
Conversion of Green Methanol to Methyl Formate


Abstract
:1. Introduction
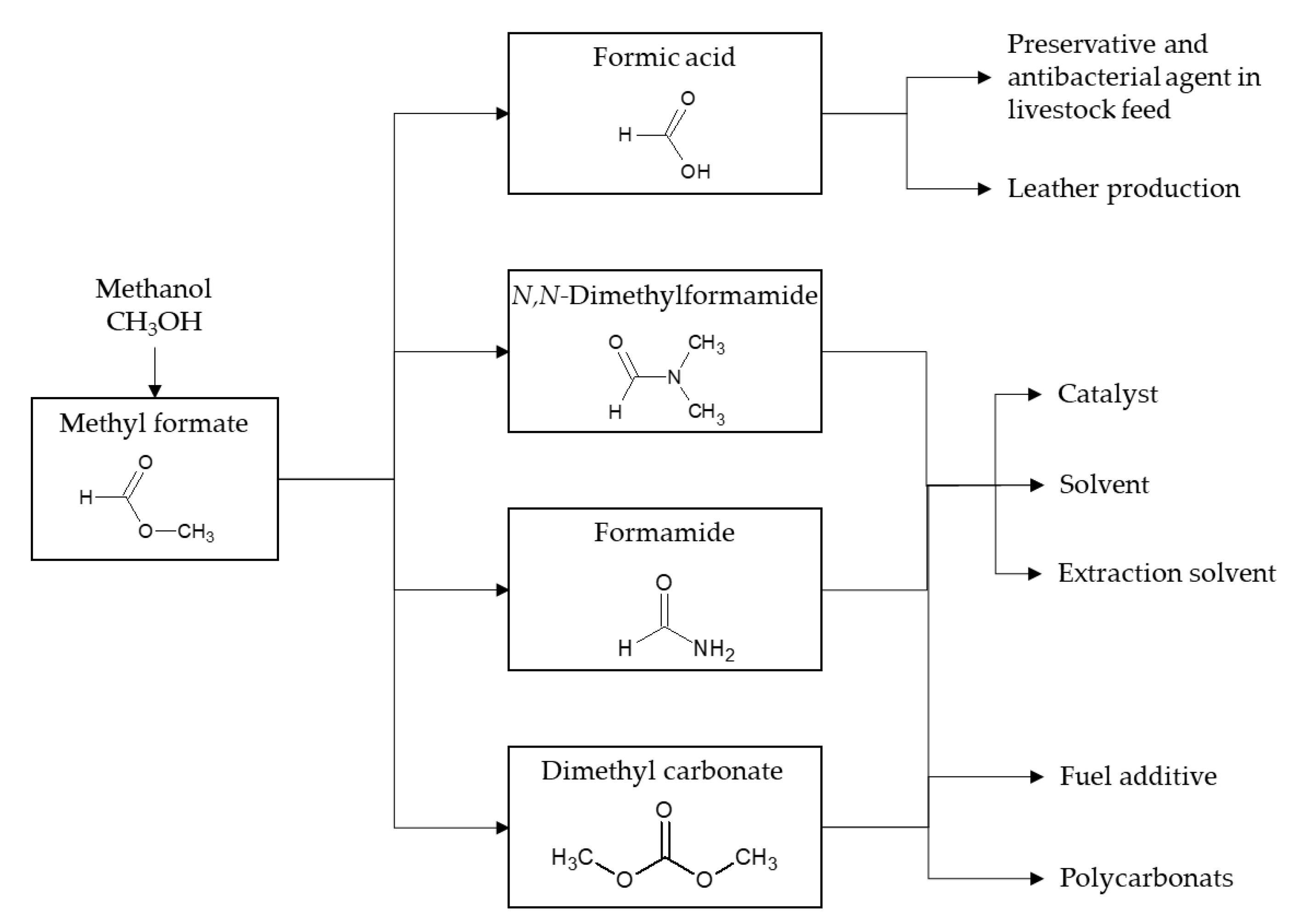
2. Why Convert Methanol into Methyl Formate?
3. Synthesis Routes
3.1. Methanol Carbonylation
3.1.1. Established Process
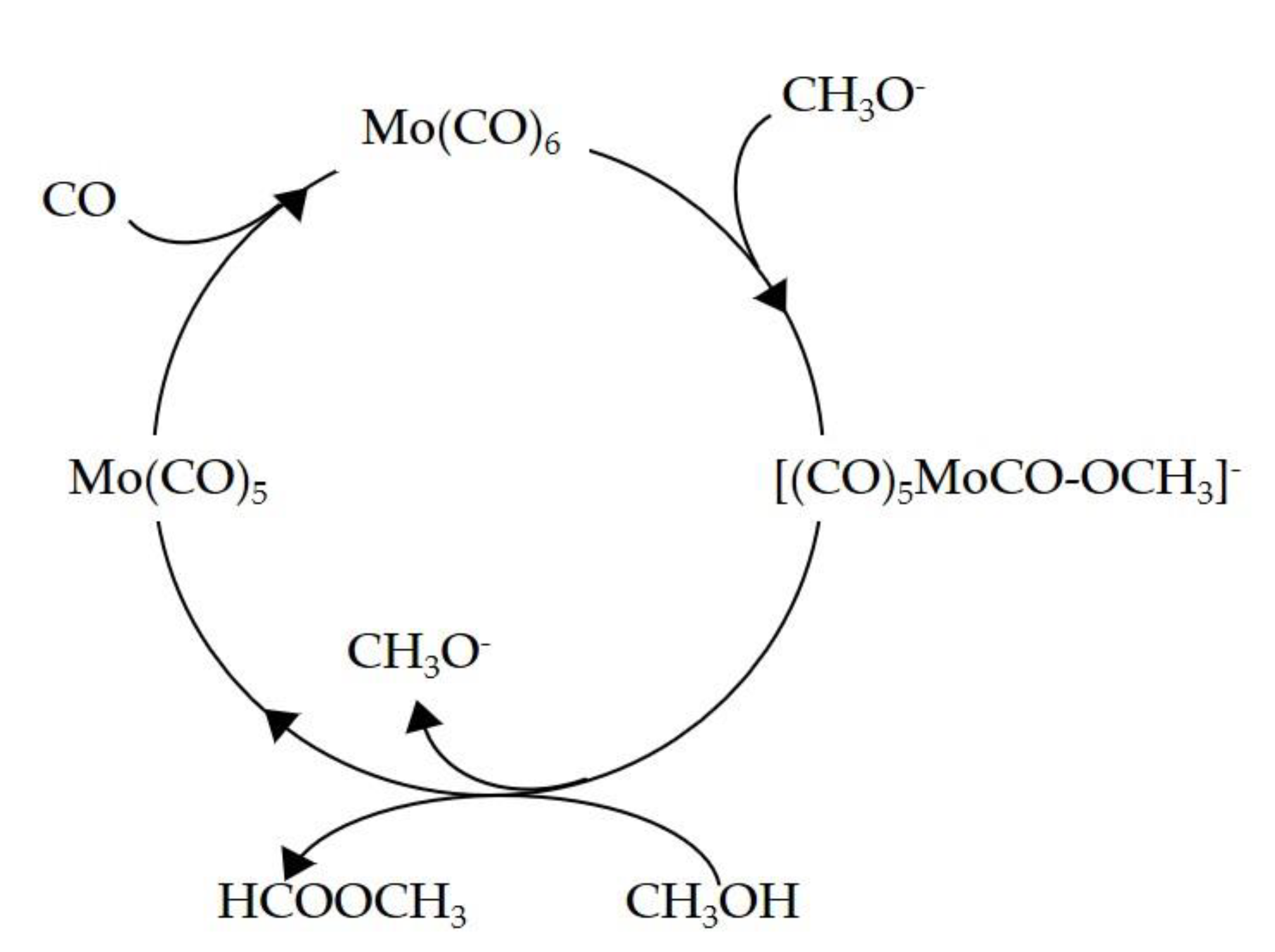
3.1.2. Mechanism


3.1.3. By-Products



3.1.4. Improvements
Homogeneous Catalysts

- (1)
- Adsorption and oxidation of methanol on Cu sites forms Cu-OCH3 and Cu-H species;
- (2)
- CO adsorption and its insertion in the Cu-O bond in Cu-OCH3 forms CH3OC=O intermediates;
- (3)
- Reductive elimination of the CH3OC=O intermediates provides MF [20].
Heterogeneous Catalysts
3.2. Dehydrogenation of Methanol
3.2.1. Process
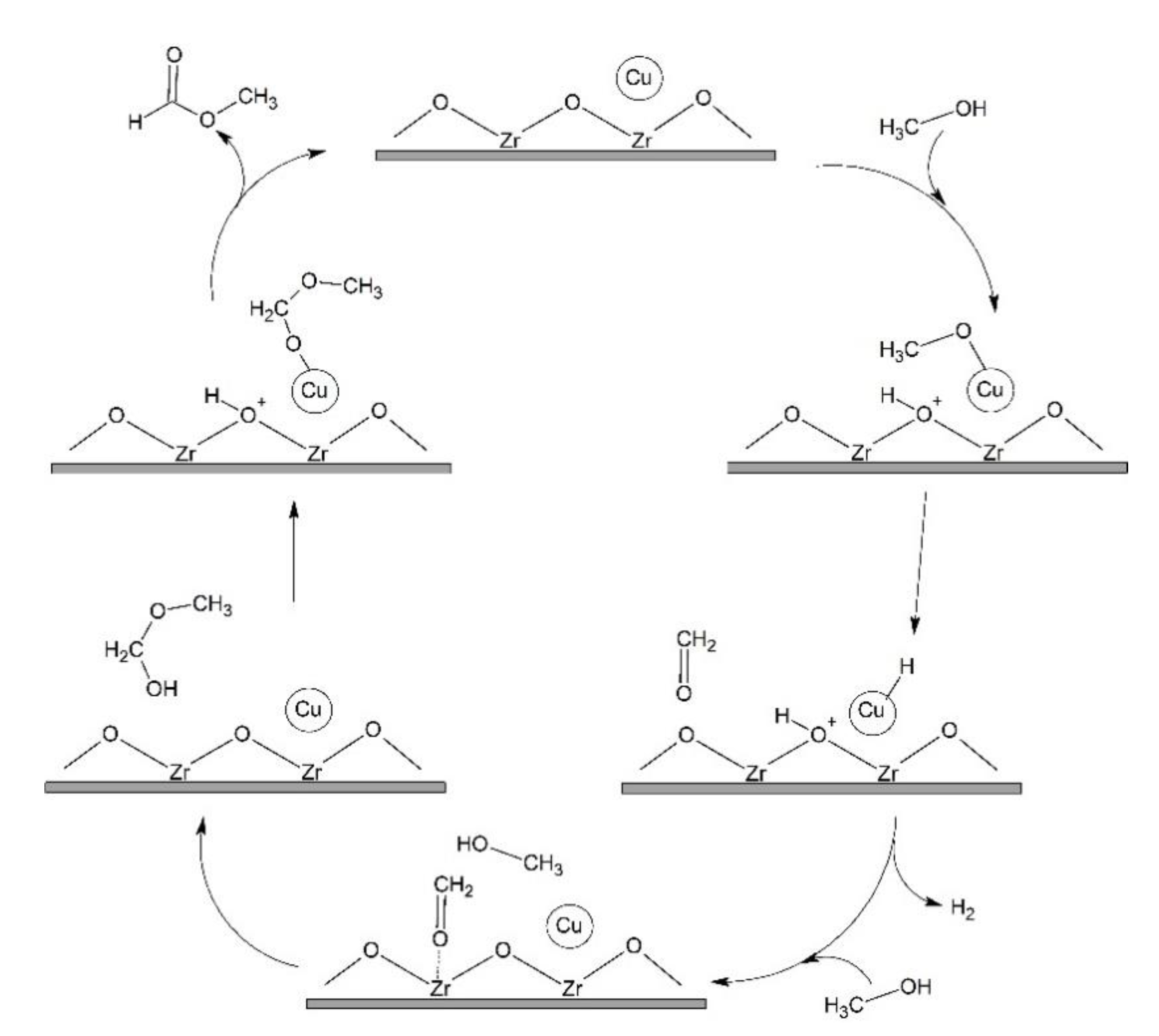
3.2.2. Mechanism
3.2.3. By-Products
3.2.4. Catalyst
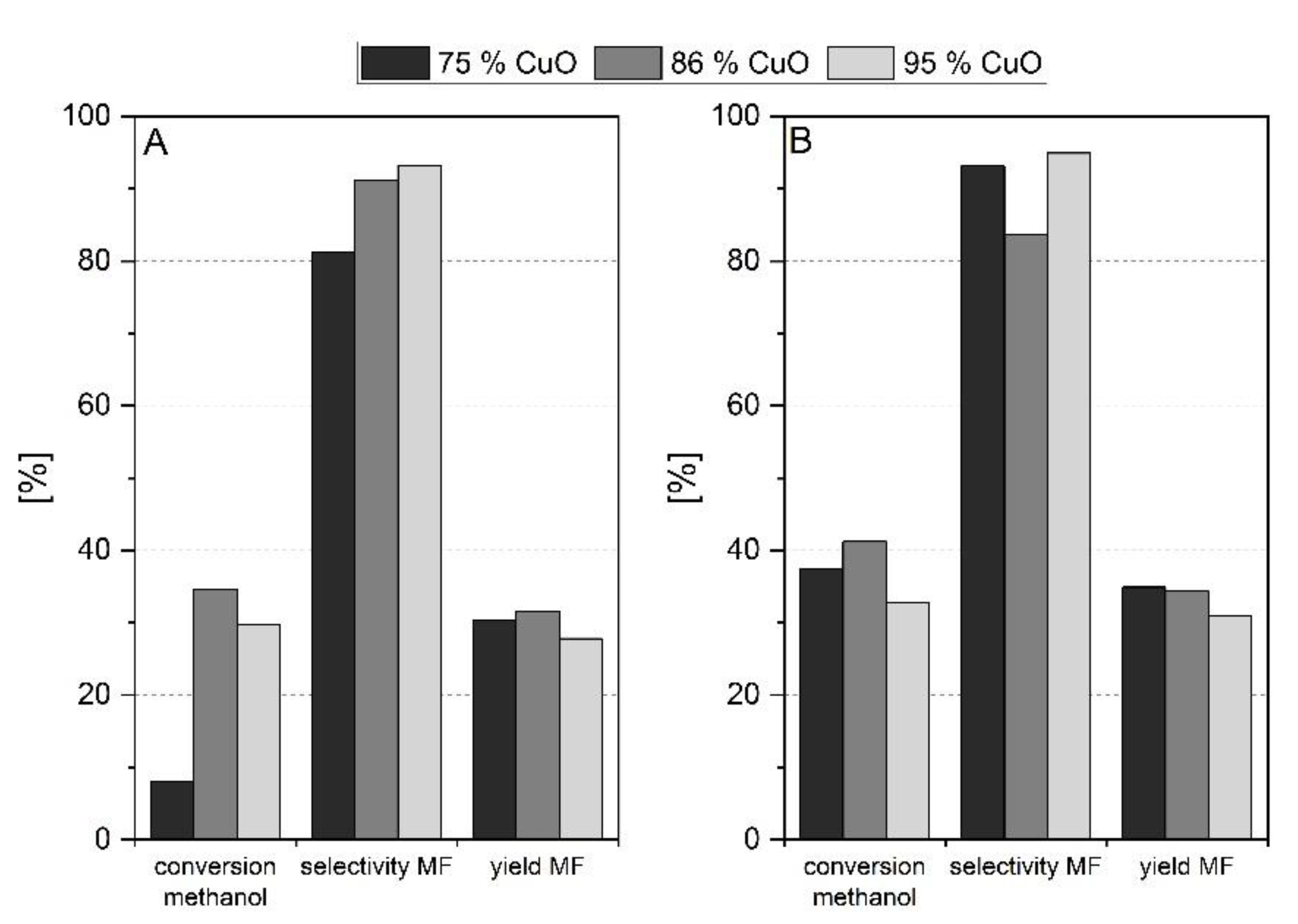
Influence of the Preparation Method
Influence of Copper Loading
Influence of Carrier Material
Addition of Further Elements
Cu-Free Catalysts
3.2.5. Reactors for Methanol Dehydrogenation
3.2.6. Coupling of Dehydrogenation and Hydrogenation Reaction



3.3. Oxidation of Methanol
Catalysts
3.4. Esterification of Methanol and Formic Acid
3.5. Hydrogenation-Condensation with Methanol
3.6. Photocatalytic Oxidation of Methanol
3.7. Electrolysis of Methanol
4. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rong, L.; Xu, Z.; Sun, J.; Guo, G. New methyl formate synthesis method: Coal to methyl formate. J. Energy Chem. 2018, 27, 238–242. [Google Scholar] [CrossRef] [Green Version]
- Hietala, J.; Vuori, A.; Johnsson, P.; Pollari, I.; Reutemann, W.; Kieczka, H. Formic Acid. Ullmann’s Encyclopedia of Industrial Chemistry; Wiley: Chichester, UK, 2010; pp. 1–22. ISBN 9783527306732. [Google Scholar]
- Bipp, H.; Kieczka, H. Formamides. Ullmann's Encyclopedia of Industrial Chemistry; Wiley: Chichester, UK, 2010; p. 5841. ISBN 3527306730. [Google Scholar]
- Jali, S.; Friedrich, H.B.; Julius, G.R. The effect of Mo(CO)6 as a co-catalyst in the carbonylation of methanol to methyl formate catalyzed by potassium methoxide under CO, syngas and H2 atmospheres. HP-IR observation of the methoxycarbonyl intermediate of Mo(CO)6. J. Mol. Catal. A Chem. 2011, 348, 63–69. [Google Scholar] [CrossRef]
- Bertau, M.; Räuchle, K.; Offermanns, H. Methanol-die Basischemikalie. Chem. Unserer. Zeit. 2015, 49, 312–329. [Google Scholar] [CrossRef]
- Klezl, P. Treibstoff für Verbrennungsmotoren und Verwendung von Methylformiat. EU Patent 91890292.5, 30 August 1995. [Google Scholar]
- Martins, J.; Brito, F.P. Alternative Fuels for Internal Combustion Engines. Energies 2020, 13, 4086. [Google Scholar] [CrossRef]
- Jacob, E. C-1 Oxygenate als nachhaltige Kraftstoffe und dren günstige Eigenschaften. In Zukünftige Kraftstoffe: Energiewende des Transports als ein weltweites Klimaziel; 1. Auflage, 2019; Maus, W., Ed.; Springer: Berlin, Germany, 2019; pp. 155–180. ISBN 978-3-662-58005-9. [Google Scholar]
- Xu, Z.-N.; Guo, G.-C.; Peng, S.-Y.; Wang, Z.-Q.; Chen, Q.-S.; Wang, M.-S.; Yao, Y.-G. A Process for Vapor-Phase Methanol Carbonylation to Methyl Formate, a Catalyst Used in the Process and a Method for Preparing the Catalyst. WO2015103851A1, 16 July 2015. [Google Scholar]
