
Physico-Chemical Changes in the KCl-MgCl2/La-FAU Composite Catalyst Induced by Oxidative Dehydrogenation of Ethane


Abstract
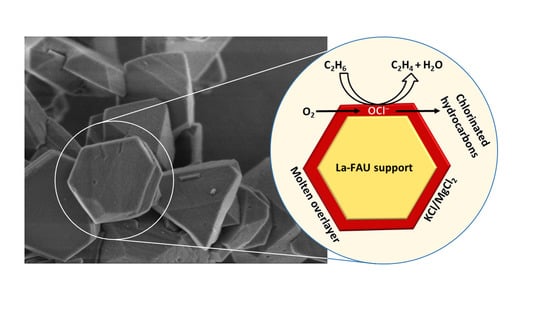
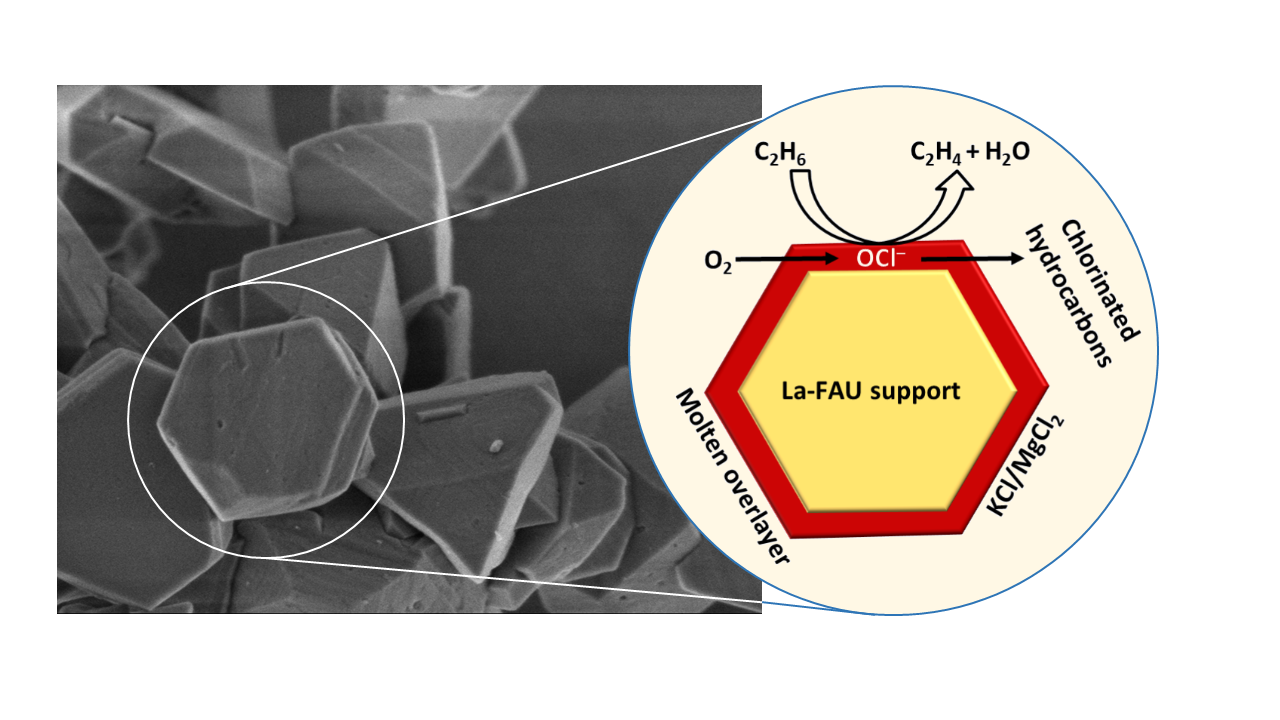
:
1. Introduction
2. Results and Discussion
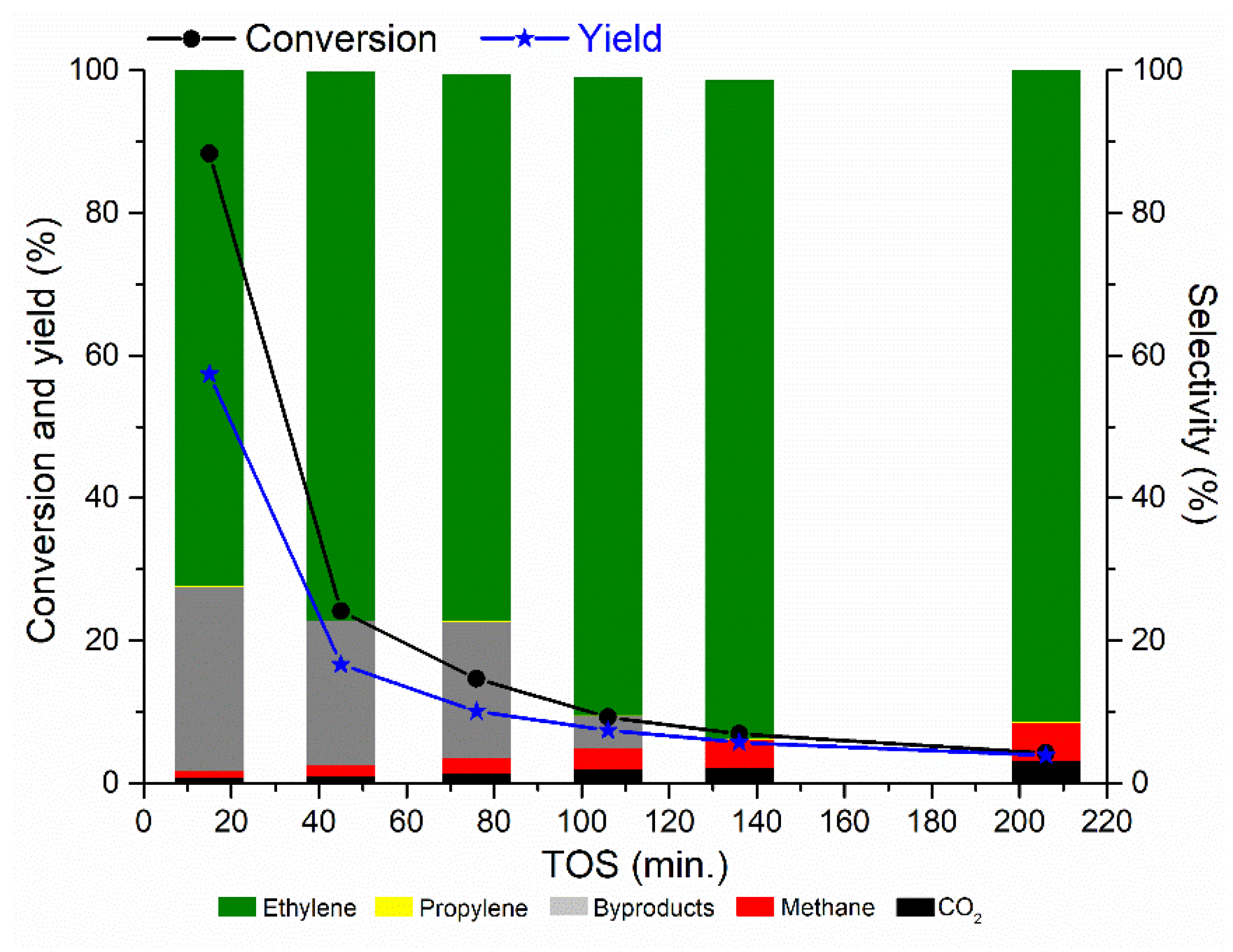
2.1. Catalytic Activity
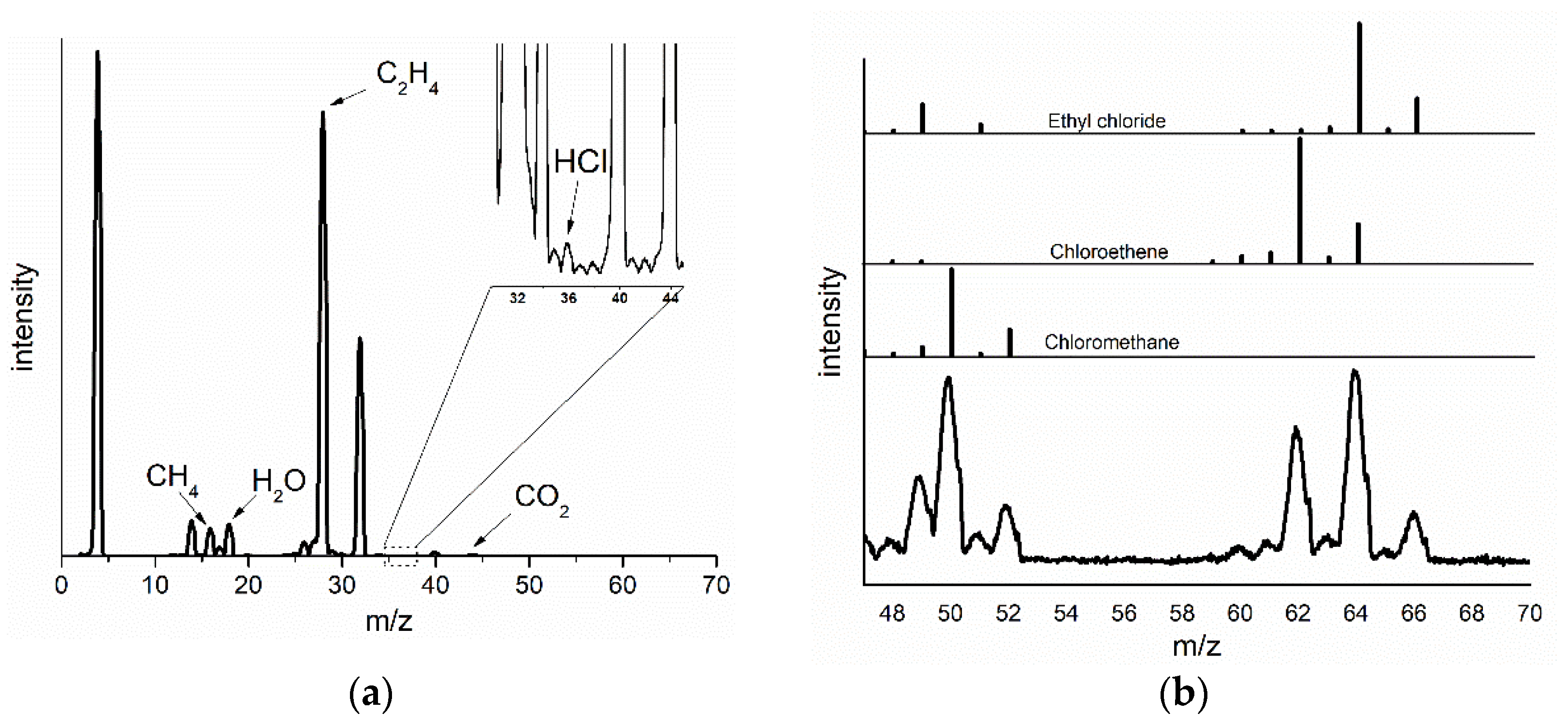
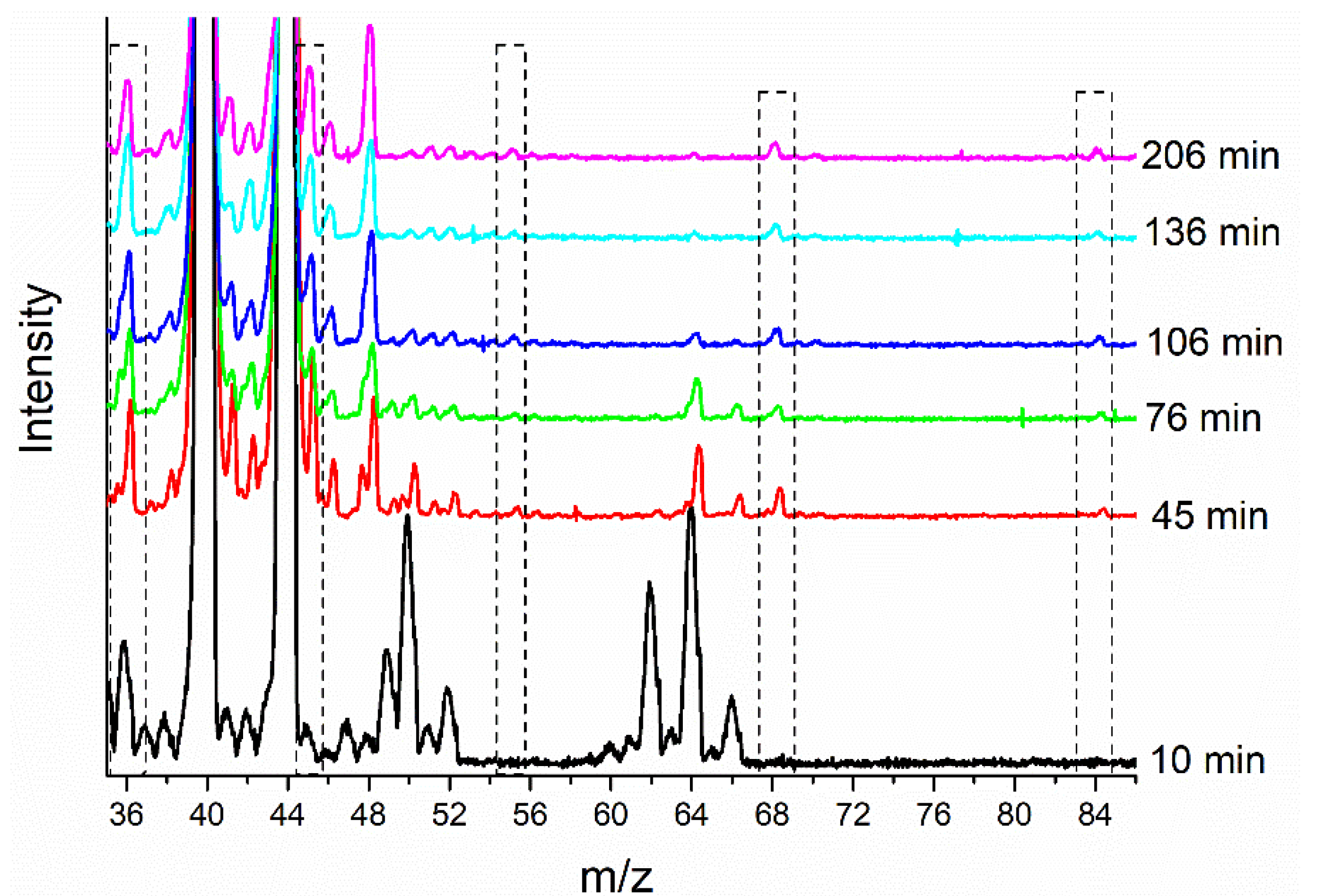
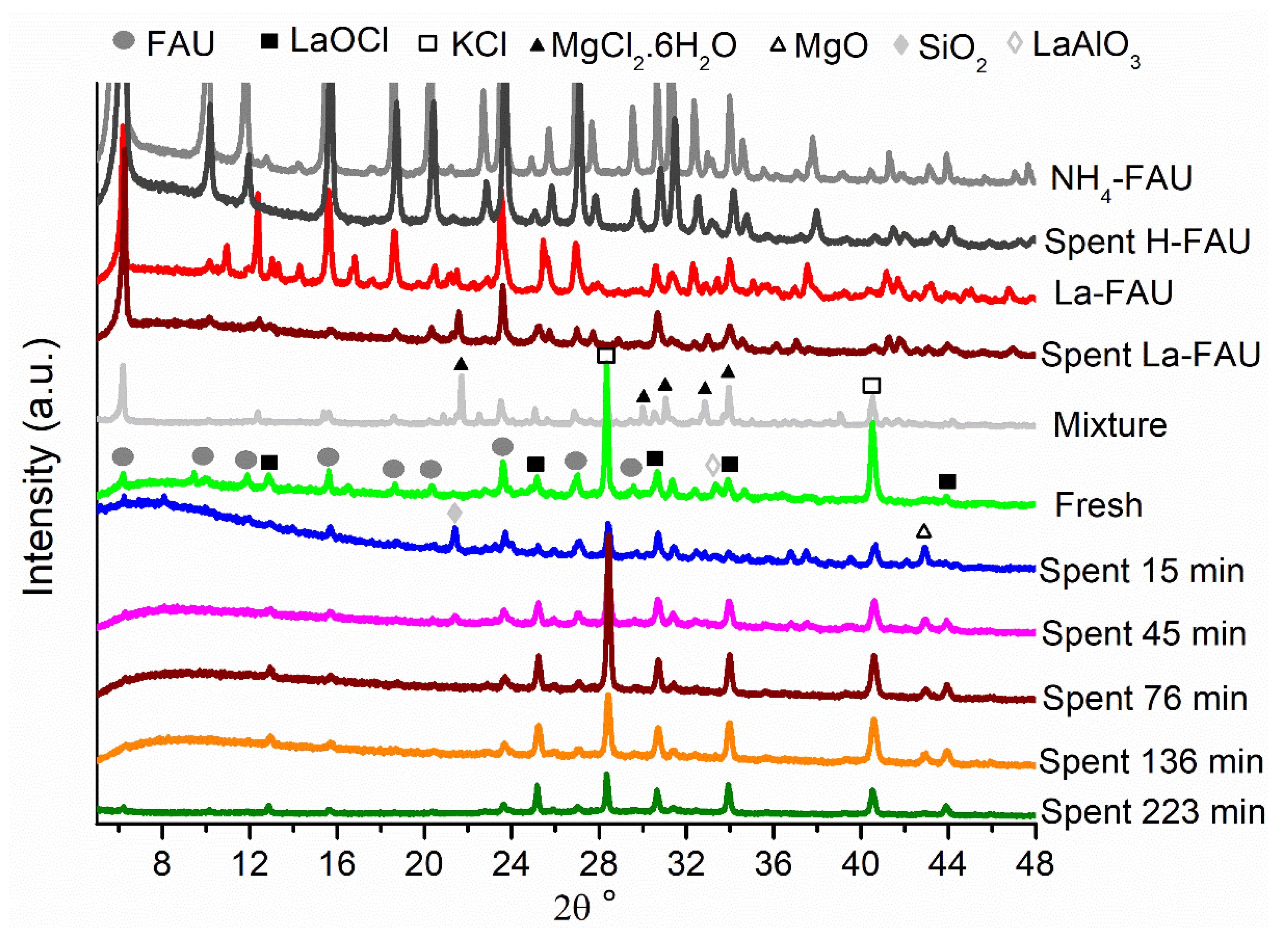
2.2. Changes in the Catalyst during Pretreatment and ODH Reaction
3. Materials and Methods
3.1. Catalysts Preparation
3.2. Catalytic Test
3.3. Characterization
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- James, O.O.; Mandal, S.; Alele, N.; Chowdhury, B.; Maity, S. Lower alkanes dehydrogenation: Strategies and reaction routes to corresponding alkenes. Fuel Process. Technol. 2016, 149, 239–255. [Google Scholar] [CrossRef]
- Sattler, J.J.H.B.; Ruiz-Martinez, J.; Santillan-Jimenez, E.; Weckhuysen, B.M. Catalytic Dehydrogenation of Light Alkanes on Metals and Metal Oxides. Chem. Rev. 2014, 114, 10613–10653. [Google Scholar] [CrossRef] [PubMed]
- Blasco, T.; Galli, A.; Nieto, J.L.; Trifiró, F. Oxidative Dehydrogenation of Ethane andn-Butane on VOx/Al2O3Catalysts. J. Catal. 1997, 169, 203–211. [Google Scholar] [CrossRef]
- Ciambelli, P.; Lisi, L.; Pirone, R.; Ruoppolo, G.; Russo, G. Comparison of behaviour of rare earth containing catalysts in the oxidative dehydrogenation of ethane. Catal. Today 2000, 61, 317–323. [Google Scholar] [CrossRef]
- Gärtner, C.A.; van Veen, A.C.; Lercher, A.J. Oxidative Dehydrogenation of Ethane: Common Principles and Mechanistic Aspects. ChemCatChem 2013, 5, 3196–3217. [Google Scholar] [CrossRef]
