
Eco-Friendly and Sustainable Process for Converting Hydrous Bioethanol to Butanol



Abstract
:
1. Introduction
2. Results and Discussion
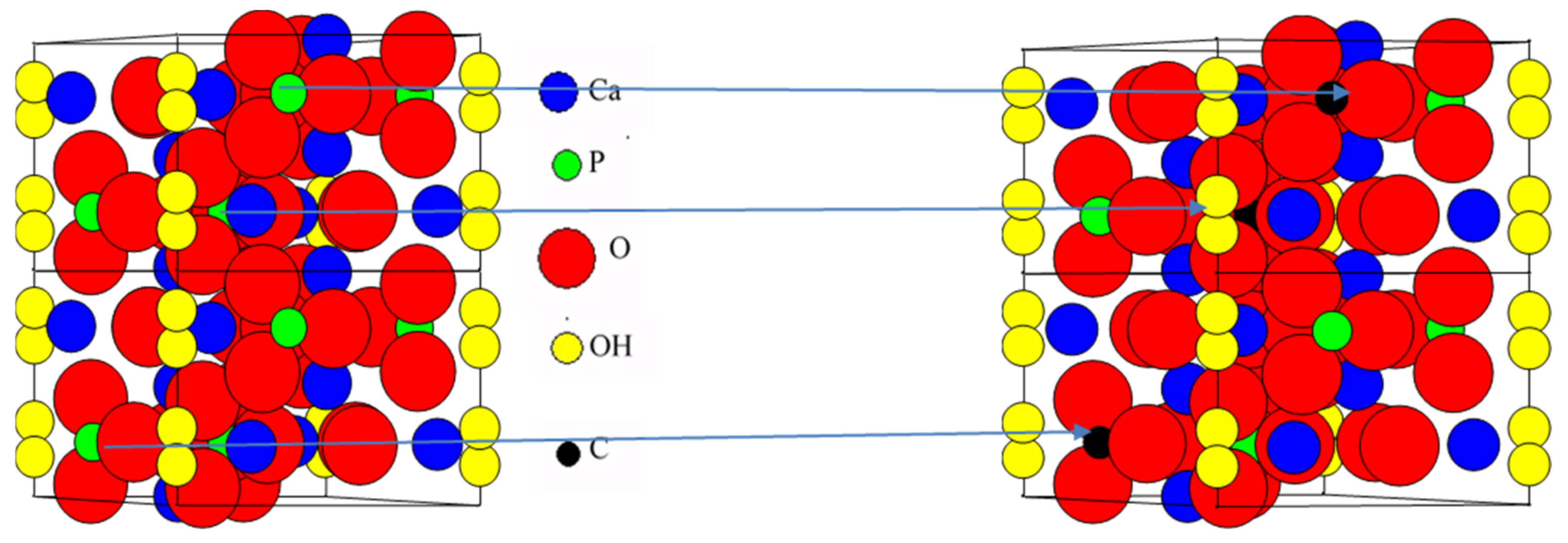
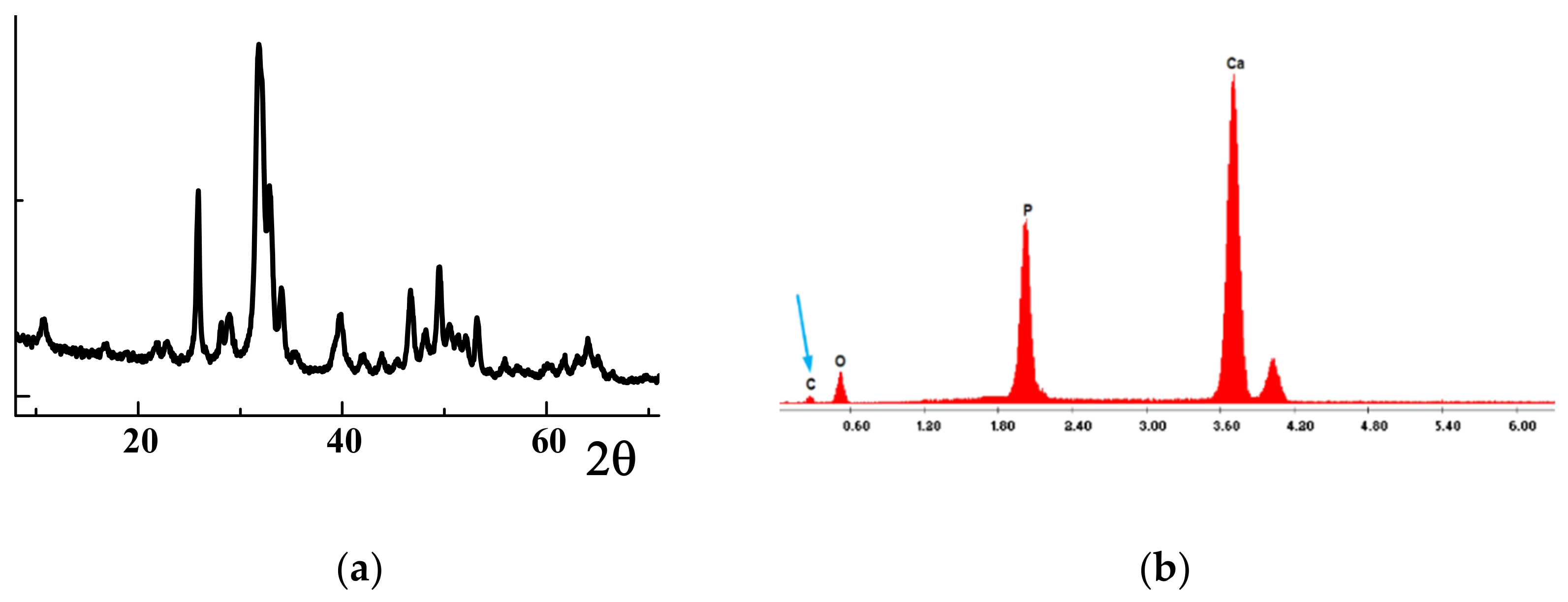
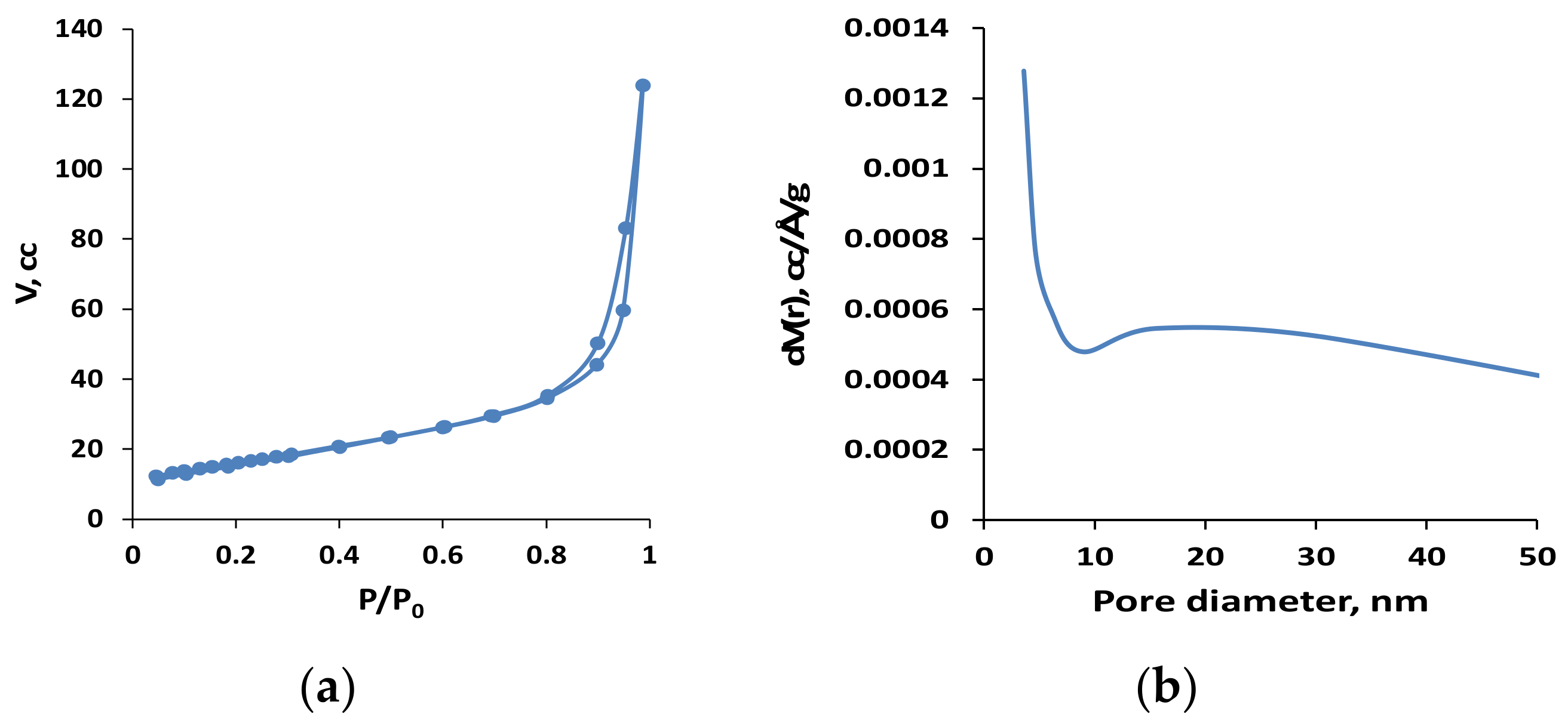
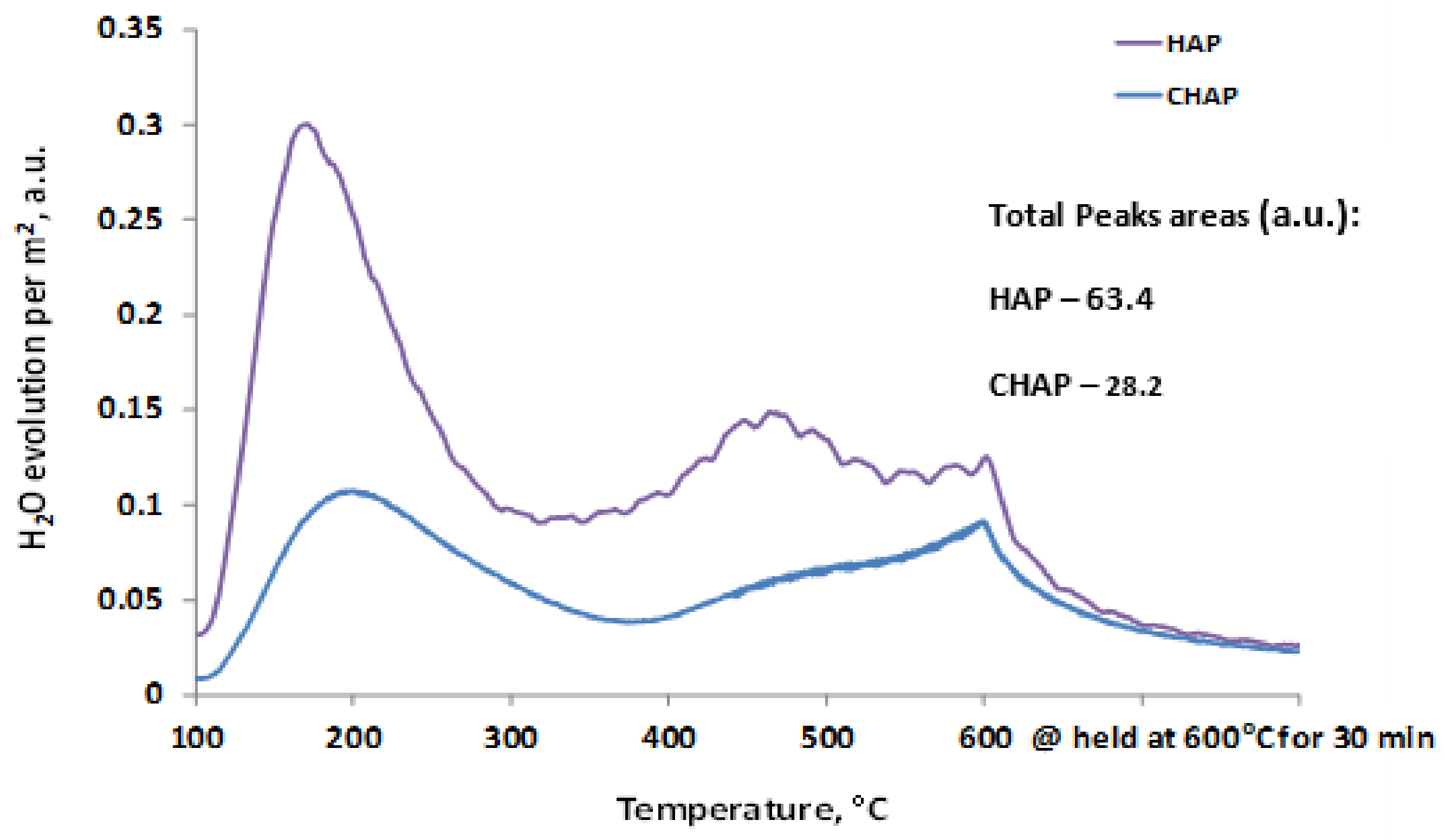
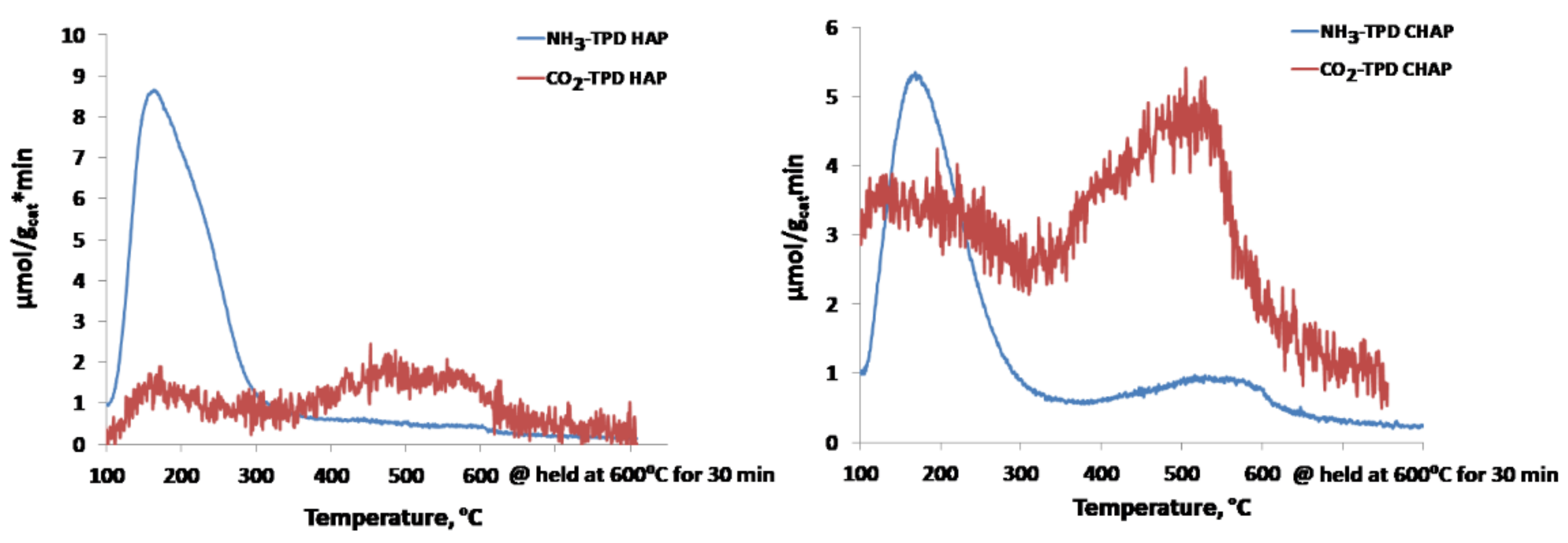
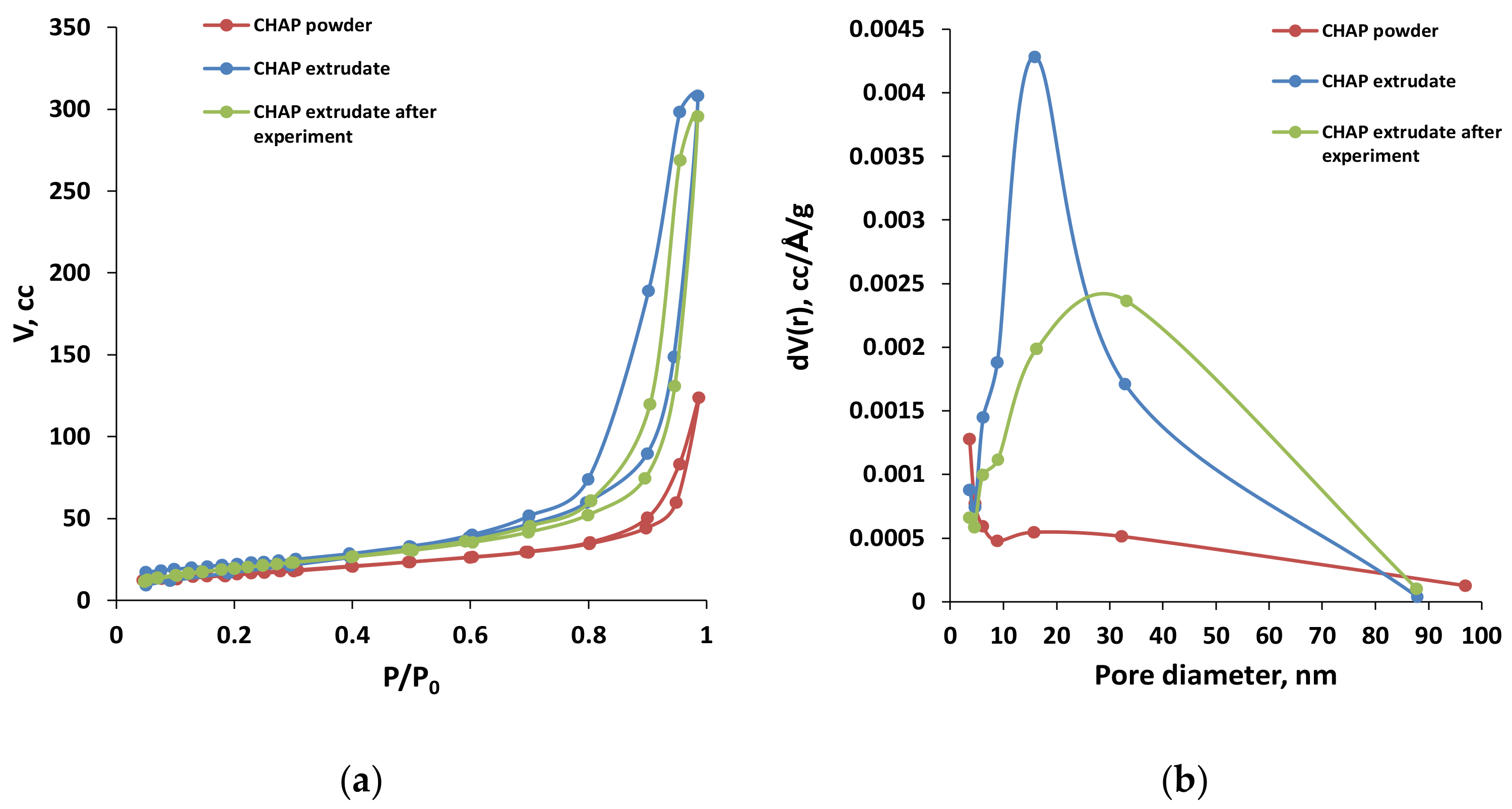
2.1. Catalyst Characterization