- Di Girolamo, M.; Lami, M.; Marchionna, M.; Sanfilippo, D. Methanol carbonylation to methyl formate catalyzed by strongly basic resins. Catal. Lett. 1996, 38, 127–131. [Google Scholar] [CrossRef]
- Jogunola, O.; Salmi, T.; Kangas, M.; Mikkola, J.-P. Determination of the kinetics and mechanism of methyl formate synthesis in the presence of a homogeneous catalyst. Chem. Eng. J. 2012, 203, 469–479. [Google Scholar] [CrossRef]
- Di Girolamo, M.; Marchionna, M. Acidic and basic ion exchange resins for industrial applications. J. Mol. Catal. A Chem. 2001, 177, 33–40. [Google Scholar] [CrossRef]
- Auer, H.; Dahlhaus, J.; Fischer, K.; Hammer, H.; Kellenbenz, J.; Schulz, M.; Thiel, J.; Vivari, M. Verfahren zur Kontinuierlichen Herstellung von Methylformiat. WO 01/07392 A1, 1 February 2001. [Google Scholar]
- Tonner, S.P.; Trimm, D.L.; Wainwright, M.S.; Cant, N.W. The base-catalysed carbonylation of higher alcohols. J. Mol. Catal. 1983, 18, 215–222. [Google Scholar] [CrossRef]
- Schneider, D.; Mohl, K.-D.; Schäfer, M.; Paschold, J.; Teles, J.H.; Rittinger, S. Verfahren zur Herstellung von Methylformiat durch Umsetzung von Methanol mit Kohlenmonoxid in Gegenwart eines Katalysatorsystems, das Alkaliformiat und Alkalialkoholat enthält. WO 2014/080026 A1, 30 May 2014. [Google Scholar]
- Gérard, E.; Götz, H.; Pellegrini, S.; Castanet, Y.; Mortreux, A. Epoxide-tertiary amine combinations as efficient catalysts for methanol carbonylation into methyl formate in the presence of carbon dioxide. Appl. Catal. A 1998, 297–306. [Google Scholar] [CrossRef]
- Sobotta, G. Verfahren zur Herstellung von Methylformiat. EP 0596483 A2, 11 May 1994. [Google Scholar]
- Ohyama, S.; Lee, E.S.; Aika, K.-I. Selective formation of methanol over nickel carbonyl with potassium methoxide. J. Mol. Catal. A Chem. 1999, 138, 305–309. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Ovalles, C. Anionic Group 6B Metal Carbonyls as Homogeneous Catalysts for Carbon Dioxide/Hydrogen Activation. The Production of Alkyl Formates. J. Am. Chem. Soc. 1984, 3750–3754. [Google Scholar] [CrossRef]
- He, L.; Liu, H.; Xiao, C.-X.; Kou, Y. Liquid-phase synthesis of methyl formate via heterogeneous carbonylation of methanol over a soluble copper nanocluster catalyst. Green Chem. 2008, 10, 619. [Google Scholar] [CrossRef]
- Guzman-Jimenez, I.Y.; van Hal, J.W.; Whitmire, K.H. Metal Cluster Catalysis: A Kinetic and Mechanistic Study of the Carbonylation of Methanol to Give Methyl Formate as Catalyzed by [Et4N]2[Fe3(CO)9E] (E = S, Se, Te). Organometallics 2003, 22, 1914–1922. [Google Scholar] [CrossRef]
- Iwase, Y.; Kobayashi, T.; Inazu, K.; Miyaji, A.; Baba, T. Reaction Kinetics of Methanol Carbonylation to Methyl Formate Catalyzed by CH3O− Exchange Resin. Catal. Lett. 2007, 118, 146–150. [Google Scholar] [CrossRef]
- Wang, Y.; Ruan, G.; Han, S. Dehydrocoupling of methanol to methyl formate overa a Cu/Cr2O3 catalyst. React. Kinet. Catal. Lett. 1999, 67, 305–310. [Google Scholar] [CrossRef]
- Yuan, D.-J.; Hengne, A.M.; Saih, Y.; Huang, K.-W. Nonoxidative Dehydrogenation of Methanol to Methyl Formate through Highly Stable and Reusable CuMgO-Based Catalysts. ACS Omega 2019, 4, 1854–1860. [Google Scholar] [CrossRef]
