
Elucidating the Influence of Electric Fields toward CO2 Activation on YSZ (111)

Abstract
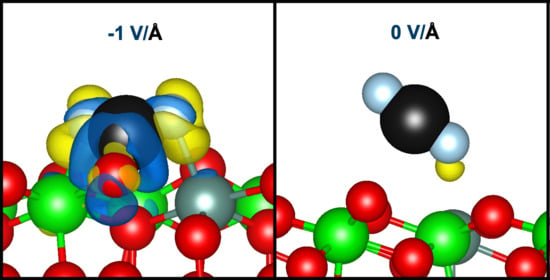
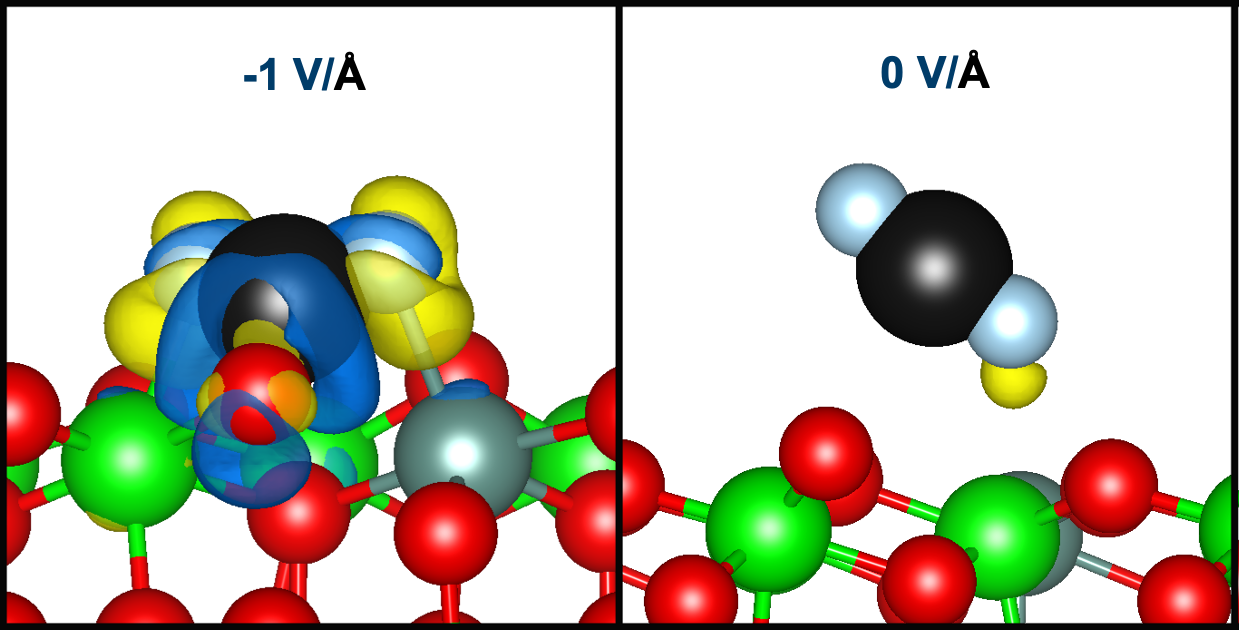
:
1. Introduction
2. Results and Discussion
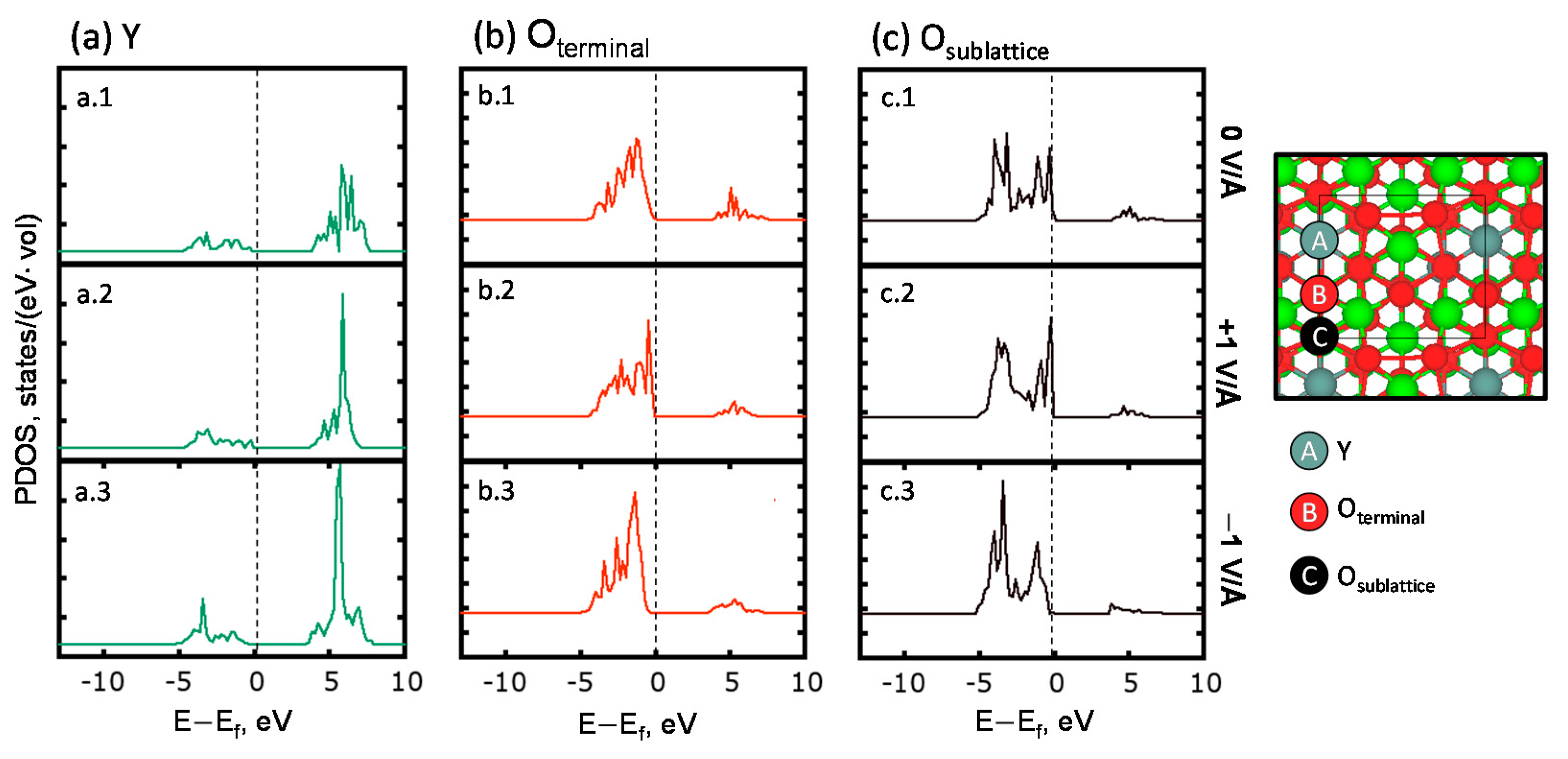
2.1. Field-Assisted Oxygen Vacancy Formation on YSZ (111)
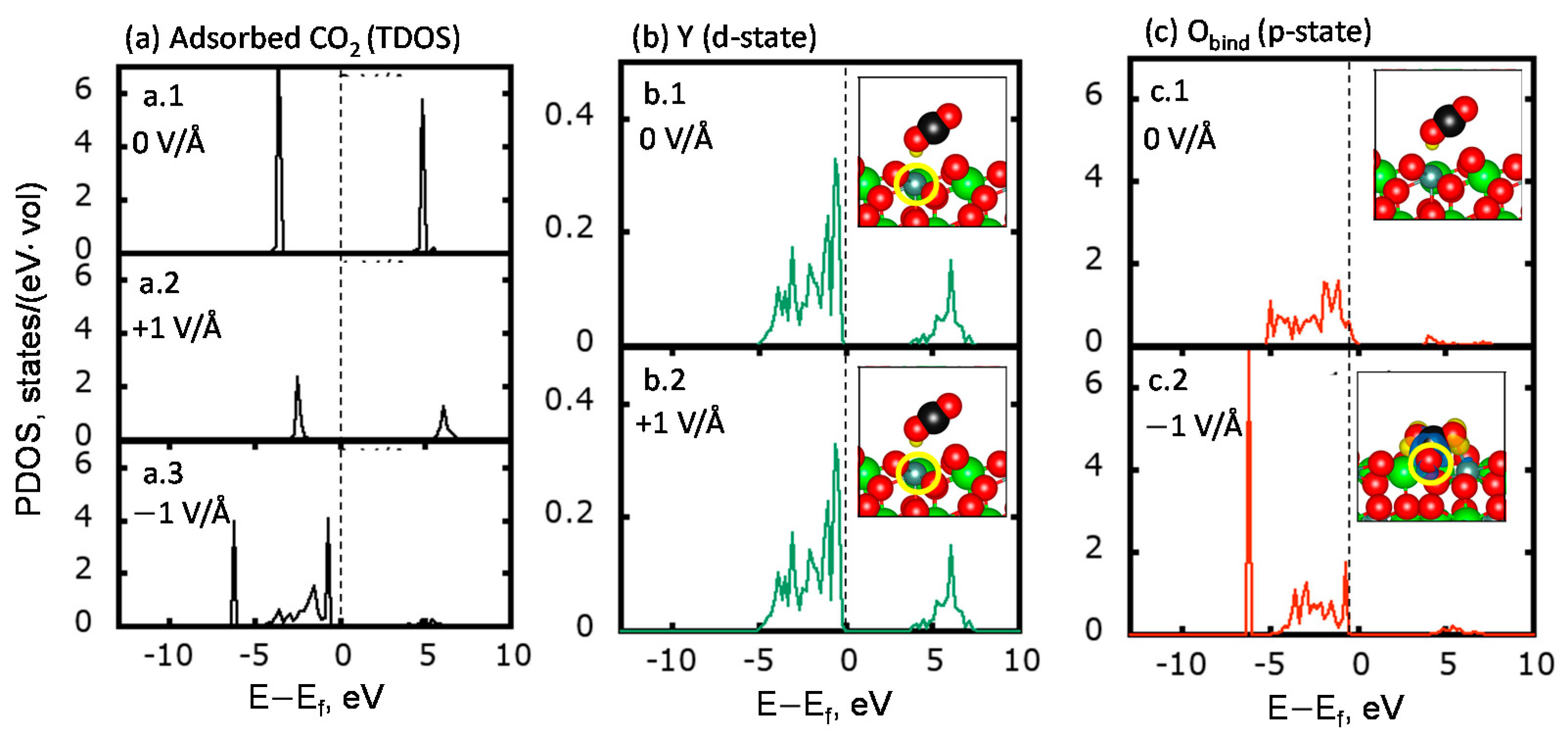
2.2. Elucidating the Influence of an Electric Field toward CO2 Activation on YSZ (111)
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pham Minh, D.; Siang, T.J.; Vo, D.-V.N.; Phan, T.S.; Ridart, C.; Nzihou, A.; Grouset, D. Chapter 4—Hydrogen Production from Biogas Reforming: An Overview of Steam Reforming, Dry Reforming, Dual Reforming, and Tri-Reforming of Methane; Azzaro-Pantel, C., Ed.; Academic Press, Elsevier: New York, NY, USA, 2018; pp. 111–166. ISBN 978-0-12-811197-0. [Google Scholar] [CrossRef] [Green Version]
- Nalbant, Y.; Colpan, C.O. An Overview of Hydrogen Production from Biogas BT—Accelerating the Transition to a 100% Renewable Energy Era; Uyar, T.S., Ed.; Springer International Publishing: Cham, Switzerland, 2020; pp. 355–373. ISBN 978-3-030-40738-4. [Google Scholar]
