
Solid-State Redox Kinetics of CeO2 in Two-Step Solar CH4 Partial Oxidation and Thermochemical CO2 Conversion


Abstract
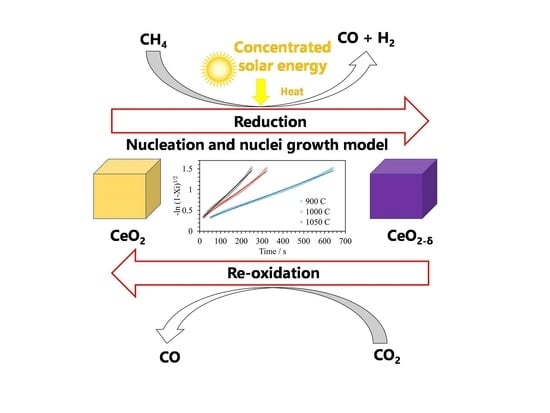
:
1. Introduction
2. Results and Discussion
3. Experimental
3.1. Material Preparation
3.2. Characterization of Materials
3.3. Redox Experiments
3.4. Kinetic Data Processing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Nomenclature
| δ | Amount of O or CO evolved during reduction or oxidation (mol·mol−1) |
| A | Pre-exponential factor (s−1) |
| Ea | Activation energy (kJ·mol−1) |
| k | Temperature-dependent overall rate constant of the reaction (s−1) |
| MCeO2 | Molar mass of CeO2 (kg·mol−1) |
| Mo | Molar mass of oxygen atom (kg·mol−1) |
| m | Initial amount of the material (kg) |
| Δmt | Mass loss during reduction (or gain during re-oxidation) at a specific time (kg) |
| Δm | Total mass variation during the reaction (kg) |
| n | Avrami exponent indicative of the reaction mechanism and growth dimension |
| t | time (s) |
| Xi | Extent of conversion |
References
- Furler, P.; Scheffe, J.R.; Steinfeld, A. Syngas production by simultaneous splitting of H2O and CO2 via ceria redox reactions in a high-temperature solar reactor. Energy Environ. Sci. 2012, 5, 6098–6103. [Google Scholar] [CrossRef]
- Scheffe, J.R.; Steinfeld, A. Thermodynamic analysis of cerium-based oxides for solar thermochemical fuel production. Energy Fuels 2012, 26, 1928–1936. [Google Scholar] [CrossRef]
- Chueh, W.C.; Falter, C.; Abbott, M.; Scipio, D.; Furler, P.; Haile, S.M.; Steinfeld, A. High-flux solar-driven thermochemical dissociation of CO2 and H2O using nonstoichiometric ceria. Science 2010, 330, 1797–1801. [Google Scholar] [CrossRef] [Green Version]
- Haeussler, A.; Abanades, S.; Jouannaux, J.; Julbe, A. Non-stoichiometric redox active perovskite materials for solar thermochemical fuel production: A review. Catalysts 2018, 8, 611. [Google Scholar] [CrossRef] [Green Version]
- Abanades, S. Metal oxides applied to thermochemical water-splitting for hydrogen production using concentrated solar energy. ChemEngineering 2019, 3, 63. [Google Scholar] [CrossRef] [Green Version]
- Charvin, P.; Abanades, S.; Bêche, E.; Lemont, F.; Flamant, G. Hydrogen production from mixed cerium oxides via three-step water-splitting cycles. Solid State Ionics 2009, 180, 1003–1010. [Google Scholar] [CrossRef]
- Haeussler, A.; Abanades, S.; Jouannaux, J.; Drobek, M.; Ayral, A.; Julbe, A. Recent progress on ceria doping and shaping strategies for solar thermochemical water and CO2 splitting cycles. AIMS Mater. Sci. 2019, 6, 657–684. [Google Scholar] [CrossRef]
- Nair, M.M.; Abanades, S. Insights into the Redox Performance of non-stoichiometric lanthanum manganite perovskites for solar thermochemical CO2 splitting. ChemistrySelect 2016, 1, 4449–4457. [Google Scholar] [CrossRef]
- Nair, M.M.; Abanades, S. Experimental screening of perovskite oxides as efficient redox materials for solar thermochemical CO2 conversion. Sustain. Energy Fuels 2018, 2, 843–854. [Google Scholar] [CrossRef]
- Haeussler, A.; Abanades, S.; Julbe, A.; Jouannaux, J.; Cartoixa, B. Solar thermochemical fuel production from H2O and CO2 splitting via two-step redox cycling of reticulated porous ceria structures integrated in a monolithic cavity-type reactor. Energy 2020, 201, 117649. [Google Scholar] [CrossRef]