- Zhang, R.; Sun, Y.-H.; Peng, S.-Y. Dehydrogenation of methanol to methyl formate over CuO−SiO2 gel catalyst. React. Kinet. Catal. Lett. 1999, 67, 95–102. [Google Scholar] [CrossRef]
- Okamoto, Y.; Fukino, K.; Imanaka, T.; Teranishi, S. Synergy between Cu and Zn for methanol conversion over Cu-ZnO cataylsts. Chem. Lett. 1984, 13, 71–74. [Google Scholar] [CrossRef] [Green Version]
- Miura, H.; Nakahara, K.; Kitajima, T.; Shishido, T. Concerted Functions of Surface Acid-Base Pairs and Supported Copper Catalysts for Dehydrogenative Synthesis of Esters from Primary Alcohols. ACS Omega 2017, 2, 6167–6173. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Yin, H.; Feng, Y.; Wang, A. Coupling reaction between methanol dehydrogenation and maleic anhydride hydrogenation over zeolite-supported copper catalysts. Can. J. Chem. Eng. 2015, 93, 1107–1118. [Google Scholar] [CrossRef]
- Guerreiro, E.D.; Gorriz, O.F.; Rivarola, J.B.; Arrúa, L.A. Characterization of Cu/SiO2 catalysts prepared by ion exchange for methanol dehydrogenation. Appl. Catal. A 1997, 165, 259–271. [Google Scholar] [CrossRef]
- Guerreiro, E.D.; Gorriz, O.F.; Larsen, G.; Arrúa, L.A. Cu/SiO2 catalysts for methanol to methyl formate dehydrogenation. Appl. Catal. A 2000, 204, 33–48. [Google Scholar] [CrossRef]
- Sodesawa, T. Dynamic change in surface area of Cu in dehydrogenation of methanol over Cu−SiO2 catalyst prepared by ion exchange method. React. Kinet. Catal. Lett. 1984, 24, 259–264. [Google Scholar] [CrossRef]
- Sodesawa, T. Effect of support on dehydrogenation of methanol to methyl formate over Cu-containing catalysts prepared by ion exchange. React. Kinet. Catal. Lett. 1986, 32, 63–69. [Google Scholar] [CrossRef]
- Sodesawa, T. Reduction behavior of Cu−SiO2 catalyst prepared by ion exchange for selective dehydrogenation to produce methyl formate from methanol. React. Kinet. Catal. Lett. 1986, 32, 51–56. [Google Scholar] [CrossRef]
- Tonner, S.P.; Trimm, D.L.; Wainwright, M.S.; Cant, N.W. Dehydrogenation of methanol to methyl formate over copper catalysts. Ind. Eng. Chem. Prod. Res. Dev. 1984, 23, 384–388. [Google Scholar] [CrossRef]
- Jung, K.-D.; Joo, O.-S. Preparation of Cu/ZnO/M2O3 (M = Al, Cr) Catalyst to Stabilize Cu/ZnO Catalyst in Methanol Dehydrogenation. Catal. Lett. 2002, 84, 21–25. [Google Scholar] [CrossRef]
- Jung, K.-D.; Joo, O.-S.; Han, S.-H.; Uhm, S.-J.; Chung, I.-J. Deactivation of Cu/ZnO catalyst during dehydrogenation of methanol. Catal. Lett. 1995, 35, 303–311. [Google Scholar] [CrossRef]
- Jung, K.-D.; Joo, O.-S.; Han, S.-H. Structural change of Cu/ZnO by reduction of ZnO in Cu/ZnO with methanol. Catal. Lett. 2000, 68, 49–54. [Google Scholar] [CrossRef]
- Spencer, M.S. α-brass formation in copper/zinc oxide catalysts. Surf. Sci. 1987, 192, 336–343. [Google Scholar] [CrossRef]
- Sato, S.; Iijima, M.; Nakayama, T.; Sodesawa, T.; Nozaki, F. Vapor-Phase Dehydrocoupling of Methanol to Methyl Formate over CuAl2O4. J. Catal. 1997, 169, 447–454. [Google Scholar] [CrossRef]
- Matsuda, T.; Yogo, K.; Pantawong, C.; Kikuchi, E. Catalytic properties of copper-exchanged clays for the dehydrogenation of methanol to methyl formate. Appl. Catal. A 1995, 126, 177–186. [Google Scholar] [CrossRef]