- Gärtner, C.A.; Van Veen, A.C.; Lercher, J.A. Oxidative Dehydrogenation of Ethane on Dynamically Rearranging Supported Chloride Catalysts. J. Am. Chem. Soc. 2014, 136, 12691–12701. [Google Scholar] [CrossRef] [PubMed]
- Bañares, M.A. Supported metal oxide and other catalysts for ethane conversion: A review. Catal. Today 1999, 51, 319–348. [Google Scholar] [CrossRef]
- Cavani, F.; Ballarini, N.; Cericola, A. Oxidative dehydrogenation of ethane and propane: How far from commercial implementation? Catal. Today 2007, 127, 113–131. [Google Scholar] [CrossRef]
- Carrero, C.; Schlögl, R.; Wachs, I.; Schomaecker, R. Critical literature review of the kinetics for the oxidative dehydrogenation of propane over well-defined supported vanadium oxide catalysts. ACS Catal. 2014, 4, 3357–3380. [Google Scholar] [CrossRef]
- Navalon, S.; Dhakshinamoorthy, A.; Alvaro, M.; Garcia, H. Carbocatalysis by Graphene-Based Materials. Chem. Rev. 2014, 114, 6179–6212. [Google Scholar] [CrossRef]
- Ronghe Lin, A.P.A.; Pérez-Ramírez, J. Halogen-Mediated Conversion of Hydrocarbons to Commodities. Chem. Rev. 2017, 117, 4182–4247. [Google Scholar]
- Burch, G.D.S.R.; Tsang, S.C. Comparative study of catalysts for the oxidative coupling of methane. Appl. Catal. A Gen. 1988, 43, 105–116. [Google Scholar] [CrossRef]
- Ueda, W.; Lin, S.-W.; Tohmoto, I. Highly selective oxidative dehydrogenation of ethane to ethene over layered complex metal chloride oxide catalysts. Catal. Lett. 1997, 44, 241–245. [Google Scholar] [CrossRef]