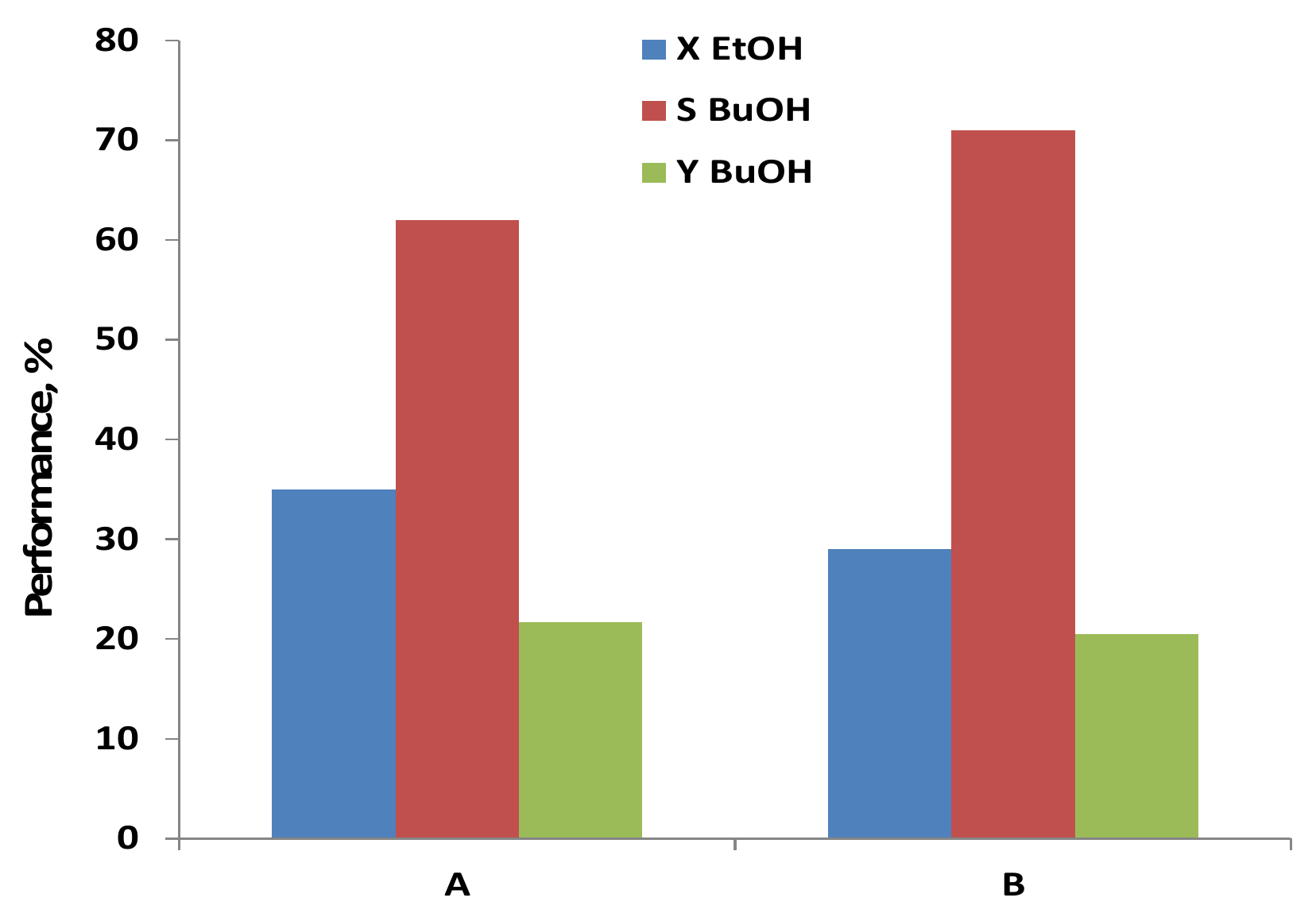
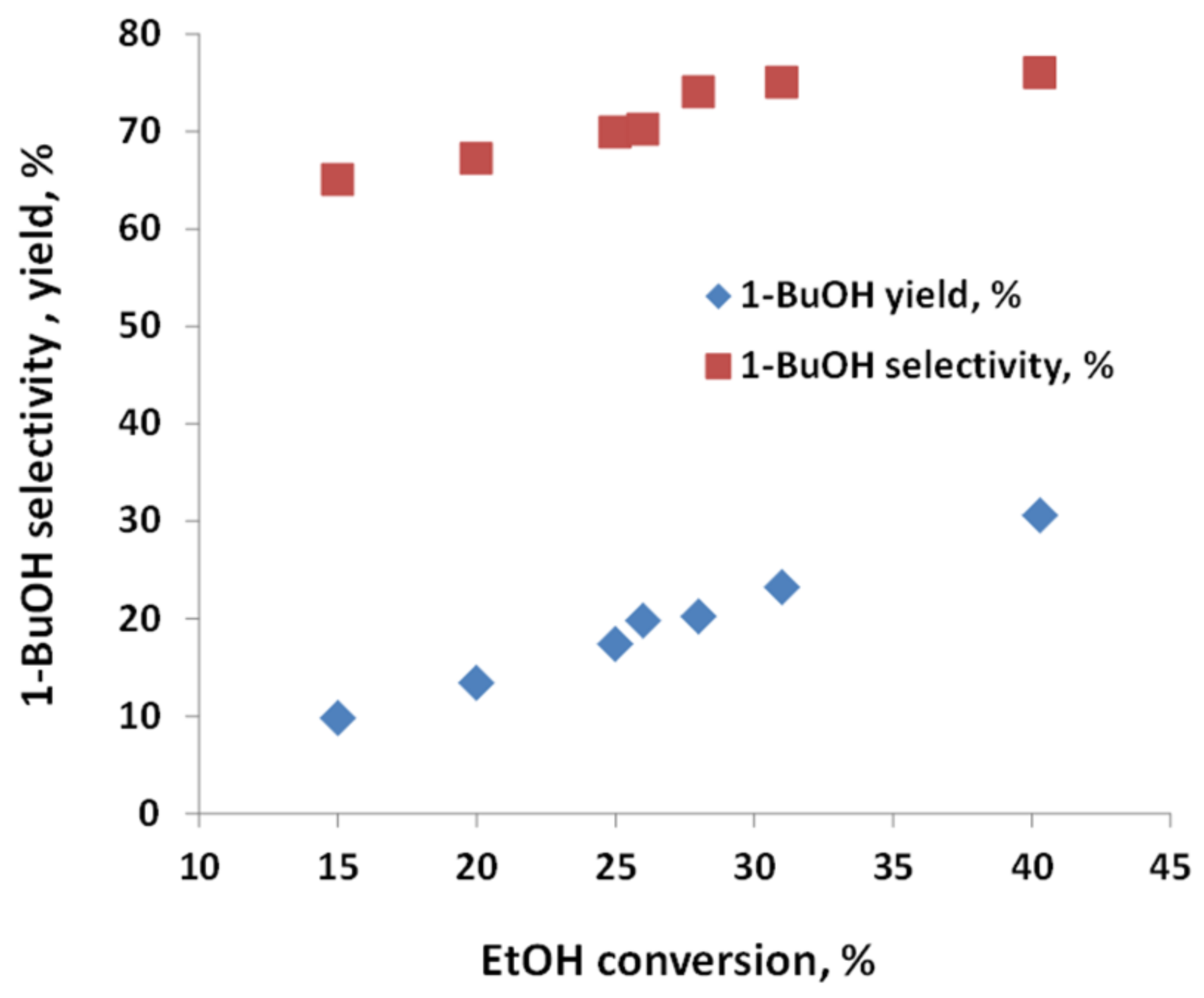
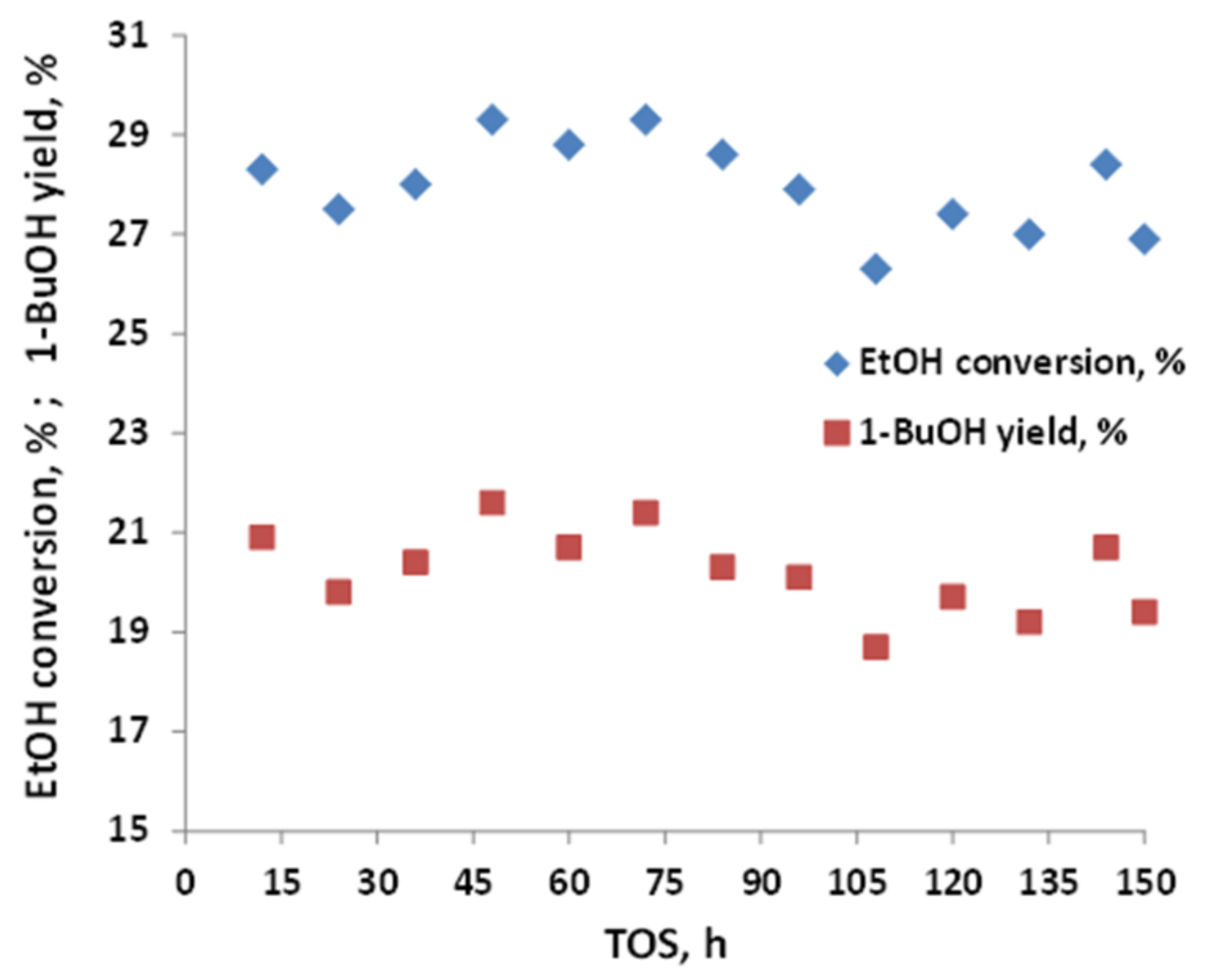
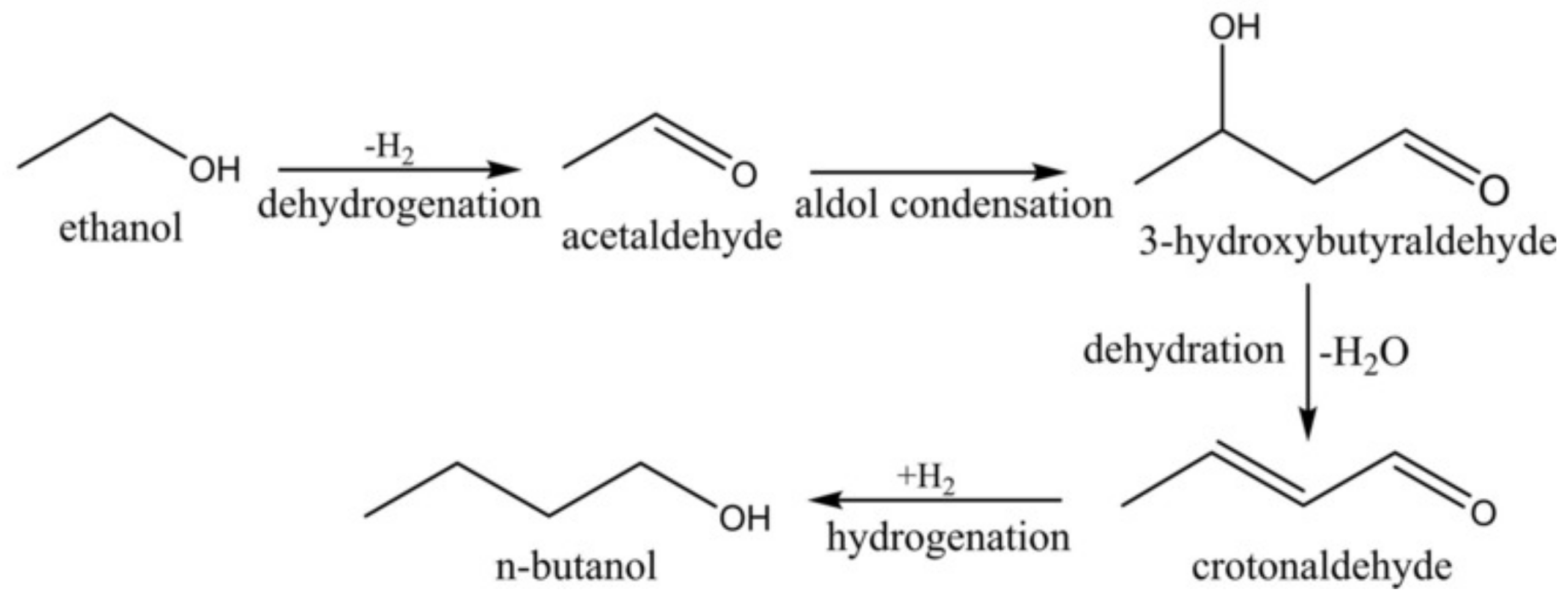
2.2. Testing of C-HAP Catalyst in Condensation of Hydrous Ethanol to 1-Butanol
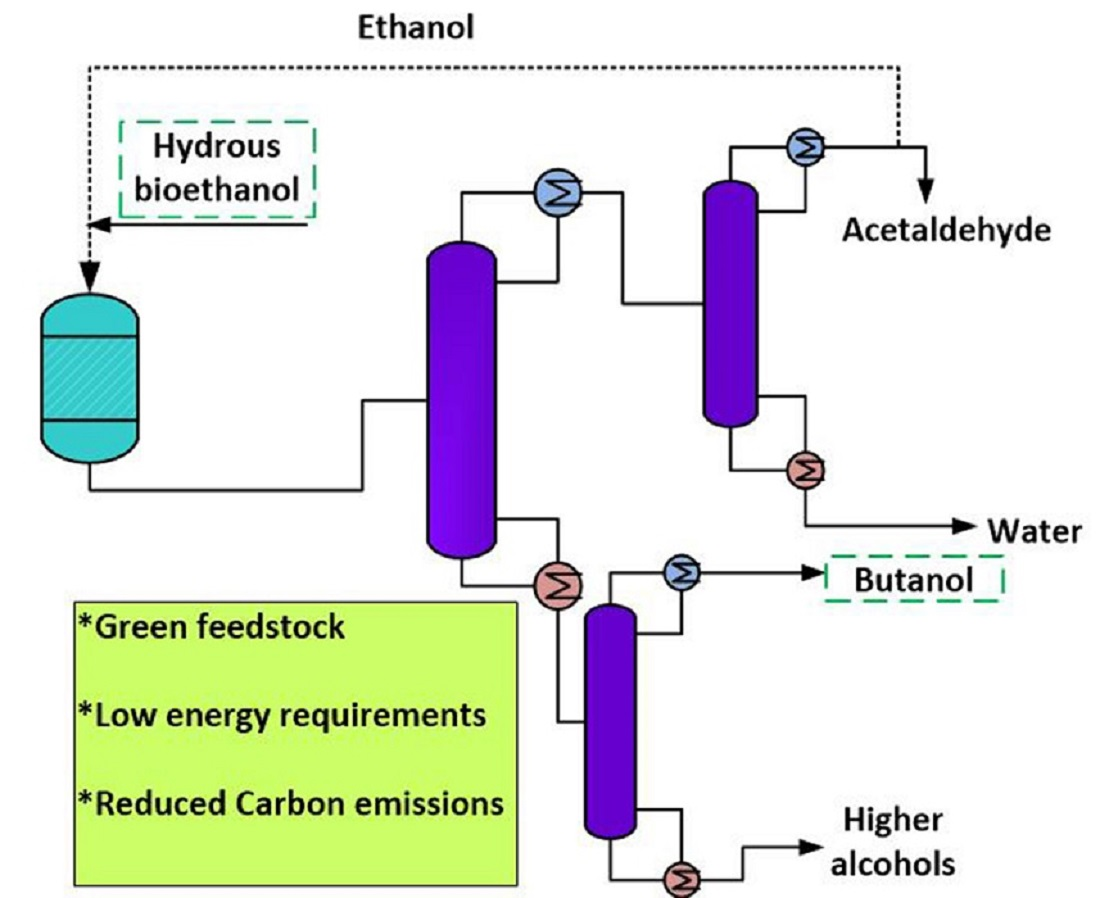
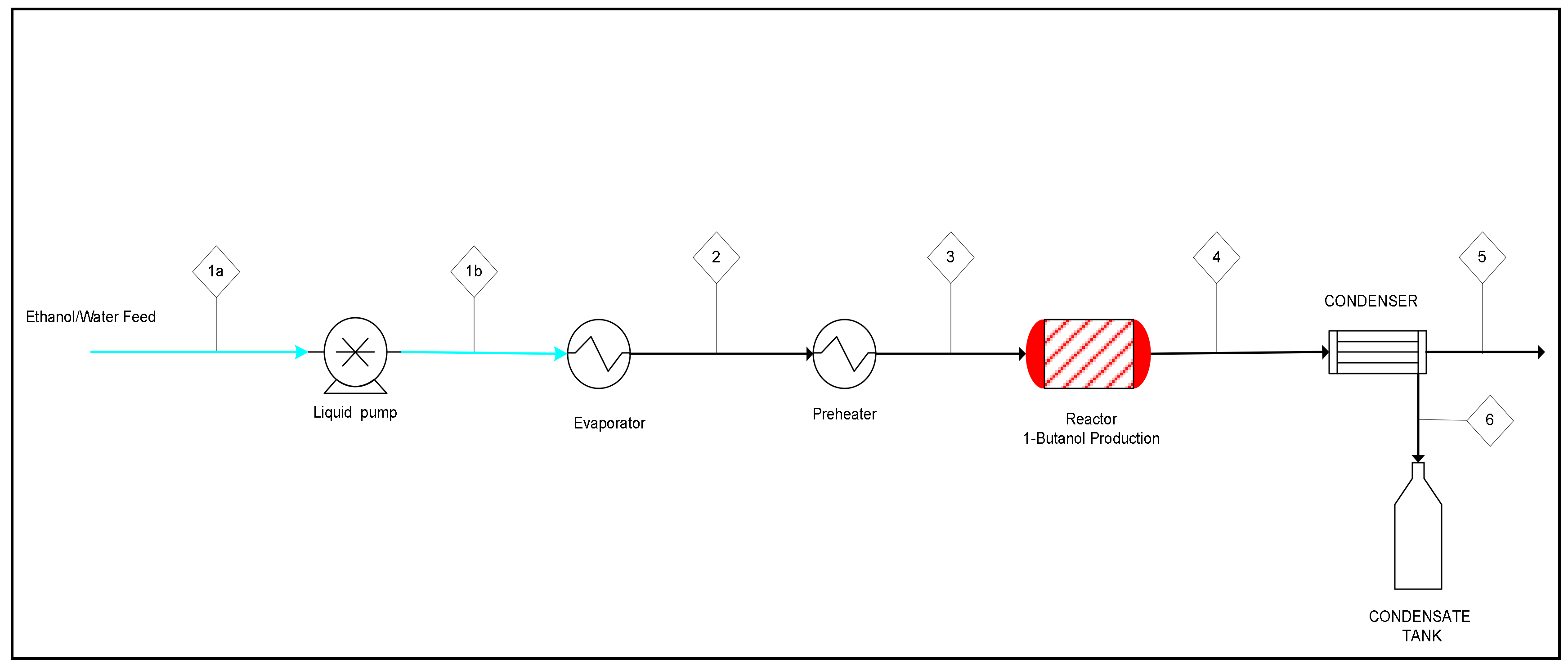
2.3. Process Simulation and Techno-Economic Analysis
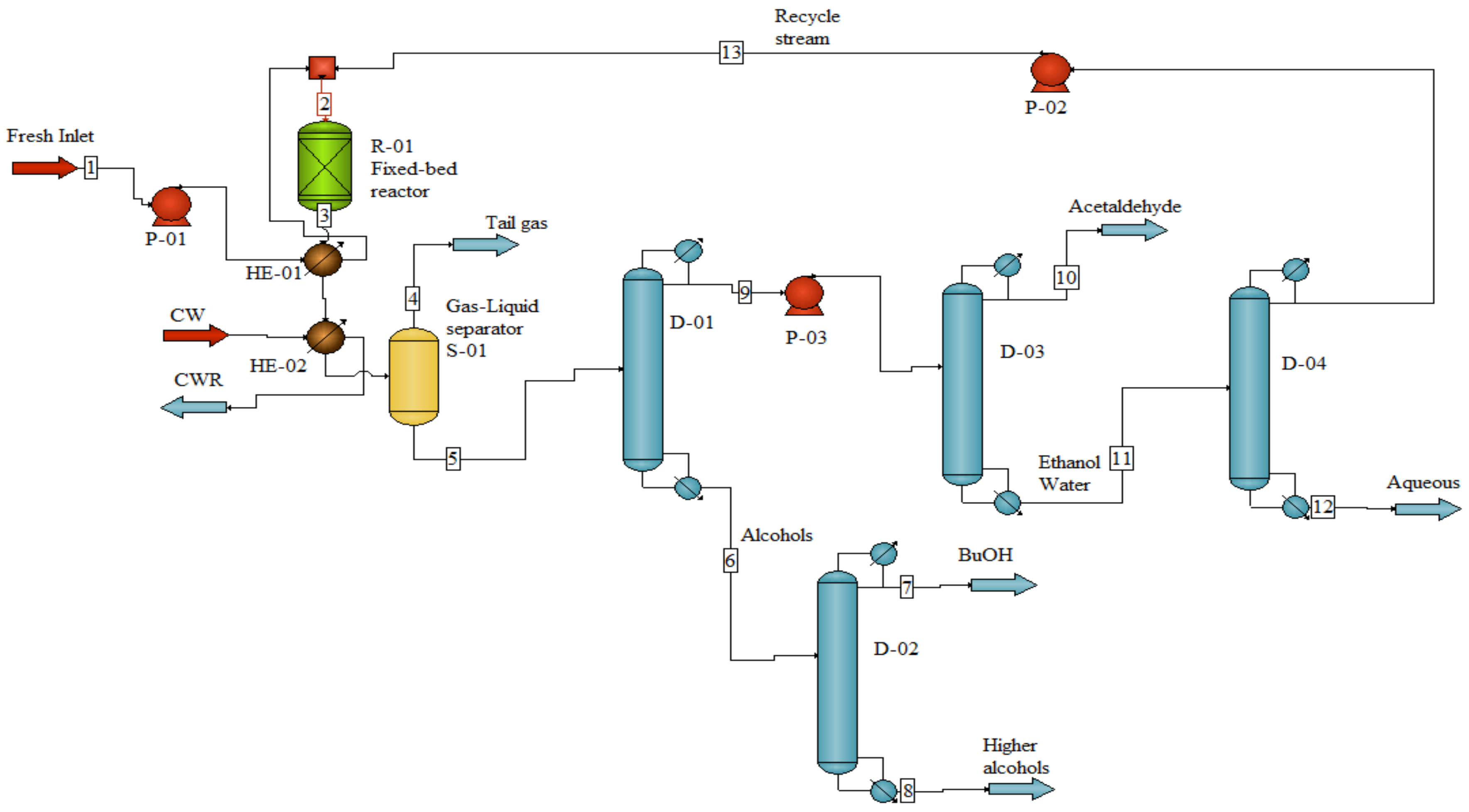