- Chen, S.-C.; Cheng, W.-J.; Lin, F.-S. Process for Producing Methyl Formate. U.S. Patent 5144062, 1 September 1992. [Google Scholar]
- Horlenko, T.; Aguilo, A. Process for Producing Methyl Formate. U.S. Patent 4480122, 30 October 1984. [Google Scholar]
- Yoneoka, M. Process for Producing Methyl Formate. U.S. Patent 4319037, 9 March 1982. [Google Scholar]
- Rodriguez-Ramos, I.; Guerrero-Ruiz, A.; Rojas, M.L.; Fierro, J.L.G. Dehydrogenation of methanol to methyl formate over copper-containing perovskite-type oxides. Appl. Catal. 1991, 68, 217–228. [Google Scholar] [CrossRef]
- Iwasa, N.; Yamamoto, O.; Akazawa, T.; Ohyama, S.; Takezawa, N.; Iwasa, N.; Ohyama, S.; Takezawa, N. Dehydrogenation of Methanol to Methyl Formate over Palladium/Zinc Oxide Catalysts//Dehydrogenation of methanol to methyl formate over palladium/zinc oxide catalysts. J. Chem. Soc. Chem. Commun. 1991, 1322–1323. [Google Scholar] [CrossRef]
- Iwasa, N. New Supported Pd and Pt Alloy Catalysts for Steam Reforming and Dehydrogenation of Methanol. Top. Catal. 2003, 22, 215–224. [Google Scholar] [CrossRef]
- Yamakawa, T.; Ohnishi, T.; Shinoda, S. Methanol dehydrogenation in the liquid phase with Cu-based solid catalysts. Catal. Lett. 1994, 23, 395–401. [Google Scholar] [CrossRef]
- Guo, Y.; Lu, G.; Mo, X.; Wang, Y. Vapor phase dehydrogenation of methanol to methyl formate in the catalytic membrane reactor with Cu/SiO2/ ceramic composite membrane. Catal. Lett. 2005, 99, 105–108. [Google Scholar] [CrossRef]
- Wang, J.; Wu, Z.; Qin, Z.; Zhu, H.; Wang, R.; Wang, G.; Fan, W. Preparation Method and Application of Au-pd Bimetallic Catalyst for Preparing Methyl Formate via Selective Oxidation of Methanol. CN 2013-10099508, 26 March 2013. [Google Scholar]
- Wittstock, A.; Biener, J.; Bäumer, M. Nanoporous gold: A new material for catalytic and sensor applications. Phys. Chem. Chem. Phys. 2010, 12, 12919–12930. [Google Scholar] [CrossRef]
- Feil, F.S.; van Ommen, J.G.; Ross, J.R.H. Infrared investigation of the adsorption and reactions of methanol on a vanadium pentoxide/titania catalyst. Langmuir 1987, 3, 668–673. [Google Scholar] [CrossRef] [Green Version]
- Forzatti, P.; Tronconi, E.; Busca, G.; Tittarelli, P. Oxidation of methanol to methyl formate over V-Ti oxide catalysts. Catal. Today 1987, 1, 209–218. [Google Scholar] [CrossRef]
- Guo, Q.; Xu, C.; Yang, W.; Ren, Z.; Ma, Z.; Dai, D.; Minton, T.K.; Yang, X. Methyl Formate Production on TiO 2 (110), Initiated by Methanol Photocatalysis at 400 nm. J. Phys. Chem. C 2013, 117, 5293–5300. [Google Scholar] [CrossRef]
- Busca, G.; Elmi, A.S.; Forzatti, P. Mechanism of selective methanol oxidation over vanadium oxide-titanium oxide catalysts: A FT-IR and flow reactor study. J. Phys. Chem. 1987, 91, 5263–5269. [Google Scholar] [CrossRef]
- Forzatti, P.; Tronconi, E.; Elmi, A.S.; Busca, G. Methanol oxidation over vanadia-based catalysts. Appl. Catal. A 1997, 157, 387–408. [Google Scholar] [CrossRef]