- Wang, S.; Murata, K.; Hayakawa, T.; Hamakawa, S.; Suzuki, K. Lithium-chloride-promoted sulfated zirconia catalysts for the oxidative dehydrogenation of ethane. Catal. Lett. 1999, 59, 173–178. [Google Scholar] [CrossRef]
- Wang, S.; Murata, K.; Hayakawa, T.; Hamakawa, S.; Suzuki, K. Oxidative Dehydrogenation of Ethane over Alkali Metal Chloride Modified Silica Catalysts. Energy Fuels 2000, 14, 899–903. [Google Scholar] [CrossRef]
- Li, M.; van Veen, A.C. Selective production of ethylene via continuous oxidative dehydrogenation of ethane in (Dy2O3/MgO)-(Li-K) Cl composite membrane reactor. Chem. Eng. J. 2019, 365, 344–350. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Chen, L.; Zou, G.; Luo, X.; Gao, R.; Chou, L.; Wang, X. A novel BaCl2–TiO2–SnO2 catalyst for the oxidative dehydrogenation of ethane. Catal. Commun. 2012, 25, 45–49. [Google Scholar] [CrossRef]
- Kristoffersen, H.H.; Metiu, H. Molten LiCl Layer Supported on MgO: Its Possible Role in Enhancing the Oxidative Dehydrogenation of Ethane. J. Phys. Chem. C 2015, 119, 8681–8691. [Google Scholar] [CrossRef]
- Zichittella, G.; Lüthi, J.; Paunović, V.; Pérez-Ramírez, J. Alkane Functionalization via Catalytic Oxychlorination: Performance as a Function of the Carbon Number. Energy Technol. 2020, 8. [Google Scholar] [CrossRef]
- Wang, D.J.; Rosynek, M.P.; Lunsford, J.H. The effect of chloride ions on a li+-mgo catalyst for the oxidative dehydrogenation of ethane. J. Catal. 1995, 151, 155–167. [Google Scholar] [CrossRef]
- Wang, S.; Murata, K.; Hayakawa, T.; Suzuki, K. Oxidative Dehydrogenation of Ethane over Zirconia-Supported Lithium Chloride Catalysts. Chem. Eng. Technol. 2000, 23, 1099–1103. [Google Scholar] [CrossRef]