2.3.1. Mass Balance and Process Simulation
2.3.2. Initial Investment and Operational Expenditures
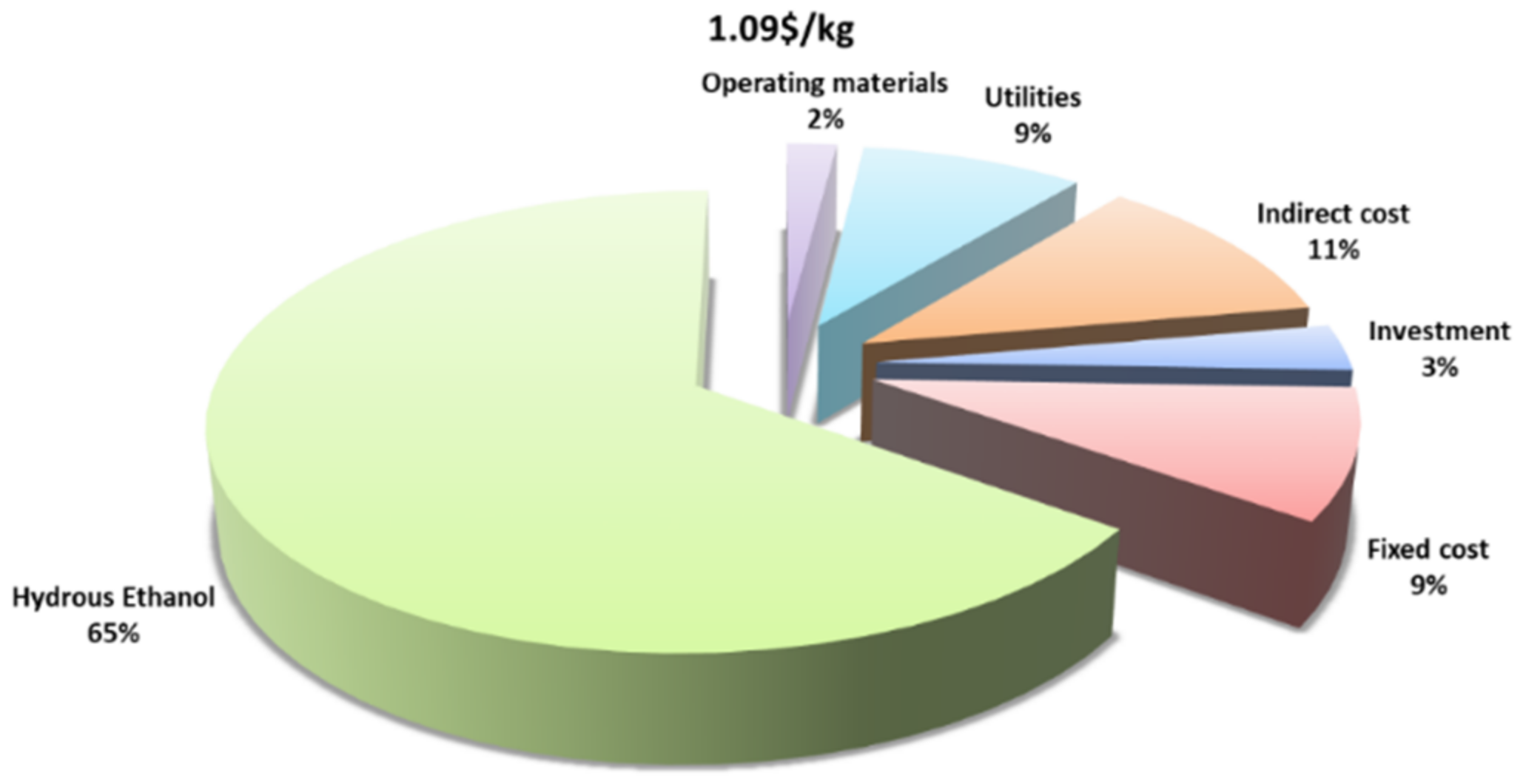
2.3.3. Process Economic Evaluation
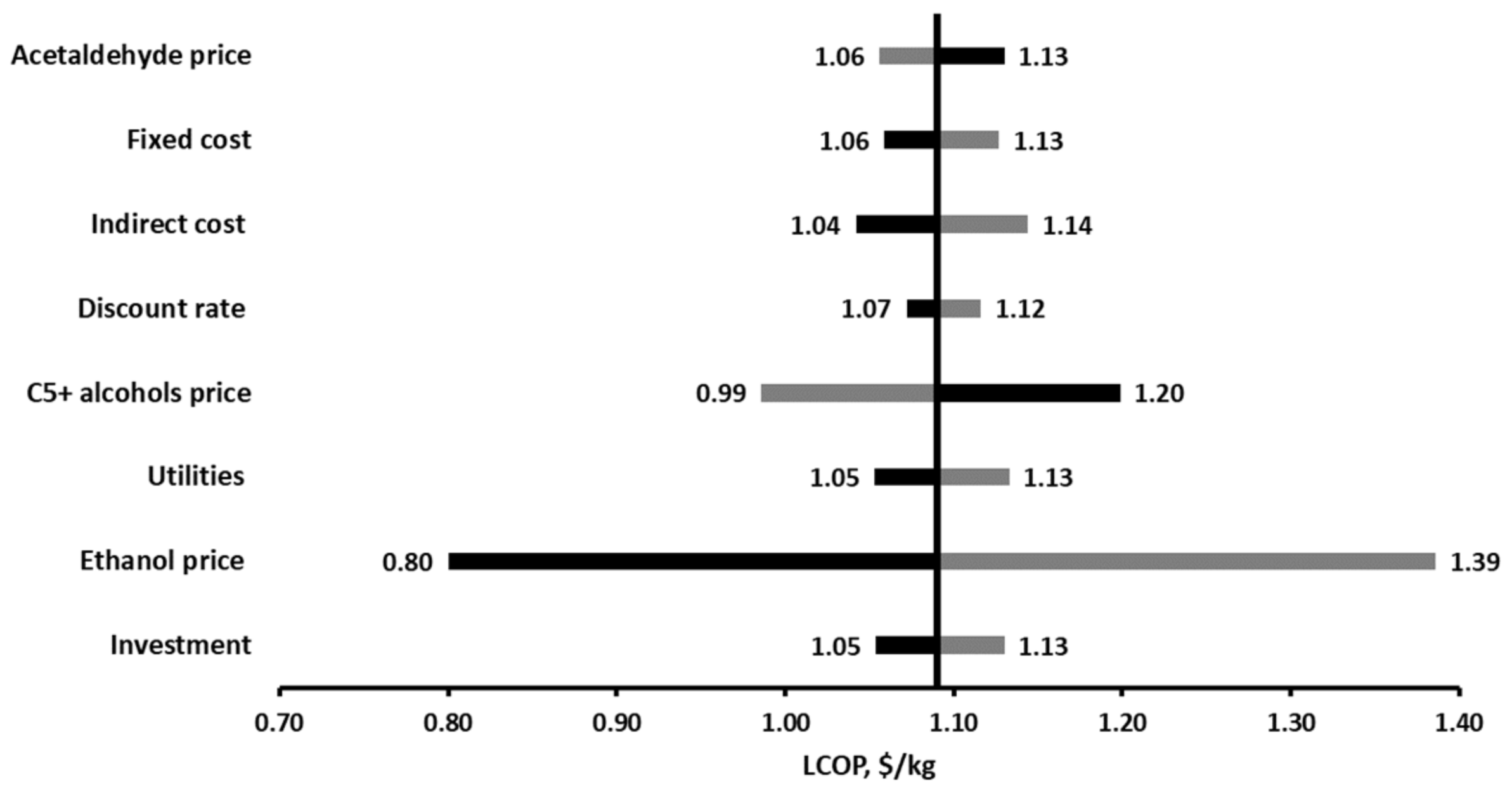
2.3.4. Sensitivity Analysis
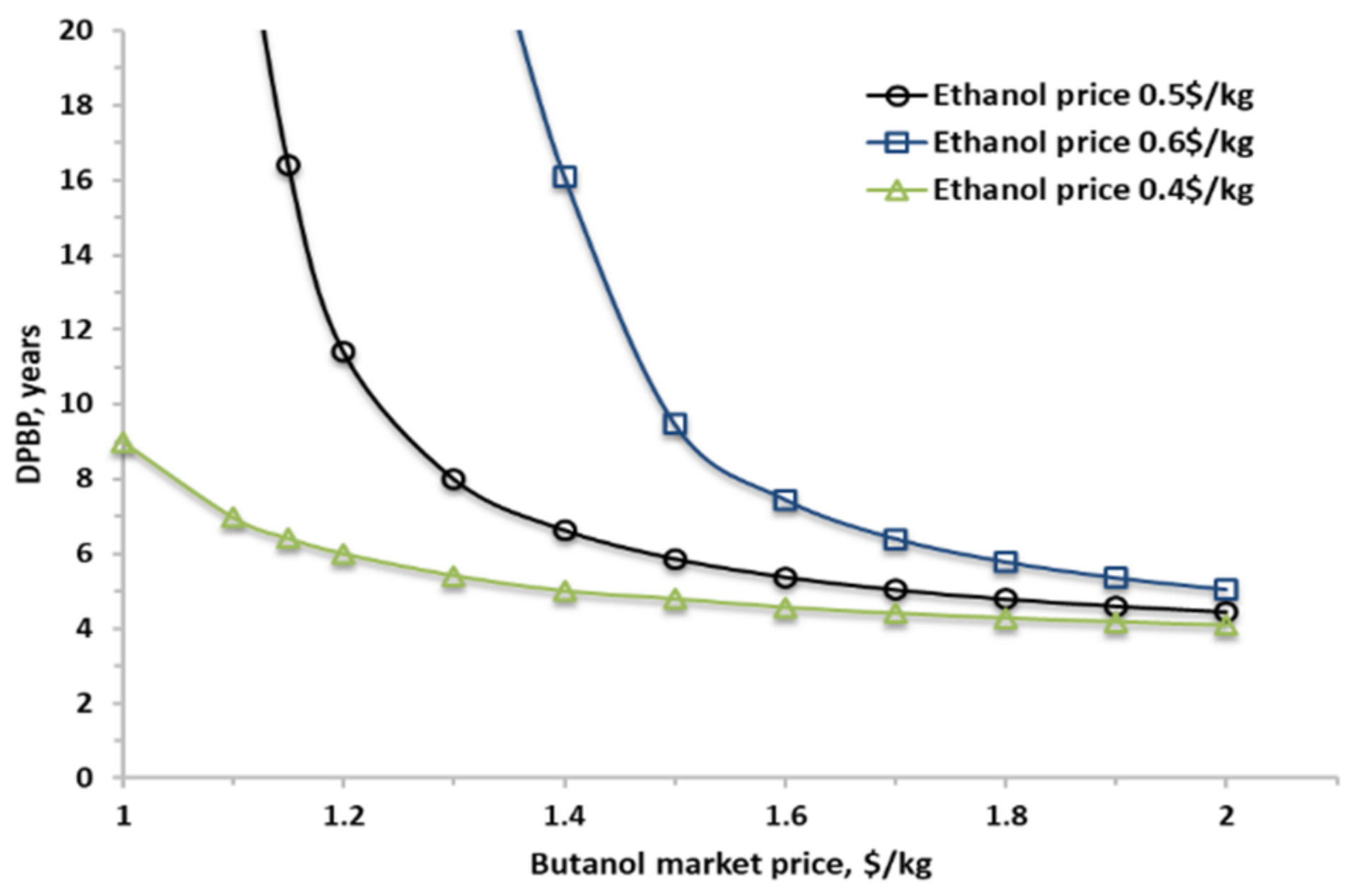
2.3.5. N-Butanol Selling Price
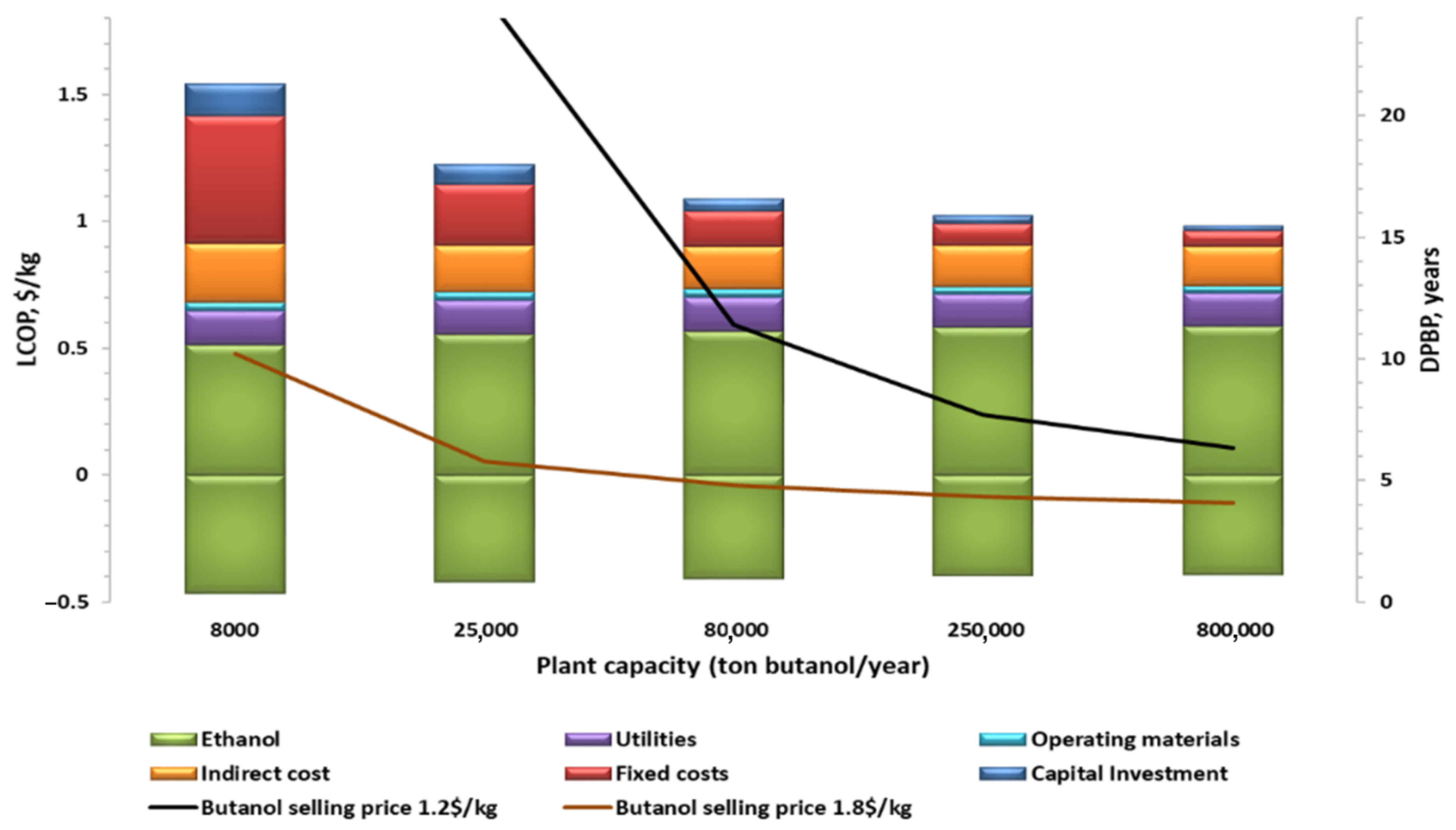
2.3.6. Impact of Plant Size
3. Materials and Methods
3.1. Preparation of Catalysts
