it was better to use 2 equivalents of NEt
3 as compared to the case with only 1 equivalent of the amine, as marked by the conversions of 80% and 68%, respectively (
Table 3/Entries 2 and 1). In the presence of CuBr, the conversion was better (83%) (
Table 3/Entry 3).
Applying bis(3,5-dimethylphenyl)phosphine oxide as the P-reagent and ligand, somewhat higher (77, 87 and 90%) conversions were detected under the conditions applied above. (Compare entries 1 and 4, entries 2 and 5, as well as entries 3 and 6 of
Table 3). Although appearance of the methyl group in position 4 of the phenyl ring slightly decreases the reactivity of the secondary phosphine oxide in the P–C coupling reaction under discussion, the steric hindrance due to the methyl groups may increase the activity of the catalyst formed, as in this case, bis-ligation is preferred [
10]. After purification, diaryl-phenylphosphine oxides
2 and
3 were obtained in yields of 78% and 84%, respectively.
It can be seen that the methyl substitution in the phenyl ring is somewhat disadvantageous in the Hirao reaction. In order to prove this directly, competitive P–C couplings were carried out applying a 1:1 mixture of Ph
2P(O)H and Ar
2P(O)H in reaction with iodobenzene. The interrupted reactions revealed a 20:12 and a 23:15 ratio of Ph
3P(O) (
1)–(4-MePh)
2P(O) (
2) and Ph
3P(O) (
1)–(3,5-diMePh)
2P(O) (
3), respectively (
Table 4/Entries 1 and 2), meaning that the secondary phosphine oxide is more reactive with phenyl groups than with 4-MePh and 3,5-diMePh substituents. The methyl group in position 4 results in larger electron density on the P atom increasing its complexation ability; at the same time, this substituent also results in a decrease in the acidity. In overall, the reactivity decreases. It was found earlier that both electron-donating and electron-withdrawing substituents decrease the reactivity of the bromoarene in P–C coupling reactions [