- Gaab, S.; Find, J.; Grasselli, R.; Lercher, J. Oxidative ethane activation over oxide supported molten alkali metal chloride catalysts. In Catalyst Deactivation, Proceedings of the 7th International Symposium, Edinburgh, UK, 24–25 May 2004; Elsevier: London, UK, 2004; Volume 147, pp. 673–678. [Google Scholar]
- Yuan, X.H.; Zhao, Y.B.; Jin, Y.X.; Weng, W.Z.; Wan, H.L. Oxidative dehydrogenation of ethane to ethylene over LiCl/SO42−-ZrO2 catalyst. Chin. J. Catal. 2006, 27, 79–85. [Google Scholar]
- Tope, B.; Zhu, Y.; Lercher, J.A. Oxidative dehydrogenation of ethane over Dy2O3/MgO supported LiCl containing eutectic chloride catalysts. Catal. Today 2007, 123, 113–121. [Google Scholar] [CrossRef]
- Kumar, C.P.; Gaab, S.; Muller, T.E.; Lercher, J.A. Oxidative Dehydrogenation of Light Alkanes on Supported Molten Alkali Metal Chloride Catalysts. Top. Catal. 2008, 50, 156–167. [Google Scholar] [CrossRef]
- Ayari, F.; Charrad, R.; Asedegbega–Nieto, E.; Mhamdi, M.; Delahay, G.; Farhat, F.; Ghorbel, A. Ethane Oxidative Dehydrogenation over ternary and binary mixtures of alkaline and alkaline earth chlorides supported on zeolites. Microporous Mesoporous Mater. 2017, 250, 65–71. [Google Scholar] [CrossRef]
- Gärtner, C.A.; Van Veen, A.C.; Lercher, J.A. Highly Selective Supported Alkali Chloride Catalysts for the Oxidative Dehydrogenation of Ethane. Top. Catal. 2014, 57, 1236–1247. [Google Scholar] [CrossRef]
- Kristoffersen, H.H.; Metiu, H. Chemistry of Solvated Electrons in Molten Alkali Chloride Salts. J. Phys. Chem. C 2018, 122, 19603–19612. [Google Scholar] [CrossRef]