3.2. Catalysts Characterization
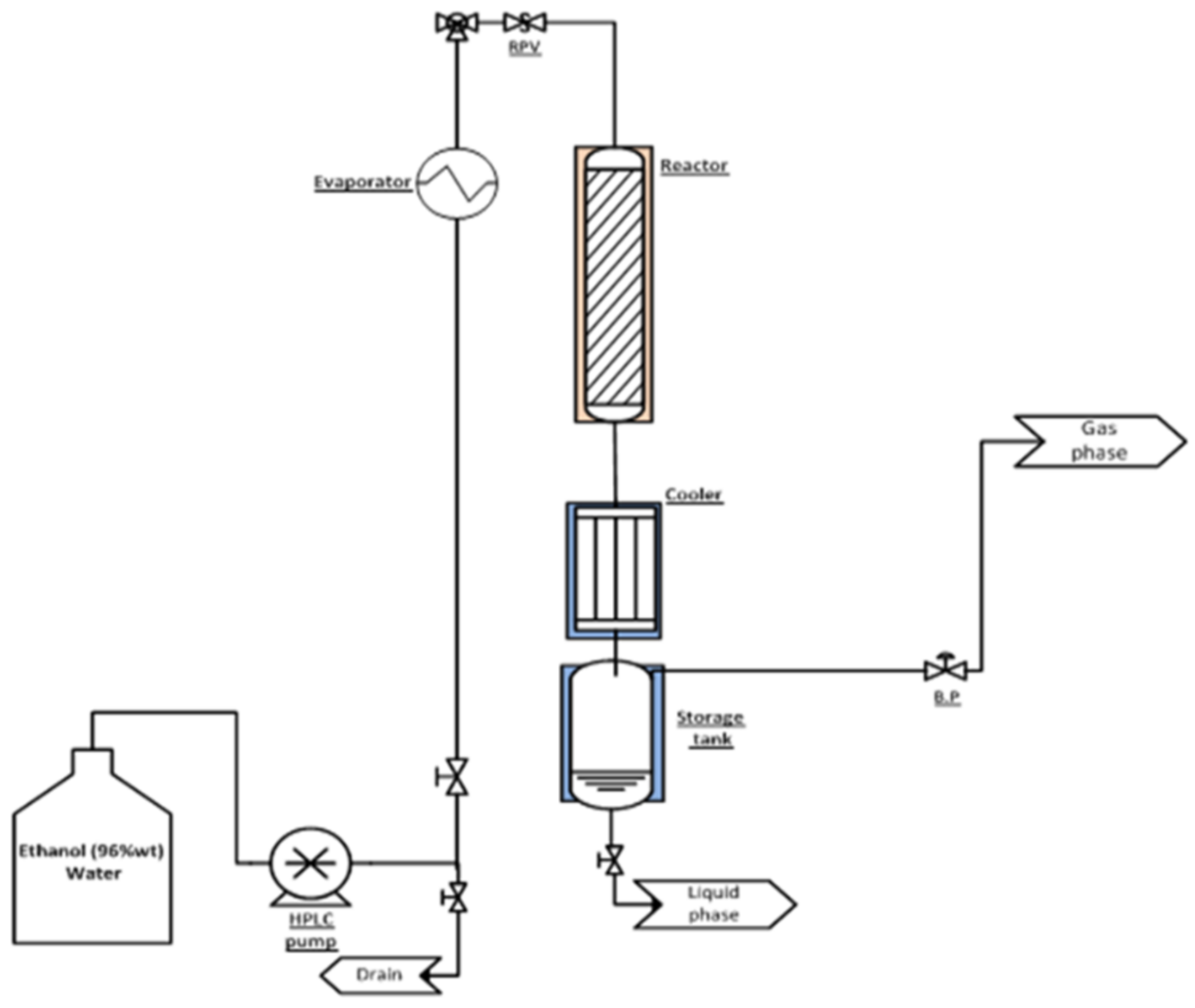
3.3. Experimental Setup and Analytical Procedures in Catalysts Testing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gavahian, M.; Munekata, P.E.S.; Eş, I.; Lorenzo, J.M.; Mousavi, K.A.; Barba, F.J. Emerging techniques in bioethanol production: From distillation to waste valorization. Green Chem. 2019, 21, 1171–1185. [Google Scholar] [CrossRef]
- Robak, K.; Balcerek, M. Review of Second-Generation Bioethanol Production from Residual Biomass. Food Technol. Biotechnol. 2018, 56, 174–187. [Google Scholar] [CrossRef] [PubMed]
- Rosales-Calderon, O.; Arantes, V. A review on commercial-scale high-value products that can be produced alongside cellulosic ethanol. Biotechnol. Biofuels 2019, 12, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grim, R.G.; To, A.T.; Farberow, C.A.; Hensley, J.E.; Ruddy, D.A.; Schaidle, J.A. Growing the Bioeconomy through Catalysis: A Review of Recent Advancements in the Production of Fuels and Chemicals from Syngas-Derived Oxygenates. ACS Catal. 2019, 9, 4145–4172. [Google Scholar] [CrossRef]
- Global Production of Bioethanol. Available online: https://link.springer.com/chapter/10.1007/978-981-15-8779-5_10 (accessed on 9 October 2020).