- Kaichev, V.V.; Popova, G.Y.; Chesalov, Y.A.; Saraev, A.A.; Zemlyanov, D.Y.; Beloshapkin, S.A.; Knop-Gericke, A.; Schlögl, R.F.; Andrushkevich, T.V.; Bukhtiyarov, V.I. Selective oxidation of methanol to form dimethoxymethane and methyl formate over a monolayer V2O5/TiO2 catalyst. J. Catal. 2014, 311, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Kaichev, V.V.; Popova, G.Y.; Chesalov, Y.A.; Saraev, A.A.; Andrushkevich, T.V.; Bukhtiyarov, V.I. Active component of supported vanadium catalysts in the selective oxidation of methanol. Kinet. Catal. 2016, 57, 82–94. [Google Scholar] [CrossRef]
- Li, N.; Wang, S.; Ren, Q.; Li, S.; Sun, Y. Catalytic Mechanisms of Methanol Oxidation to Methyl Formate on Vanadia–Titania and Vanadia–Titania–Sulfate Catalysts. J. Phys. Chem. C 2016, 120, 29290–29301. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, R.; Wang, S.; Li, N.; Sima, R.; Liu, G.; Wu, P.; Zeng, G.; Li, S.; Sun, Y. Highly Efficient and Stable Vanadia–Titania–Sulfate Catalysts for Methanol Oxidation to Methyl Formate: Synthesis and Mechanistic Study. J. Phys. Chem. C 2016, 120, 6591–6600. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, G.; Shi, L.; Wu, P.; Zeng, G.; Zhang, C.; Yang, N.; Li, S.; Sun, Y.; Zhang, Y.; et al. Quantitative Conversion of Methanol to Methyl Formate on Graphene-Confined Nano-Oxides. iScience 2020, 23, 101157. [Google Scholar] [CrossRef]
- Haruta, M. New generation of gold catalysts: Nanoporous foams and tubes--is unsupported gold catalytically active? ChemPhysChem 2007, 8, 1911–1913. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhu, C.; Yang, G.; Sun, Y.; Wang, D.; Liu, J. High-performance microstructured Au-Ag bimetallic catalyst for oxidative coupling of methanol to methyl formate. Catal. Commun. 2019, 129, 105741. [Google Scholar] [CrossRef]
- Han, C.; Yang, X.; Gao, G.; Wang, J.; Lu, H.; Liu, J.; Tong, M.; Liang, X. Selective oxidation of methanol to methyl formate on catalysts of Au–Ag alloy nanoparticles supported on titania under UV irradiation. Green Chem. 2014, 16, 3603–3615. [Google Scholar] [CrossRef]
- Wang, R.; Wu, Z.; Chen, C.; Qin, Z.; Zhu, H.; Wang, G.; Wang, H.; Wu, C.; Dong, W.; Fan, W.; et al. Graphene-supported Au-Pd bimetallic nanoparticles with excellent catalytic performance in selective oxidation of methanol to methyl formate. Chem. Commun. 2013, 49, 8250–8252. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-B.; Shi, R.-P.; Qin, Z.; Liu, H.; Li, Z.-K.; Zhu, H.; Zhao, Y.-X.; Wang, J. Selective oxidation of methanol to methyl formate over bimetallic Au-Pd nanoparticles supported on SiO2. J. Fuel Chem. Technol. 2019, 47, 780–790. [Google Scholar] [CrossRef]
- Whiting, G.T.; Kondrat, S.A.; Hammond, C.; Dimitratos, N.; He, Q.; Morgan, D.J.; Dummer, N.F.; Bartley, J.K.; Kiely, C.J.; Taylor, S.H.; et al. Methyl Formate Formation from Methanol Oxidation Using Supported Gold–Palladium Nanoparticles. ACS Catal. 2015, 5, 637–644. [Google Scholar] [CrossRef]
- Colmenares, J.C.; Lisowski, P.; Łomot, D.; Chernyayeva, O.; Lisovytskiy, D. Sonophotodeposition of bimetallic photocatalysts Pd-Au/TiO2: Application to selective oxidation of methanol to methyl formate. ChemSusChem 2015, 8, 1676–1685. [Google Scholar] [CrossRef]
- Baldovino-Medrano, V.G.; Pollefeyt, G.; Bliznuk, V.; Van Driessche, I.; Gaigneaux, E.M.; Ruiz, P.; Wojcieszak, R. Synergetic Behavior of TiO2-Supported Pd(z)Pt(1−z) Catalysts in the Green Synthesis of Methyl Formate. ChemCatChem 2016, 8, 1157–1166. [Google Scholar] [CrossRef]