- Haeussler, A.; Abanades, S.; Julbe, A.; Jouannaux, J.; Cartoixa, B. Two-step CO2 and H2O splitting using perovskite-coated ceria foam for enhanced green fuel production in a porous volumetric solar reactor. J. CO2 Util. 2020, 41, 101257. [Google Scholar] [CrossRef]
- Chuayboon, S.; Abanades, S. Solar-driven chemical looping methane reforming using ZnO oxygen carrier for syngas and Zn production in a cavity-type solar reactor. Catalysts 2020, 10, 1356. [Google Scholar] [CrossRef]
- Zhang, Z.; André, L.; Abanades, S. Experimental assessment of oxygen exchange capacity and thermochemical redox cycle behavior of Ba and Sr series perovskites for solar energy storage. Sol. Energy 2016, 134, 494–502. [Google Scholar] [CrossRef]
- Vieten, J.; Bulfin, B.; Huck, P.; Horton, M.; Guban, D.; Zhu, L.; Lu, Y.; Persson, K.A.; Roeb, M.; Sattler, C. Materials design of perovskite solid solutions for thermochemical applications. Energy Environ. Sci. 2019, 12, 1369–1384. [Google Scholar] [CrossRef]
- Vieten, J.; Bulfin, B.; Call, F.; Lange, M.; Schmucker, M.; Francke, A.; Roeb, M.; Sattler, C. Perovskite oxides for application in thermochemical air separation and oxygen storage. J. Mater. Chem. A 2016, 4, 13652–13659. [Google Scholar] [CrossRef]
- Sarshar, Z.; Kleitz, F.; Kaliaguine, S. Novel oxygen carriers for chemical looping combustion: La1-xCexBO3(B = Co, Mn) perovskites synthesized by reactive grinding and nanocasting. Energy Environ. Sci. 2011, 4, 4258–4269. [Google Scholar] [CrossRef]
- Zeng, L.; Cheng, Z.; Fan, J.A.; Fan, L.; Gong, J. Metal oxide redox chemistry for chemical looping processes. Nat. Rev. Chem. 2018, 2, 349–364. [Google Scholar] [CrossRef]
- Muhich, C.L.; Evanko, B.W.; Weston, K.C.; Lichty, P.; Liang, X.; Martinek, J.; Musgrave, C.B.; Weimer, A.W. Efficient generation of H2 by splitting water with an isothermal redox cycle. Science 2013, 341, 540–542. [Google Scholar] [CrossRef]
- Krenzke, P.T.; Davidson, J.H. Thermodynamic analysis of syngas production via the solar thermochemical cerium oxide redox cycle with methane-driven reduction. Energy Fuels 2014, 28, 4088–4095. [Google Scholar] [CrossRef]
- Krenzke, P.T.; Fosheim, J.R.; Davidson, J.H. Solar fuels via chemical-looping reforming. Sol. Energy 2017, 156, 48–72. [Google Scholar] [CrossRef]
- Nair, M.M.; Abanades, S. Tailoring hybrid nonstoichiometric ceria redox cycle for combined solar methane reforming and thermochemical conversion of CO2/H2O. Energy Fuels 2016, 30, 6050–6058. [Google Scholar] [CrossRef]
- Chuayboon, S.; Abanades, S.; Rodat, S. Solar chemical looping reforming of methane combined with isothermal H2O/CO2 splitting using ceria oxygen carrier for syngas production. J. Energy Chem. 2020, 41, 60–72. [Google Scholar] [CrossRef] [Green Version]
- Chuayboon, S.; Abanades, S.; Rodat, S. Syngas production via solar-driven chemical looping methane reforming from redox cycling of porous foam in a volumetric solar reactor. Chem. Eng. J. 2019, 356, 756–770. [Google Scholar] [CrossRef]
- Chuayboon, S.; Abanades, S.; Rodat, S. Stepwise solar methane reforming and water-splitting via lattice oxygen transfer in iron and cerium oxides. Energy Technol. 2020, 8, 1900415. [Google Scholar] [CrossRef]
- Chuayboon, S.; Abanades, S.; Rodat, S. High-purity and clean syngas and hydrogen production from two-step CH4 reforming and H2O splitting through isothermal ceria redox cycle using concentrated sunlight. Front. Energy Res. 2020, 8, 128. [Google Scholar] [CrossRef]