- Stansfield, P.A.; Bisordi, A.; OudeNijeweme, D.; Williams, J.; Gold, M.; Ali, R. The performance of a modern vehicle on a variety of alcohol-gasoline fuel blends. SAE Int. J. Fuels Lubr. 2012, 5, 813–822. [Google Scholar] [CrossRef]
- Liu, H.; Wang, X.; Zhang, D.; Dong, F.; Liu, X.; Yang, Y.; Huang, H.; Wang, Y.; Wang, Q.; Zheng, Z. Investigation on Blending Effects of Gasoline Fuel with N-Butanol, DMF, and Ethanol on the Fuel Consumption and Harmful Emissions in a GDI Vehicle. Energies 2019, 12, 1845. [Google Scholar] [CrossRef] [Green Version]
- Mascal, M. Chemicals from butanol: Technologies and markets. Biofuels Bioprod. Biorefining 2012, 6, 483–0493. [Google Scholar] [CrossRef]
- Wu, X.; Fang, G.; Tong, Y.; Jiang, D.; Liang, Z.; Leng, W.; Liu, L.; Tu, P.; Wang, H.; Ni, J.; et al. Catalytic Upgrading of Ethanol to n-Butanol: Progress in Catalyst Development. ChemSusChem 2018, 11, 71–85. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, K.; An, Z.; Zhu, Y.; Shu, X.; Song, H.; Xiang, X.; He, J. Acid–Base Promoted Dehydrogenation Coupling of Ethanol on Supported Ag Particles. Ind. Eng. Chem. Res. 2020, 59, 3342–3350. [Google Scholar] [CrossRef]
- Larina, O.V.; Valihura, K.V.; Kyriienko, P.I.; Vlasenko, N.V.; Balakin, D.Y.; Khalakhan, I.; Čendak, T.; Soloviev, S.O.; Orlyk, S.M. Successive vapour phase Guerbet condensation of ethanol and 1-butanol over Mg-Al oxide catalysts in a flow reactor. Appl. Catal. A Gen. 2019, 588, 117265. [Google Scholar] [CrossRef]
- Scheid, A.J.; Barbosa-Coutinho, E.; Schwaab, M.; Salau, N.P.G. Mechanism and Kinetic Modeling of Ethanol Conversion to 1-Butanol over Mg and Al Oxide Derived from Hydrotalcites. Ind. Eng. Chem. Res. 2019, 58, 12981–12995. [Google Scholar] [CrossRef]
- Ljpez-Olmos, C.; Virtudes Morales, M.; Guerrero-Ruiz, A.; Ramirez-Barria, C.; Asedegbega-Nieto, E.; Rodriguez-Ramos, I. Continuous Gas-Phase Condensation of Bioethanol to 1-Butanol over Bifunctional Pd/Mg and Pd/Mg–Carbon Catalysts. ChemSusChem 2018, 11, 3502–3511. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Olmos, C.; Guerrero-Ruiz, A.; Rodríguez-Ramos, I. Optimization of Cu-Ni-Mn-catalysts for the conversion of ethanol to butanol. Catal. Today 2020, 357, 132–142. [Google Scholar] [CrossRef]
- Nezam, I.; Peereboom, L.; Miller, D.J. Continuous condensed-phase ethanol conversion to higher alcohols: Experimental results and techno-economic analysis. J. Clean. Prod. 2019, 209, 1365–1375. [Google Scholar] [CrossRef]
- Eagan, N.M.; Lanci, M.P.; Huber, G.W. Kinetic Modeling of Alcohol Oligomerization over Calcium Hydroxyapatite. ACS Catal. 2020, 10, 2978–2989. [Google Scholar] [CrossRef]
- Marcu, I.-C.; Tichit, D.; Fajula, F.; Tanchoux, N. Catalytic valorization of bioethanol over Cu-Mg-Al mixed oxide catalysts. Catal. Today 2009, 147, 231–238. [Google Scholar] [CrossRef]
- Petrolini, D.D.; Eagan, N.; Ball, M.R.; Burt, S.P.; Hermans, I.; Huber, G.W.; Dumesic, J.A.; Martins, L. Ethanol condensation at elevated pressure over copper on AlMgO and AlCaO porous mixed oxide supports. Catal. Sci. Technol. 2019, 9, 2032–2042. [Google Scholar] [CrossRef]
- Hanspal, S.; Young, Z.D.; Prillaman, J.T.; Davis, R.J. Influence of surface acid and base sites on the Guerbet coupling of ethanol to butanol over metal phosphate catalysts. J. Catal. 2017, 352, 182–190. [Google Scholar] [CrossRef]
- Corno, M.; Busco, C.; Bolis, V.; Tosoni, S.; Ugliengo, P. Water Adsorption on the Stoichiometric (001) and (010) Surfaces of Hydroxyapatite: A Periodic B3LYP Study. Langmuir 2009, 25, 2188–2198. [Google Scholar] [CrossRef]
- Bolis, V.; Busco, C.; Gianmario, M.; Bertinetti, L.; Sakhno, Y.; Ugliengo, P.; Chiatti, F.; Corno, M.; Roveri, N. Coordination chemistry of Ca sites at the surface of nanosized hydroxyapatite: Interaction with H2O and CO. Philos. Trans. R. Soc. A 2012, 370, 1313–13236. [Google Scholar] [CrossRef] [Green Version]
- Diallo-Garcia, S.; Osman, M.B.; Krafft, J.-M.; Casale, S.; Thomas, C.; Kubo, J.; Costentin, G. Identification of Surface Basic Sites and Acid−Base Pairs of Hydroxyapatite. J. Phys. Chem. C 2014, 118, 12744–12757. [Google Scholar] [CrossRef]
- Fubini, B.; Bolis, V.; Bailes, M.; Stone, F.S. The reactivity of oxides with water vapor. Solid State Ion. 1989, 32–33, 258–272. [Google Scholar] [CrossRef]
- Hanspal, S.; Young, Z.D.; Shou, H.; Davis, R.J. Multiproduct Steady-State Isotopic Transient Kinetic Analysis of the Ethanol Coupling Reaction over Hydroxyapatite and Magnesia. ACS Catal. 2015, 5, 1737–1746. [Google Scholar] [CrossRef]
- Osman, M.B.; Krafft, J.-M.; Thomas, C.; Yoshioka, T.; Kubo, J.; Costentin, G. Importance of the Nature of the Active Acid/Base Pairs of Hydroxyapatite Involved in the Catalytic Transformation of Ethanol to n-Butanol Revealed by Operando DRIFTS. ChemCatChem 2019, 11, 1765–1778. [Google Scholar] [CrossRef]
- Silvester, L.; Lamonier, J.-F.; Faye, J.; Capron, M.; Vannier, R.-N.; Lamonier, C.; Dubois, J.-L.; Couturier, J.-L.; Calais, C.; Dumeignil, F. Reactivity of ethanol over hydroxyapatite-based Ca-enriched catalysts with various carbonate contents. Catal. Sci. Technol. 2015, 5, 2994–3006. [Google Scholar] [CrossRef]
- Fagan, P.J.; Calvarese, T.G.; Davis, R.J.; Ozer, R. Conversion of Ethanol to a Reaction Product Comprising 1-Butanol Using Hydroxyapatite Catalysts. U.S. Patent 8.962.896 B2, 24 February 2015. [Google Scholar]
- Tsuchida, T.; Sakuma, S.; Takeguchi, T.; Ueda, W. Direct Synthesis of n-Butanol from Ethanol over Nonstoichiometric Hydroxyapatite. Ind. Eng. Chem. Res. 2006, 45, 8634–8642. [Google Scholar] [CrossRef]
- Gounder, R. Hydrophobic microporous and mesoporous oxides as Brønsted and Lewis acid catalysts for biomass conversion in liquid water. Catal. Sci. Technol. 2014, 4, 2877–2886. [Google Scholar] [CrossRef]
- Elliott, J.C. Structure and Chemistry of the Apatites and Other Calcium Orthophosphates; Elsevier: Amsterdam, The Netherlands, 1994. [Google Scholar]
- Resende, N.S.; Nele, M.; Salim, V.M.M. Effects of anion substitution on the acid properties of hydroxdyapatite. Thermochim. Acta 2006, 451, 16–21. [Google Scholar] [CrossRef]
- Landi, E.; Celotti, G.; Logroscino, G.; Tampieri, A. Carbonated hydroxyapatite as bone substitute. J. Eur. Ceram. Soc. 2003, 23, 2931–2937. [Google Scholar] [CrossRef]
- Kozlowski, J.T.; Davis, R.J. Heterogeneous catalysts for the Guerbet coupling of alcohols. ACS Catal. 2013, 3, 1588–1600. [Google Scholar] [CrossRef]
- Lovon-Quintana, J.J.; Rodrigues-Guerrero, J.K.; Valenca, P.G. Carbonate hydroxyapatite as a catalyst for ethanol conversion to hydrocarbon fuels. Appl. Catal. A 2017, 542, 136–145. [Google Scholar] [CrossRef]
- Ohayon Dahan, H.; Landau, M.V.; Herskowitz, M. Effect of surface acidity-basicity balance in modified ZnxZryOz catalyst on its performance in the conversion of hydrous ethanol to hydrocarbons. J. Ind. Eng. Chem. 2021, 95, 156–169. [Google Scholar] [CrossRef]
- Zhang, M.; Yu, Y. Dehydration of ethanol to ethylene. Ind. Eng. Chem. Res. 2013, 52, 9505–9514. [Google Scholar] [CrossRef]
- Vlasenko, N.V.; Kyriienko, P.I.; Valihura, K.V.; Yanushevska, O.I.; Soloviev, S.O.; Strizhak, P.E. Effect of modifying additives on the catalytic properties of zirconium dioxide in the conversion of ethanol to 1-butanol. Theor. Exp. Chem. 2019, 55, 43–49. [Google Scholar] [CrossRef]
- Michaels, W.; Zhang, H.; Luyben, W.L.; Baltrusaitis, J. Design of a separation section in an ethanol-to-butanol process. Biomass Bioenergy 2018, 109, 231–238. [Google Scholar] [CrossRef]
- Sinnott, R. Chemical Engineering Design: Chemical Engineering; Elsevier: Amsterdam, The Netherlands, 2005; Volume 6. [Google Scholar]
- Sinnott, R.; Towler, G. Principles, Practice and Economics of Plant and Process Design, Chemical Engineering Design; Elsevier: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Peters, M.S.; Timmerhaus, K.D.; West, R.E. Plant Design and Economics for Chemical Engineers; McGraw-Hill: New York, NY, USA, 2003. [Google Scholar]
- Grim, R.G.; Huang, Z.; Guarnieri, M.T.; Ferrell, J.R.; Tao, L.; Schaidle, J.A. Transforming the carbon economy: Challenges and opportunities in the convergence of low-cost electricity and reductive CO2 utilization. Energy Environ. Sci. 2020, 13, 472–494. [Google Scholar] [CrossRef]
- EIA. Natural Gas Prices. 2020. Available online: https://www.eia.gov/dnav/ng/ng_pri_sum_dcu_nus_a.htm (accessed on 30 June 2020).