- Lisowski, P.; Colmenares, J.C.; Łomot, D.; Chernyayeva, O.; Lisovytskiy, D. Preparation by sonophotodeposition method of bimetallic photocatalysts Pd–Cu/TiO2 for sustainable gaseous selective oxidation of methanol to methyl formate. J. Mol. Catal. A Chem. 2016, 411, 247–256. [Google Scholar] [CrossRef]
- Wisniewska, J.; Yang, C.-M.; Ziolek, M. Changes in bimetallic silver–platinum catalysts during activation and oxidation of methanol and propene. Catal. Today 2019, 333, 89–96. [Google Scholar] [CrossRef]
- Wojcieszak, R.; Gaigneaux, E.M.; Ruiz, P. Direct Methyl Formate Formation from Methanol over Supported Palladium Nanoparticles at Low Temperature. ChemCatChem 2013, 5, 339–348. [Google Scholar] [CrossRef]
- Wojcieszak, R.; Karelovic, A.; Gaigneaux, E.M.; Ruiz, P. Oxidation of methanol to methyl formate over supported Pd nanoparticles: Insights into the reaction mechanism at low temperature. Catal. Sci. Technol. 2014, 4, 3298–3305. [Google Scholar] [CrossRef]
- Li, W.; Liu, H.; Iglesia, E. Structures and properties of zirconia-supported ruthenium oxide catalysts for the selective oxidation of methanol to methyl formate. J. Phys. Chem. B 2006, 110, 23337–23342. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Li, W.; Liu, H. Effect of treatment temperature on structures and properties of zirconia-supported ruthenium oxide catalysts for selective oxidation of methanol to methyl formate. Catal. Today 2012, 183, 58–64. [Google Scholar] [CrossRef]
- Liu, J.; Zhan, E.; Cai, W.; Li, J.; Shen, W. Methanol Selective Oxidation to Methyl Formate over ReOx/CeO2 Catalysts. Catal. Lett. 2008, 120, 274–280. [Google Scholar] [CrossRef]
- Indu, B.; Ernst, W.R.; Gelbaum, L.T. Methanol-formic acid esterification equilibrium in sulfuric acid solutions: Influence of sodium salts. Ind. Eng. Chem. Res. 1993, 32, 981–985. [Google Scholar] [CrossRef]
- Westhues, N.; Belleflamme, M.; Klankermayer, J. Base-Free Hydrogenation of Carbon Dioxide to Methyl Formate with a Molecular Ruthenium-Phosphine Catalyst. ChemCatChem 2019, 11, 5269–5274. [Google Scholar] [CrossRef]
- Süss-Fink, G.; Soulié, J.-M.; Rheinwald, G.; Stoeckli-Evans, H.; Sasaki, Y. Hydrocondensation of Carbon Dioxide with Methanol Catalyzed by Anionic Ruthenium Complexes: Isolation, Structural Characterization, and Catalytic Implications of the Dinuclear Anion [Ru2(CO)4(µ2-η2-CO2CH3)2(µ2-OCH3)Cl2]−. Organometallicy 1996, 15, 3416–3422. [Google Scholar] [CrossRef]
- Jessop, P.G.; Hsiao, Y.; Ikariya, T.; Noyori, R. Methyl formate synthesis by hydrogenation of supercritical carbon dioxide in the presence of methanol. J. Chem. Soc. Chem. Commun. 1995, 707. [Google Scholar] [CrossRef]
- Kröcher, O.; Köppel, R.A.; Baiker, A. Highly active ruthenium complexes with bidentate phosphine ligands for the solvent-free catalytic synthesis of N,N-dimethylformamide and methyl formate. Chem. Commun. 1997, 453–454. [Google Scholar] [CrossRef]