- Gaab, S.; Machli, M.; Find, J.; Grasselli, R.; Lercher, J. Oxidative Dehydrogenation of Ethane Over Novel Li/Dy/Mg Mixed Oxides: Structure–Activity Study. Top. Catal. 2003, 23, 151–158. [Google Scholar] [CrossRef]
- Grant, J.T.; Venegas, J.M.; McDermott, W.P.; Hermans, I. Aerobic Oxidations of Light Alkanes over Solid Metal Oxide Catalysts. Chem. Rev. 2018, 118, 2769–2815. [Google Scholar] [CrossRef]
- Bulánek, R.; Čičmanec, P.; Sheng-Yang, H.; Knotek, P.; Čapek, L.; Setnička, M. Effect of preparation method on nature and distribution of vanadium species in vanadium-based hexagonal mesoporous silica catalysts: Impact on catalytic behavior in propane ODH. Appl. Catal. A: Gen. 2012, 415–416, 29–39. [Google Scholar] [CrossRef]
- Zichittella, G.; Aellen, N.; Paunović, V.; Amrute, A.P.; Pérez-Ramírez, J. Olefins from Natural Gas by Oxychlorination. Angew. Chem. Int. Ed. 2017, 56, 13670–13674. [Google Scholar] [CrossRef] [PubMed]
- Leveles, L.; Fuchs, S.; Seshan, K.; Lercher, J.A.; Lefferts, L. Oxidative conversion of light alkanes to olefins over alkali promoted oxide catalysts. Appl. Catal. A: Gen. 2002, 227, 287–297. [Google Scholar] [CrossRef]
- Conway, S.J.; Lunsford, J.H. The oxidative dehydrogenation of ethane over chlorine-promoted lithium-magnesium oxide catalysts. J. Catal. 1991, 131, 513–522. [Google Scholar] [CrossRef]
- Yu, F.; Wu, X.; Zhang, Q.; Wang, Y. Oxidative dehydrogenation of ethane to ethylene in the presence of HCl over CeO2-based catalysts. Chin. J. Catal. 2014, 35, 1260–1266. [Google Scholar] [CrossRef]
- Gaab, S.; Find, J.; Muller, T.E.; Lercher, J.A. Kinetics and mechanism of the oxidative dehydrogenation of ethane over Li/Dy/Mg/O/(Cl) mixed oxide catalysts. Top Catal. 2007, 46, 101–110. [Google Scholar] [CrossRef]