7].
2.3. Complexation
The complexation process is rather complicated, as Cu
+ may be ligated with both the tautomeric form of the reagent (Ph
2POH,
1a) and the trialkylamine that is present as base. There is a multiple possibility for the formation of “Cu
+P
4”, “Cu
+N
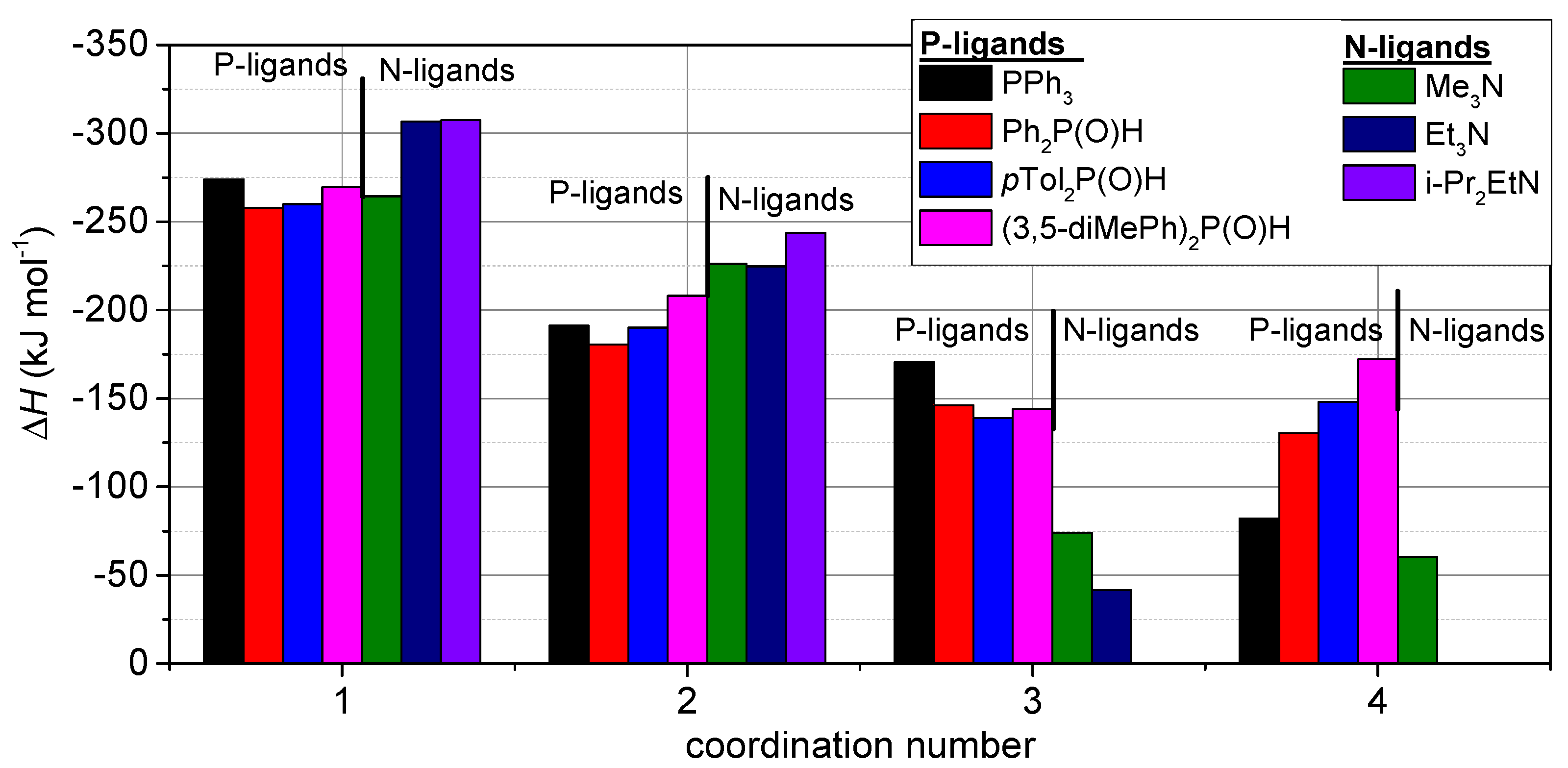
4” and the mixed complexes as well. At first, examining only the “homogeneous” complexation, a series of P- and N-ligands were considered up to four ligations. For P-ligation, the complex formation is beneficial in all steps up to the fourth ligation. However, the step-by-step enthalpy values are somewhat decreasing in the order of ligation(s) marked by numbers 1, 2, 3, 4. This is in contrast to the analogous complexation with Pd(0), where due to steric hindrance, the fourth ligation is already unfavorable. It means that despite the steric hindrance, the most preferred form for Cu