- Federsel, C.; Boddien, A.; Jackstell, R.; Jennerjahn, R.; Dyson, P.J.; Scopelliti, R.; Laurenczy, G.; Beller, M. A well-defined iron catalyst for the reduction of bicarbonates and carbon dioxide to formates, alkyl formates, and formamides. Angew. Chem. Int. Ed. Engl. 2010, 49, 9777–9780. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.; Linehan, J.C.; Karkamkar, A.J.; van der Eide, E.; Heldebrant, D.J. Homogeneous hydrogenation of CO2 to methyl formate utilizing switchable ionic liquids. Inorg. Chem. 2014, 53, 9849–9854. [Google Scholar] [CrossRef]
- Sun, R.; Kann, A.; Hartmann, H.; Besmehn, A.; Hausoul, P.J.C.; Palkovits, R. Direct Synthesis of Methyl Formate from CO2 With Phosphine-Based Polymer-Bound Ru Catalysts. ChemSusChem 2019, 12, 3278–3285. [Google Scholar] [CrossRef]
- Krocher, O.; Kröcher, O.; Köppel, R.A.; Fröba, M.; Baiker, A. Silica Hybrid Gel Catalysts Containing Group(VIII) Transition Metal Complexes: Preparation, Structural, and Catalytic Properties in the Synthesis of N,N-Dimethylformamide and Methyl Formate from Supercritical Carbon Dioxide//Silica Hybrid Gel Catalysts Containing Group(VIII) Transition Metal Complexes: Preparation, Structural, and Catalytic Properties in the Synthesis ofN,N-Dimethylformamide and Methyl Formate from Supercritical Carbon Dioxide. J. Catal. 1998, 178, 284–298. [Google Scholar] [CrossRef]
- Yu, K.M.K.; Yeung, C.M.Y.; Tsang, S.C. Carbon dioxide fixation into chemicals (methyl formate) at high yields by surface coupling over a Pd/Cu/ZnO nanocatalyst. J. Am. Chem. Soc. 2007, 129, 6360–6361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerry Yu, K.M.; Tsang, S.C. A Study of Methyl Formate Production from Carbon Dioxide Hydrogenation in Methanol over a Copper Zinc Oxide Catalyst. Catal. Lett. 2011, 141, 259–265. [Google Scholar] [CrossRef]
- Wu, C.; Zhang, Z.; Zhu, Q.; Han, H.; Yang, Y.; Han, B. Highly efficient hydrogenation of carbon dioxide to methyl formate over supported gold catalysts. Green Chem. 2015, 17, 1467–1472. [Google Scholar] [CrossRef]
- Corral-Pérez, J.J.; Bansode, A.; Praveen, C.S.; Kokalj, A.; Reymond, H.; Comas-Vives, A.; Vande Vondele, J.; Copéret, C.; von Rohr, P.R.; Urakawa, A. Decisive Role of Perimeter Sites in Silica-Supported Ag Nanoparticles in Selective Hydrogenation of CO2 to Methyl Formate in the Presence of Methanol. J. Am. Chem. Soc. 2018, 140, 13884–13891. [Google Scholar] [CrossRef] [Green Version]
- Corral-Pérez, J.J.; Copéret, C.; Urakawa, A. Lewis acidic supports promote the selective hydrogenation of carbon dioxide to methyl formate in the presence of methanol over Ag catalysts. J. Catal. 2019, 380, 153–160. [Google Scholar] [CrossRef]
- Liu, G.; Wang, L.; Yang, H.G.; Cheng, H.-M.; Lu, G.Q. Titania-based photocatalysts—Crystal growth, doping and heterostructuring. J. Mater. Chem. 2010, 20, 831–843. [Google Scholar] [CrossRef]
- Czelej, K.; Cwieka, K.; Colmenares, J.C.; Kurzydlowski, K.J.; Xu, Y.-J. Toward a Comprehensive Understanding of Enhanced Photocatalytic Activity of the Bimetallic PdAu/TiO2 Catalyst for Selective Oxidation of Methanol to Methyl Formate. ACS Appl. Mater. Interfaces 2017, 9, 31825–31833. [Google Scholar] [CrossRef]
- Li, C.; Yang, X.; Gao, G.; Li, Y.; Zhang, W.; Chen, X.; Su, H.; Wang, S.; Wang, Z. Copper on the inner surface of mesoporous TiO 2 hollow spheres: A highly selective photocatalyst for partial oxidation of methanol to methyl formate. Catal. Sci. Technol. 2019, 9, 6240–6252. [Google Scholar] [CrossRef]