- NIST Mass Spectrometry Data Center, W.E.W. Director, “Mass Spectra”. In NIST Chemistry WebBook; NIST Standard Reference Database Number, 69; Linstrom, P.J., Mallard, W.G., Eds.; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2005; p. 20899. retrieved March 18, 2021. [Google Scholar] [CrossRef]
- Wang, S.B.; Murata, K.; Hayakawa, T.; Hamakawa, S.; Suzuki, K. Performance of metal-oxide-promoted LiCl/sulfated-zirconia catalysts in the ethane oxidative dehydrogenation into ethene. Catal. Lett. 1999, 62, 191–195. [Google Scholar] [CrossRef]
- Schüßler, F.; Pidko, E.A.; Kolvenbach, R.; Sievers, C.; Hensen, E.J.M.; Van Santen, R.A.; Lercher, J.A. Nature and Location of Cationic Lanthanum Species in High Alumina Containing Faujasite Type Zeolites. J. Phys. Chem. C 2011, 115, 21763–21776. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | X * | S † (C2H4) | T # | Y ‡ | TOS | Ref. |
|---|---|---|---|---|---|---|
| S-ACl | 80 | 72 | 500 | 57 | 15 | This work |
| NaY-La(Cl)-(NaMg) | 2 | 100 | 500 | 2 | - | [26] |
| NaY-La(Cl)-(KMg) | 27 | 100 | 500 | 27 | - | [26] |
| NaY-La(Cl)-(RbMg) | 40 | 100 | 500 | 40 | - | [26] |
| NaY-La(Cl)-(CsMg) | 7 | 100 | 500 | 7 | - | [26] |
| NaY-Bin(NaMg) | 3 | 100 | 500 | 3 | - | [26] |
| NaY-La(Cl)-(NaKMg) | 20 | 100 | 500 | 20 | - | [26] |
| LiCl/SiO2 | 99 | 79 | 600 | 78 | 5 | [15] |
| NaCl/SiO2 | 88 | 69 | 550 | 61 | 5 | [15] |
| KCl/SiO2 | 70 | 75 | 550 | 52 | 5 | [15] |
| LiCl/SZ (sulfated zirconia) | 98 | 70 | 650 | 68 | 1 | [14] |
| LiCl/AZ (amorphous zirconia) | 87 | 60 | 650 | 52 | 1 | [21] |
| LiCl/ZrON | 95 | 71 | 650 | 68 | 1 | [21] |