+ is the tetra-coordinated complex. Even triphenylphosphine may be involved in tetracoordination as shown in
Figure 2. In case of N-ligation, the sterically less hindered NMe
3 may participate in tetraligation, but the sterically more requiring NEt
3 and N
iPr
3 may be involved in only triple- and bisligation, respectively. The computed values are summarized in
Table 6 and visually displayed in
Figure 2. According to the computed data, the N-ligation is stronger in the first two steps (1 and 2), but at higher coordinations (such as 3 and 4), the P-ligation overcomes N-ligation.
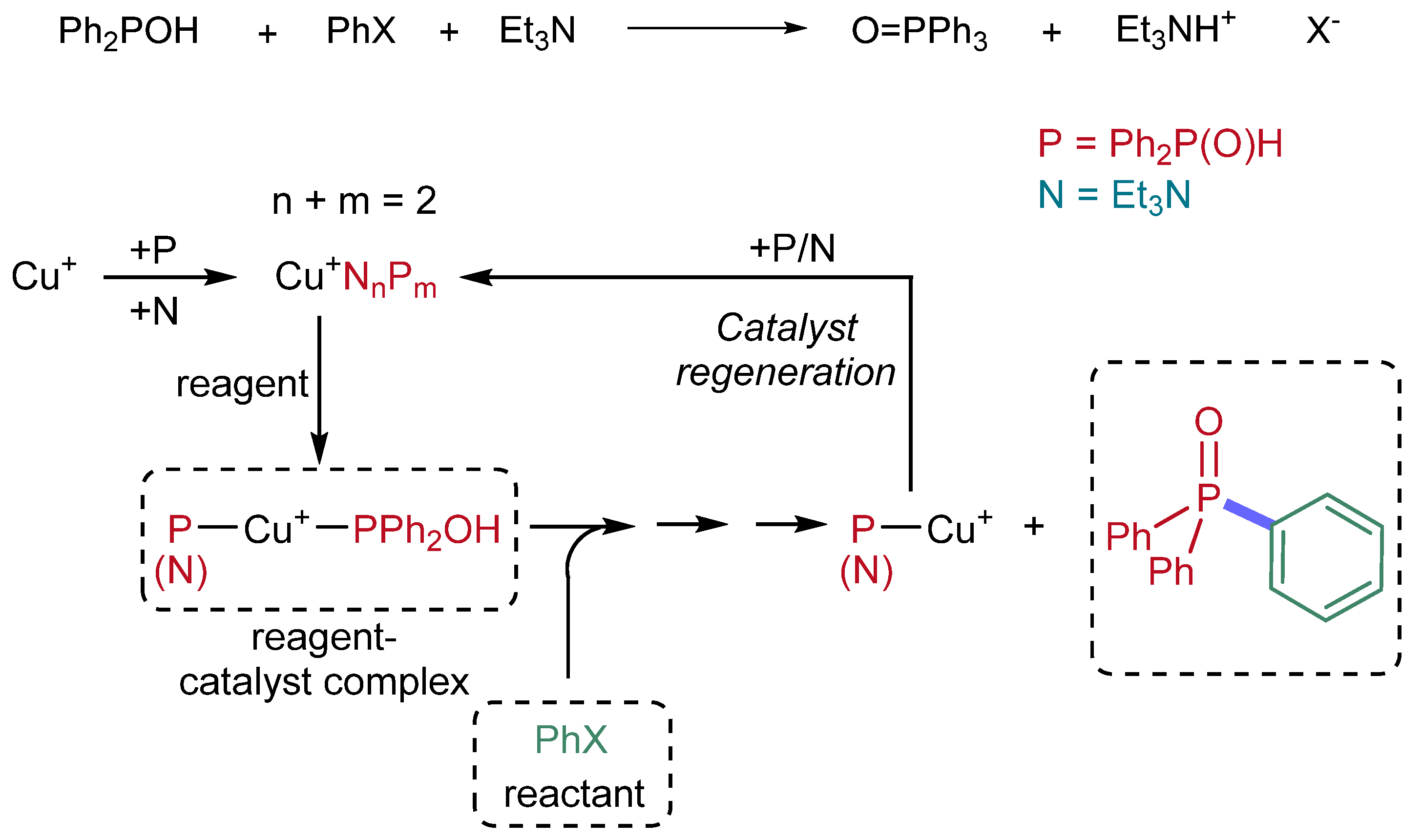
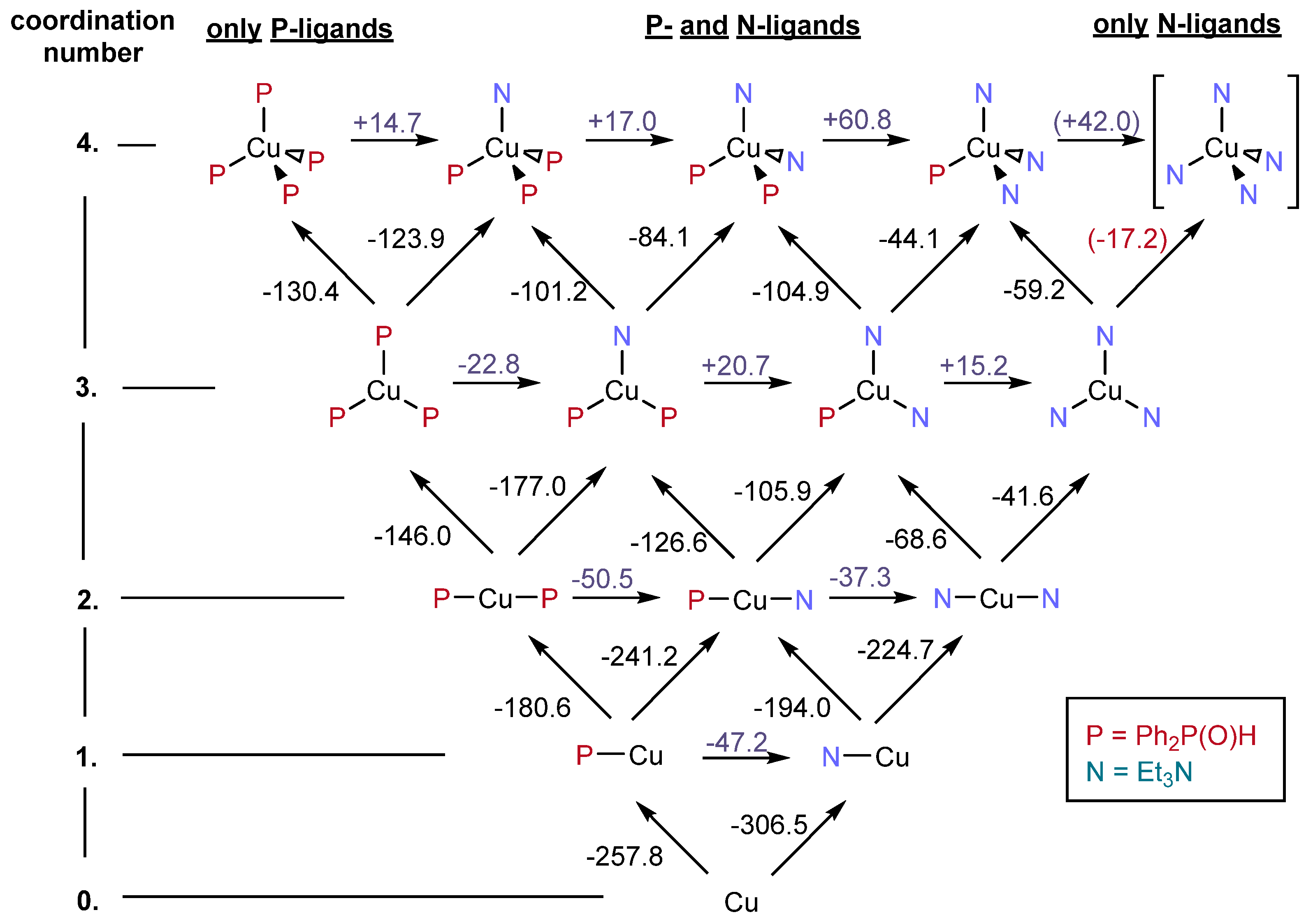
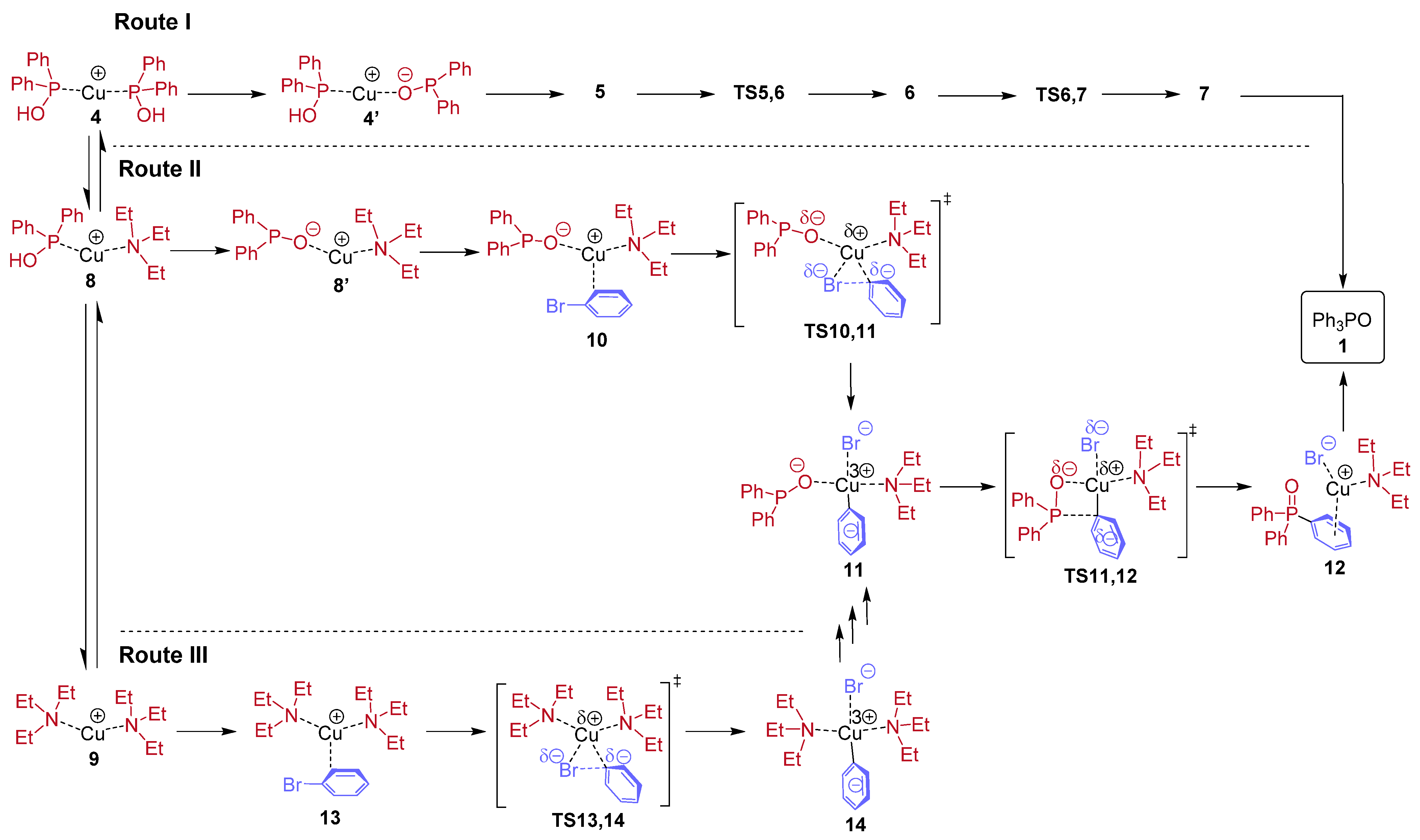
Extending the study to “heterogeneous” complexation, where NEt
3 and Ph
2P(O)H are present at the same time and compete with each other; overall, 14 species may be deduced. The map of the possible complexations is presented in
Figure 3 together with the stepwise enthalpies computed.