- Kishi, R.; Ogihara, H.; Yoshida-Hirahara, M.; Shibanuma, K.; Yamanaka, I.; Kurokawa, H. Green Synthesis of Methyl Formate via Electrolysis of Pure Methanol. ACS Sustain. Chem. Eng. 2020, 8, 11532–11540. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gasoline | Methanol | Methyl Formate | |
|---|---|---|---|
| RON | 97.7 | 108.7–115 | 115 |
| MON | 89 | 88.6 | 114.8 |
| ρ [kg/m3] | 720–780 | 800 | 957 |
| TBoiling [°C] | 25–210 | 64.6 | 31.5 |
| Flash Point [°C] | −40 | 12 | −19 |
| Lower heating value (LHV) [MJ/kg] | 44 | 19.7 | 15.8 |
| Dispersity [%] | Selectivity MF [%] | TOF [s−1] | |||
|---|---|---|---|---|---|
| 190 °C | 250 °C | 190 °C | 250 °C | ||
| 1.5 wt.% Cu/SiO2 | 75.5 | 83 | 25 | 0.033 | 0.086 |
| 1.5 wt.% Cu/ZrO2 | 60.7 | 83 | 63 | 0.018 | 0.10 |
| 1.5 wt.% Cu/TiO2 | 23.0 | 92 | 91 | 0.0077 | 0.18 |
| Reaction Temperature [°C] | Methanol Conversion [%] | Selectivity MF [%] | Selectivity CO [%] | Selectivity CO2 [%] | Selectivity DME [%] |
|---|---|---|---|---|---|
| 270 | 26.2 | 95.4 | 4.6 | 0 | 0 |
| 310 | 63.6 | 57.6 | 15.9 | 6.8 | 20.3 |
| Noble Metal Composition | Support | Temperature [°C] | Methanol Conversion [%] | Selectivity MF [%] | Reference |
|---|---|---|---|---|---|
| Au1.0-Ag0.2 | Al-fiber | 170 | 42.0 | 82.0 | [62] |
| Au1.0-Ag1.0 | TiO2 | 35; UV | 82.5 | 87.5 | [63] |
| Au2.0-Pd1.0 | Graphene | 70 | 90.2 | 100.0 | [64] |
| Au2.0-Pd1.0 | SiO2 | 130 | 57.0 | 72.7 | [65] |
| Au1-0-Pd1.0 | TiO2 | 30 | 15.0 | 70.0 | [66] |
| Au0.5-Pd0.5 | TiO2 | 30; UV | 85.0 | 70.0 | [67] |
| Pd0.65-Pt0.35 | TiO2 | 50 | 78.0 | 67.0 | [68] |
| Pd1.0-Cu1.0 | SiO2 | 30; UV | 53.0 | ~80.0 | [69] |
| Ag2.0-Pt0.5 | SiO2 | 100 | 99.5 | 58.9 | [70] |
| Catalyst | Reaction Condition | TON | MF Selectivity | Reference | |
|---|---|---|---|---|---|
| homogeneous | RuCl2(PMe3)4 | 80 °C, 64 h, 20 MPa | 3500 | 34% | [79] |
| [Ru(N-triphosCy)(tmm)] | 60 °C, 18 h, 12 MPa | 9542 | 94% | [77] | |
| [RuCl2(dppe)2] | 80 °C, 15.5 h, 13 MPa | 12,900 | n.d. | [80] | |
| Fe(BF4)2/triphos 1 | 100 °C, 20 h, 3 MPa | 292 | 56% | [81] | |
| RuCl2(PPh3)3/DBU | 140 °C, 40 h, 2 MPa | 1510 | 59% | [82] | |
| pDPPE | 160 °C, 12 h, 8 MPa | 3401 | n.d. | [83] | |
| RuCl2(PMe2(CH2)2Si(OEt)3]3 | 100 °C, 64 h, 13 MPa | 3180 | n.d. | [84] | |
| heterogeneous | Pd/Cu/ZnO/Al2O3 | 140 °C, 0.5 h, 14 MPa | 109 | n.d. | [85,86] |
| Cu/ZnO/Al2O3 | 140 °C, 0.5 h, 14 MPa | 131 | n.d. | [86] | |
| Au/ZrO2, Au/CeO2, Au/TiO2 | 140 °C, 1 h, 16 MPa | 204 | >99.9% | [87] | |
| Ag/SiO2, Au/SiO2, Cu/SiO2 | 140–260 °C, 3 MPa | n.d. | >99.9% | [88,89] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaiser, D.; Beckmann, L.; Walter, J.; Bertau, M. Conversion of Green Methanol to Methyl Formate. Catalysts 2021, 11, 869. https://doi.org/10.3390/catal11070869
Kaiser D, Beckmann L, Walter J, Bertau M. Conversion of Green Methanol to Methyl Formate. Catalysts. 2021; 11(7):869. https://doi.org/10.3390/catal11070869
Chicago/Turabian StyleKaiser, Doreen, Luise Beckmann, Jan Walter, and Martin Bertau. 2021. "Conversion of Green Methanol to Methyl Formate" Catalysts 11, no. 7: 869. https://doi.org/10.3390/catal11070869