| LiCl/ZrOCl | 28 | 97 | 650 | 27 | 1 | [21] |
| LiCl/ZrSO4 | 89 | 83 | 650 | 74 | 1 | [21] |
| LiCl/ SZ (sulfated zirconia) | 53 | 90 | 650 | 48 | 1 | [38] |
| BaCl2–TiO2–SnO2 | 66 | 93 | 720 | 60 | 30 | [17] |
| Li/Dy/Mg/O/C1 | - | - | 650 | 77 | - | [22] |
| (Li-K)Cl-(Dy2O3/MgO) membrane | 34 | 97 | 700 | 33 | - | [16] |
| (Li-K)Cl-(Dy2O3/MgO) membrane | 85 | 75 | 750 | 64 | - | [16] |
| S-ACl | 22 | 77 | 500 | 17 | 45 | This work |
| PbBi3O4Cl3 | 51 | 88 | 660 | 45 | 60 | [13] |
| S-ACl | 8 | 89 | 500 | 7 | 106 | This work |
| Li/K/Cl-MgO/Dy2O3 | 5 | 94 | 550 | 5 | 120 | [25] |
| Li/Na/Cl-MgO/Dy2O3 | 2 | 75 | 500 | 2 | 120 | [25] |
| Li/Cl-MgO/Dy2O3 | 2 | 60 | 500 | 1 | 120 | [25] |
| K/Cl-MgO/Dy2O3 | 2 | 40 | 500 | 1 | 120 | [25] |
| Na/Cl-MgO/Dy2O3 | 2 | 37 | 500 | 1 | 120 | [25] |
| S-ACl | 4 | 91 | 500 | 4 | 206 | This work |
| SrBi3O4Cl3 | 20 | 89 | 660 | 17 | 360 | [13] |
| SrBi3O4Cl3 + SrCl2 | 35 | 89 | 660 | 31 | 360 | [13] |
| SrBi3O4Cl3 + 2SrCl2 | 44 | 90 | 660 | 39 | 360 | [13] |
| SrBi3O4Cl3 + KCl | 36 | 96 | 660 | 34 | 360 | [13] |
| SrBi3O4Cl3 + SrCl2 + KCl | 45 | 92 | 660 | 42 | 360 | [13] |
| SrBi3O4Cl3 + SrCl2 + LiCl | 42 | 94 | 660 | 40 | 360 | [13] |
| SrBi3O4Cl3 + SrCl2 + NaCl | 41 | 95 | 660 | 39 | 360 | [13] |
| LiCl/ SZ (sulfated zirconia) | 70 | 66 | 650 | 46 | 900 | [14] |
| SBET (m2/g) a | Sext. (m2/g) b | Vmic. (cm3/g) c | Vp (cm3/g) d | |
|---|---|---|---|---|
| H-FAU | 965 | 16 | 0.34 | 0.38 |
| Spent H-FAU | 769 | 16 | 0.30 | 0.33 |
| La-FAU | 450 | 8 | 0.17 | 0.19 |
| Spent La-FAU | 442 | 16 | 0.17 | 0.21 |
| Mechanical mixture | 179 | 4 | 0.07 | 0.10 |
| Fresh | 7.7 | 7.7 | 0 | 0.015 |