- Bulfin, B.; Lowe, A.J.; Keogh, K.A.; Murphy, B.E.; Lubben, O.; Krasnikov, S.A.; Shvets, I.V. Analytical model of CeO2 oxidation and reduction. J. Phys.Chem. C 2013, 117, 24129–24137. [Google Scholar] [CrossRef]
- Zhao, Z.; Uddi, M.; Tsvetkov, N.; Yildiz, B.; Ghoniem, A.F. Redox kinetics study of fuel reduced ceria for chemical-looping water splitting. J. Phys. Chem. C 2016, 120, 16271–16289. [Google Scholar] [CrossRef] [Green Version]
- Hosseini, S.Y.; Khosravi-Nikou, M.R.; Shariati, A. Kinetic study of the reduction step for chemical looping steam methane reforming by CeO2-Fe2O3 oxygen carriers. Chem. Eng. Technol. 2020, 43, 540–552. [Google Scholar] [CrossRef]
- Leveque, G.; Abanades, S. Design and operation of a solar-driven thermogravimeter for high temperature kinetic analysis of solid-gas thermochemical reactions in controlled atmosphere. Sol. Energy 2014, 105, 225–235. [Google Scholar] [CrossRef]
- Ackermann, S.; Sauvin, L.; Castiglioni, R.; Rupp, J.L.M.; Scheffe, J.R.; Steinfeld, A. Kinetics of CO2 reduction over nonstoichiometric ceria. J. Phys. Chem. C 2015, 119, 16452–16461. [Google Scholar] [CrossRef] [Green Version]
- Nair, M.M.; Abanades, S. Solid-state reoxidation kinetics of A/B-site substituted LaMnO3 during solar thermochemical CO2 conversion. Energy Technol. 2021, 9, 2000885. [Google Scholar] [CrossRef]
- Monazam, E.R.; Breault, R.W.; Siriwardane, R. Kinetics of hematite to wustite by hydrogen for chemical looping combustion. Energy Fuels 2014, 28, 5406–5414. [Google Scholar] [CrossRef]
- Kharatyan, S.L.; Chatilyan, H.A.; Manukyan, K.V. Kinetics and mechanism of nickel oxide reduction by methane. J. Phys. Chem. C 2019, 123, 21513–21521. [Google Scholar] [CrossRef]
- Dudek, R.B.; Tian, Y.; Jin, G.; Blivin, M.; Li, F. Reduction kinetics of perovskite oxides for selective hydrogen combustion in the context of olefin production. Energy Technol. 2020, 8, 1900738. [Google Scholar] [CrossRef]
- Dubey, A.K.; Samanta, A.; Sarkar, P.; Dey, R.; Saxena, C.K. Performance and kinetic evaluation of synthesized CuO/SBA-15 oxygen carrier for chemical looping with oxygen uncoupling. Energy Technol. 2019, 7, 1900407. [Google Scholar] [CrossRef]
- Hossain, M.M.; de Lasa, H.I. Reduction and oxidation kinetics of Co-Ni/Al2O3 oxygen carrier involved in a chemical-looping combustion cycles. Chem. Eng. Sci. 2010, 65, 98–106. [Google Scholar] [CrossRef]
- Dai, X.; Cheng, J.; Li, Z.; Liu, M.; Ma, Y.; Zhang, X. Reduction kinetics of lanthanum ferrite perovskite for the production of synthesis gas by chemical looping methane reforming. Chem. Eng. Sci. 2016, 153, 236–245. [Google Scholar] [CrossRef]
- Abanades, S.; Flamant, G. Hydrogen production from solar thermal dissociation of methane in a high-temperature fluid-wall chemical reactor. Chem. Eng. Process. Process Intensif. 2008, 47, 490–498. [Google Scholar] [CrossRef]
- Abanades, S.; Kimura, H.; Otsuka, H. Hydrogen production from CO2-free thermal decomposition of methane: Design and on-sun testing of a tube-type solar thermochemical reactor. Fuel Process. Technol. 2014, 122, 153–162. [Google Scholar] [CrossRef]
- Rodat, S.; Abanades, S.; Flamant, G. High temperature solar methane dissociation in a multi-tubular cavity-type reactor in the temperature range 1823–2073 K. Energy Fuels 2009, 23, 2666–2674. [Google Scholar] [CrossRef]