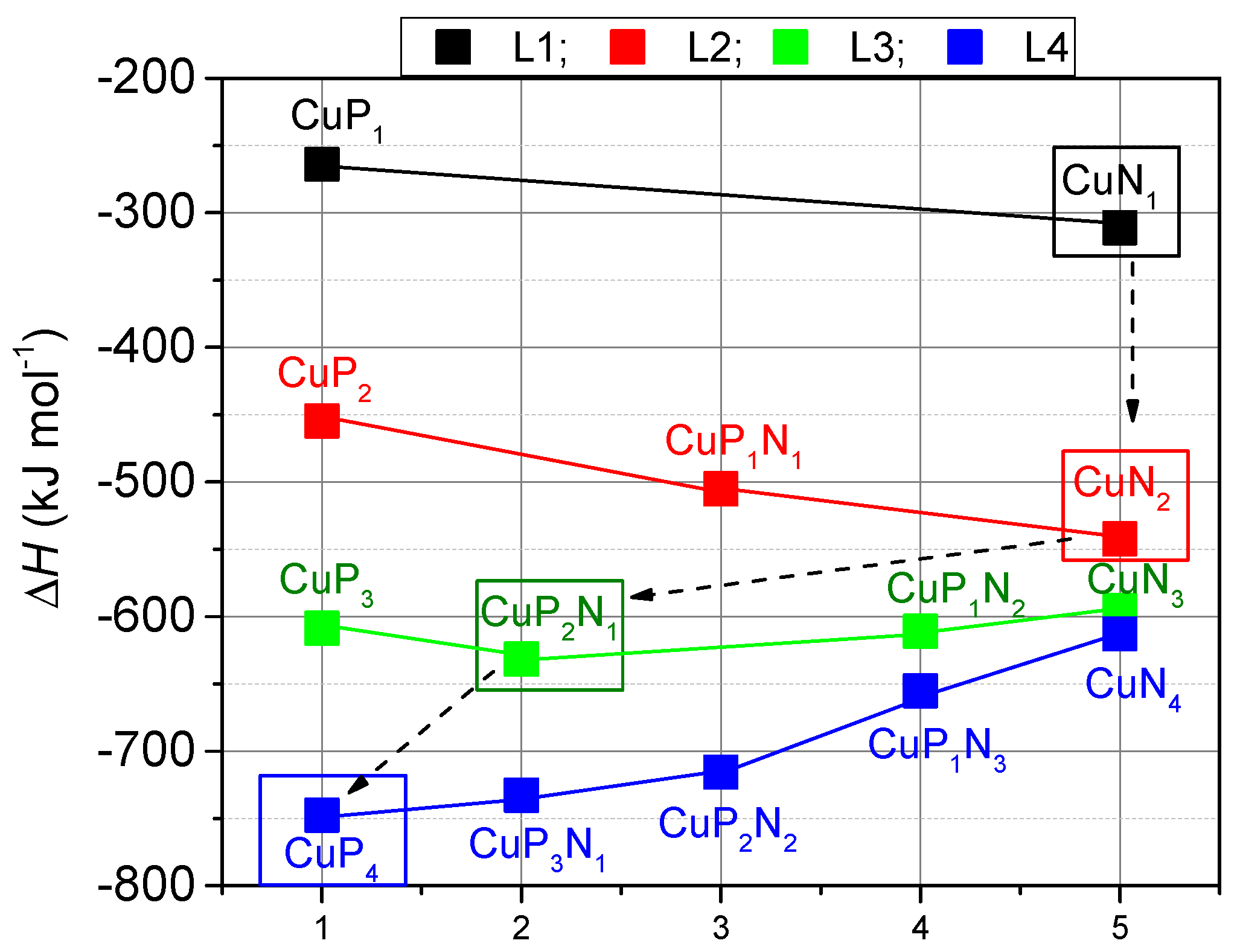
Figure 4 summarizes the situation showing that in case of higher ligations, the complexation with P-ligands is more beneficial than that with N-ligands.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