| TOS: 15 min. | 5.9 | 5.9 | 0 | 0.010 |
| TOS: 45 min. | 7.9 | 7.9 | 0 | 0.016 |
| TOS: 76 min. | 6.8 | 6.8 | 0 | 0.014 |
| TOS: 136 min. | 7.8 | 7.8 | 0 | 0.016 |
| TOS: 223 min. | 10 | 10 | 0 | 0.03 |
| Spent Catalysts, Different TOS (Min.) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Element | Nominal | NH4-FAU | La-FAU | Fresh | 15 | 45 | 76 | 136 | 223 |
| O | 44.3 | 64.4 | 64.2 | 52.7 | 57.8 | 57.6 | 56.3 | 57.6 | 60.0 |
| Na | 0.1 | 1.4 | 1.1 | 0.7 | 0.9 | 0.9 | 0.7 | 0.7 | 0.7 |
| Mg | 8.3 | 0.0 | 0.0 | 9.4 | 11.6 | 10.4 | 13.1 | 13.2 | 9.7 |
| Al | 3.8 | 7.9 | 7.1 | 3.9 | 4.4 | 4.5 | 4.2 | 4.0 | 4.3 |
| Si | 9.8 | 20.2 | 17.9 | 9.1 | 11.1 | 11.4 | 10.8 | 10.4 | 11.8 |
| Cl | 24.3 | 0.0 | 5.9 | 11.5 | 11.1 | 7.6 | 7.1 | 6.6 | 6.3 |
| K | 7.6 | 0.0 | 0.0 | 5.8 | 5.5 | 5.2 | 5.4 | 5.1 | 5.4 |
| La | 1.7 | 0.0 | 3.7 | 1.7 | 2.6 | 2.5 | 2.4 | 2.5 | 1.9 |
| N | 0 | 6.1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sajad, M.; Bulánek, R.; Šlang, S. Physico-Chemical Changes in the KCl-MgCl2/La-FAU Composite Catalyst Induced by Oxidative Dehydrogenation of Ethane. Catalysts 2021, 11, 392. https://doi.org/10.3390/catal11030392
Sajad M, Bulánek R, Šlang S. Physico-Chemical Changes in the KCl-MgCl2/La-FAU Composite Catalyst Induced by Oxidative Dehydrogenation of Ethane. Catalysts. 2021; 11(3):392. https://doi.org/10.3390/catal11030392
Chicago/Turabian StyleSajad, Mehran, Roman Bulánek, and Stanislav Šlang. 2021. "Physico-Chemical Changes in the KCl-MgCl2/La-FAU Composite Catalyst Induced by Oxidative Dehydrogenation of Ethane" Catalysts 11, no. 3: 392. https://doi.org/10.3390/catal11030392