- Hancock, J.D.; Sharp, J.H. Method of comparing solid-state kinetic data and its application to the decomposition of Kaolinite, Brucite, and BaCO3. J. Am. Ceram. Soc. 1972, 55, 74. [Google Scholar] [CrossRef]
- Zhou, Z.; Han, L.; Bollas, G.M. Kinetics of NiO reduction by H2 and Ni oxidation at conditions relevant to chemical-looping combustion and reforming. Int. J. Hydrog. Energy 2014, 39, 8535–8556. [Google Scholar] [CrossRef]
- Plascencia, G.; Utigard, T.A. The reduction of Tokyo and Sinter 75 nickel oxides with hydrogen. Chem. Eng. Sci. 2009, 64, 3879–3888. [Google Scholar] [CrossRef]
- Utigard, T.A.; Wu, M.; Plascencia, G.; Martin, T. Reduction kinetics of Goro nickel oxide using hydrogen. Chem. Eng. Sci. 2005, 60, 2061–2068. [Google Scholar] [CrossRef]
- Abad, A.; Garcia-Labiano, F.; de Diego, L.F.; Gayan, P.; Adanez, J. Reduction kinetics of Cu-, Ni-, and Fe-based oxygen carriers using syngas (CO+H2) for chemical-looping combustion. Energy Fuels 2007, 21, 1843–1853. [Google Scholar] [CrossRef] [Green Version]
- Abad, A.; Adanez, J.; Garcia-Labiano, F.; de Diego, L.F.; Gayan, P.; Celaya, J. Mapping of the range of operational conditions for Cu-, Fe-, and Ni-based oxygen carriers in chemical-looping combustion. Chem. Eng. Sci. 2007, 62, 533–549. [Google Scholar] [CrossRef] [Green Version]
- Ksepko, E.; Sciazko, M.; Babinski, P. Studies on the redox reaction kinetics of Fe2O3-CuO/Al2O3 and Fe2O3/TiO2 oxygen carriers. Appl. Energy 2014, 115, 374–383. [Google Scholar] [CrossRef]
- Ksepko, E.; Babinski, P.; Evdou, A.; Nalbandian, L. Studies on the redox reaction kinetics of selected, naturally occurring oxygen carrier. J. Therm. Anal. Calorim. 2016, 124, 137. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Model | T (°C) | Rate Constant (s−1) | Ea (kJ·mol−1) a | A (s−1) b | |||
|---|---|---|---|---|---|---|---|
| Reduction | Re-Oxidation | Reduction | Re-Oxidation | Reduction | Re-Oxidation | ||
| 2D NGM | 900 | 1.9 × 10−3 | 2.7 × 10−3 | 79.61 | 10.5 | 6.69 | 127.0 |
| 1000 | 3.6 × 10−3 | 2.9 × 10−3 | |||||
| 1050/1070 | 4.8 × 10−3 | 3.1 × 10−3 | |||||
| 3D NGM | 900 | 2.4 × 10−3 | 3.5 × 10−3 | 80.28 | 8.2 | 9.07 | 123.6 |
| 1000 | 4.6 × 10−3 | 3.7 × 10−3 | |||||
| 1050/1070 | 6.1 × 10−3 | 3.9 × 10−3 | |||||
| 2D SCM | 900 | 1.1 × 10−3 | 1.5 × 10−3 | 76.68 | 9.5 | 2.86 | 252.9 |
| 1000 | 2.0 × 10−3 | 1.6 × 10−3 | |||||
| 1050/1070 | 2.7 × 10−3 | 1.7 × 10−3 | |||||
| 3D SCM | 900 | 8.0 × 10−4 | 1.2 × 10−3 | 83.49 | 11.7 | 4.21 | 252.3 |
| 1000 | 1.6 × 10−3 | 1.3 × 10−3 | |||||
| 1050/1070 | 2.1 × 10−3 | 1.4 × 10−3 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nair, M.M.; Abanades, S. Solid-State Redox Kinetics of CeO2 in Two-Step Solar CH4 Partial Oxidation and Thermochemical CO2 Conversion. Catalysts 2021, 11, 723. https://doi.org/10.3390/catal11060723
Nair MM, Abanades S. Solid-State Redox Kinetics of CeO2 in Two-Step Solar CH4 Partial Oxidation and Thermochemical CO2 Conversion. Catalysts. 2021; 11(6):723. https://doi.org/10.3390/catal11060723
Chicago/Turabian StyleNair, Mahesh Muraleedharan, and Stéphane Abanades. 2021. "Solid-State Redox Kinetics of CeO2 in Two-Step Solar CH4 Partial Oxidation and Thermochemical CO2 Conversion" Catalysts 11, no. 6: 723. https://doi.org/10.3390/catal11060723