- ICIS. Americas Chemicals Outlook; Reed Business Information Ltd.: London, UK, 2019. [Google Scholar]
- US Grains Council. Ethanol Market and Pricing Data; US Grains Council: Washington, DC, USA, 2019. [Google Scholar]
- ICIS. Indicative Chemicals Prices A-Z. 2020. Available online: https://www.icis.com/explore/chemicals/channel-info-chemicals-a-z/ (accessed on 30 June 2020).
- Madhu, K.; Zilberman, D. Handbook of Bioenergy Economics and Policy: Volume II; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- ICIS. Europe Chemicals Outlook; Reed Business Information Ltd.: London, UK, 2019. [Google Scholar]
- ICIS. Asia Chemicals Outlook; Reed Business Information Ltd.: London, UK, 2019. [Google Scholar]
- Tecnon OrbiChem, Chemical Market Insight and Foresight—On a Single Page: N-Butanol. 2013. Available online: http://www.orbichem.com/userfiles/CNFSamples/act_13_11.pdf (accessed on 30 June 2020).
- Renewable Fuels Association. 2019 Ethanol Industry Outlook. 2019. Available online: https://ethanolrfa.org/wp-content/uploads/2019/02/RFA2019Outlook.pdf (accessed on 30 June 2020).
- BBI International. Fuel Ethanol Industry Directory, 19th ed.; BBI International: Grand Forks, ND, USA, 2019. [Google Scholar]
- Herskowitz, M.; Landau, M.V.; Vidruk, R.; Amoyal, M. Catalyst Composition and Catalytic Process for Producing Liquid Hydrocarbons. U.S. Patent 10,589,257, 17 March 2020. [Google Scholar]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Surface Area, m2/g | Pore Volume, cm3/g | Average Pore Diameter, nm |
|---|---|---|---|
| C-HAP | 57 | 0.192 | 14 |
| HAP | 49 | 0.348 | 28 |
| Catalyst | Texture Parameters | ||
|---|---|---|---|
| Surface Area, m2/g | Pore Volume, cm3/g | Average Pore Diameter, nm | |
| C-HAP-SiO2 | 77 | 0.377 | 19 |
| C-HAP-ZrO2 | 35 | 1.18 | 94 |
| C-HAP-Al2O3 | 96 | 0.49 | 19 |
| Binding Material | Testing Results of C-HAP Catalyst Pellets | ||
|---|---|---|---|
| EtOH Conversion, % | 1-BuOH Selectivity, % | 1-BuOH Yield, % | |
| SiO2 | 27.5 | 72.2 | 19.8 |
| Al2O3 | 49.2 | 12.5 | 6.1 |
| ZrO2 | 56.1 | 26.3 | 14.5 |
| Catalyst | Surface Area, m2/g | Pore Volume, cm3/g | Average Pore Diameter, nm |
|---|---|---|---|
| C-HAP-30%SiO2 pellets as prepared | 77 | 0.377 | 19 |
| C-HAP-30%SiO2 pellets after testing: TOS = 150 h | 65 | 0.357 | 22 |
| WHSV, h−1; TOS, h | Feed, g/h | Products Out, g/h | Performance, % | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| EtOH | H2O | EtOH | BuOH | H2O * | Acetaldehyde | Heavier Alcohols | H2 | EtOH Conversion | 1-BuOH Selectivity | 1-BuOH Yield | |
| 0.5; 24 | 31.2 | 1.3 | 21.7 | 5.8 | 3.0 | 0.7 | 1.6 | 0.03 | 30.5 | 74.4 | 22.7 |
| 1.0; 24 | 62.4 | 2.6 | 44.4 | 10.6 | 5.8 | 1.0 | 3.4 | 0.04 | 28.8 | 73.2 | 21.1 |
| 2.8; 24 | 174.7 | 7.3 | 133.2 | 22.1 | 14.6 | 4.0 | 8.2 | 0.02 | 23.7 | 66.6 | 15.8 |
| 4.0; 98 | 250 | 10.4 | 202.9 | 24.0 | 18.1 | 5.2 | 9.8 | 0.02 | 18.8 | 63.3 | 11.9 |
| Stream No. | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| Name | Fresh feed | Reactor Inlet | Reactor Outlet | Tail Gas | D-01 Inlet | Alcohols | BuOH |
| Temperature, °C | 25 | 400 | 435 | 40 | 40 | 122 | 118 |
| Total Mass rate, kg/h | 19,135.0 | 67,351.8 | 67,351.8 | 242.4 | 67,109.4 | 12,853.5 | 10,010.0 |
| Ethanol rate, kg/h | 18,369.5 | 63,211.1 | 45,074.9 | 140.8 | 44,934.1 | - | - |
| Butanol rate, kg/h | - | - | 10,678.9 | 5.4 | 10,673.5 | 10,033.1 | 10,000.0 |
| Water rate, kg/h | 765.5 | 4032.7 | 7622.2 | 19.4 | 7602.8 | 1.3 | 1.3 |
| Acetaldehyde rate, kg/h | - | 108.0 | 1110.8 | 30.9 | 1079.9 | - | - |
| Hydrogen rate, kg/h | - | - | 45.9 | 45.9 | - | - | - |
| C5+ alcohols | - | - | 2819.1 | 0.0 | 2819.1 | 2819.1 | 8.7 |
| Stream No. | 8 | 9 | 10 | 11 | 12 | 13 | |
| Name | High alcohols | D-03 Inlet | Acetaldehyde | D-04 Inlet | Aqueous | Recycle stream | |
| Temperature, °C | 157 | 77 | 41 | 97 | 110 | 96 | |
| Total Mass rate, kg/h | 2843.5 | 54,255.9 | 974.8 | 53,281.1 | 5064.3 | 48,216.8 | |
| Ethanol rate, kg/h | - | 44,934.1 | 2.6 | 44,931.5 | 89.9 | 44,841.6 | |
| Butanol rate, kg/h | 33.1 | 640.4 | - | 640.4 | 640.4 | - | |
| Water rate, kg/h | - | 7601.5 | 0.3 | 7601.2 | 4334.0 | 3267.2 | |
| Acetaldehyde rate, kg/h | - | 1079.9 | 971.9 | 108.0 | - | 108.0 | |
| Hydrogen rate, kg/h | - | - | - | - | - | - | |
| C5+ alcohols | 2810.4 | - | - | - | - | - | |
| Item | Factor | Cost, $K |
|---|---|---|
| Purchased Equipment Cost | 5829 | |
| Equipment erection | 0.40 | 2332 |
| Piping | 0.70 | 4080 |
| Buildings and site development | 0.60 | 3497 |
| Electrical | 0.20 | 1166 |
| Instruments and control | 0.30 | 1749 |
| Storages | 0.15 | 874 |
| Total Direct Plant Cost | 3.35 | 19,527 |
| Contractor’s fee | 0.05 | 976 |
| Design and Engineering | 0.30 | 5858 |
| Contingency allowance | 0.10 | 1953 |
| Capex | 4.86 | 28,314 |
| Working capital a | 2831 | |
| Total Capital Investment (TCI) | 31,146 |
| Utility/Material | Cost | Units | Quantity, tonne/year | Units | Cost $K/year | Ref. |
|---|---|---|---|---|---|---|
| Cooling water | 0.03 | $/tonne | 51,840,000 | tonne/year | 1555 | [40] |
| Electricity | 0.06 | $/kWh | 2400 | MWh/year | 144 | [42] |
| Thermal energy | 3.9 | $/MMBTU c | 2,240,000 | MMBTU/year | 8736 | [43] |
| Hydrous Ethanol | 500 | $/tonne | 153,080 | tonne/year | 76,540 | [44,45] |
| Waste water treatment | 2 | $/tonne | 40,520 | tonne/year | 81 | [40] |
| Acetaldehyde | 1000 | $/tonne | 7800 | tonne/year | 7800 | [46] |
| C5+ alcohol mixture | 1000 a | $/tonne | 22,552 | tonne/year | 22,552 | - |
| Mg catalyst | 30,000 a | $/tonne | 68 b | tonne/year | 2040 | - |
| Fixed Expenses | $K |
|---|---|
| Maintenance a | 2265 |
| Operating labour b | 1200 |
| Laboratory costs c | 300 |
| Supervision d | 300 |
| Plant overheads e | 600 |
| Capital charges f | 1133 |
| Local taxes g | 566 |
| Insurance h | 566 |
| Total fixed expenses | 6930 |
| Variable expenses | |
| Hydrous Ethanol | 76,540 |
| Catalyst& Operating materials i | 2351 |
| Utilities | 10,516 |
| Acetaldehyde credit | −7800 |
| C5+ alcohols credit | −22,552 |
| Total Variable expenses | 59,056 |
| Total direct costs | 65,986 |
| Indirect costs j | 13,197 |
| Annual production expenses | 79,183 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Landau, M.V.; Hos, T.; Vidruk Nehemya, R.; Nomikos, G.; Herskowitz, M. Eco-Friendly and Sustainable Process for Converting Hydrous Bioethanol to Butanol. Catalysts 2021, 11, 498. https://doi.org/10.3390/catal11040498
Landau MV, Hos T, Vidruk Nehemya R, Nomikos G, Herskowitz M. Eco-Friendly and Sustainable Process for Converting Hydrous Bioethanol to Butanol. Catalysts. 2021; 11(4):498. https://doi.org/10.3390/catal11040498
Chicago/Turabian StyleLandau, Miron V., Tomy Hos, Roxana Vidruk Nehemya, George Nomikos, and Moti Herskowitz. 2021. "Eco-Friendly and Sustainable Process for Converting Hydrous Bioethanol to Butanol" Catalysts 11, no. 4: 498. https://doi.org/10.3390/catal11040498