- Nehring, R. Traversing the mountaintop: World fossil fuel production to 2050. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 3067–3079. [Google Scholar] [CrossRef] [PubMed]
- Lelieveld, J.; Klingmüller, K.; Pozzer, A.; Burnett, R.T.; Haines, A.; Ramanathan, V. Effects of fossil fuel and total anthropogenic emission removal on public health and climate. Proc. Natl. Acad. Sci. USA 2019, 116, 7192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoel, M.; Kverndokk, S. Depletion of fossil fuels and the impacts of global warming. Resour. Energy Econ. 1996, 18, 115–136. [Google Scholar] [CrossRef]
- Pareek, A.; Dom, R.; Gupta, J.; Chandran, J.; Adepu, V.; Borse, P.H. Insights into renewable hydrogen energy: Recent advances and prospects. Mater. Sci. Energy Technol. 2020, 3, 319–327. [Google Scholar] [CrossRef]
- Staffell, I.; Scamman, D.; Velazquez Abad, A.; Balcombe, P.; Dodds, P.E.; Ekins, P.; Shah, N.; Ward, K.R. The role of hydrogen and fuel cells in the global energy system. Energy Environ. Sci. 2019, 12, 463–491. [Google Scholar] [CrossRef] [Green Version]
- Marcoberardino, G.; Vitali, D.; Spinelli, F.; Binotti, M.; Manzolini, G. Green Hydrogen Production from Raw Biogas: A Techno-Economic Investigation of Conventional Processes Using Pressure Swing Adsorption Unit. Processes 2018, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Alves, H.J.; Bley Junior, C.; Niklevicz, R.R.; Frigo, E.P.; Frigo, M.S.; Coimbra-Araújo, C.H. Overview of hydrogen production technologies from biogas and the applications in fuel cells. Int. J. Hydrogen Energy 2013, 38, 5215–5225. [Google Scholar] [CrossRef]
- Gray, J.T.; Che, F.; McEwen, J.-S.; Ha, S. Field-assisted suppression of coke in the methane steam reforming reaction. Appl. Catal. B Environ. 2020, 260, 118132. [Google Scholar] [CrossRef]
- Che, F.; Gray, J.T.; Ha, S.; Kruse, N.; Scott, S.L.; McEwen, J.-S. Elucidating the Roles of Electric Fields in Catalysis: A Perspective. ACS Catal. 2018, 8, 5153–5174. [Google Scholar] [CrossRef]
- Che, F.; Gray, J.T.; Ha, S.; McEwen, J.-S. Improving Ni Catalysts Using Electric Fields: A DFT and Experimental Study of the Methane Steam Reforming Reaction. ACS Catal. 2017, 7, 551–562. [Google Scholar] [CrossRef]
- Che, F.; Gray, J.T.; Ha, S.; McEwen, J.-S. Catalytic water dehydrogenation and formation on nickel: Dual path mechanism in high electric fields. J. Catal. 2015, 332, 187–200. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.S.; Lee, H.H.; Hong, S.C. A study on the effect of support’s reducibility on the reverse water-gas shift reaction over Pt catalysts. Appl. Catal. A Gen. 2012, 423–424, 100–107. [Google Scholar] [CrossRef]
- Bernal, S.; Blanco, G.; Gatica, J.M.; Larese, C.; Vidal, H. Effect of Mild Re-oxidation Treatments with CO2 on the Chemisorption Capability of a Pt/CeO2 Catalyst Reduced at 500 °C. J. Catal. 2001, 200, 411–415. [Google Scholar] [CrossRef]
- Zhu, M.; Ge, Q.; Zhu, X. Catalytic Reduction of CO2 to CO via Reverse Water Gas Shift Reaction: Recent Advances in the Design of Active and Selective Supported Metal Catalysts. Trans. Tianjin Univ. 2020, 26, 172–187. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Chen, W.; Zhang, D.; Du, Y.; Amal, R.; Qiao, S.; Wu, J.; Yin, Z. Surface strategies for catalytic CO2 reduction: From two-dimensional materials to nanoclusters to single atoms. Chem. Soc. Rev. 2019, 48, 5310–5349. [Google Scholar] [CrossRef] [PubMed]
- Köck, E.M.; Kogler, M.; Bielz, T.; Klötzer, B.; Penner, S. In situ FT-IR spectroscopic study of CO2 and CO adsorption on Y2O3, ZrO2, and yttria-stabilized ZrO2. J. Phys. Chem. C 2013, 117, 17666–17673. [Google Scholar] [CrossRef] [PubMed]
- Goguet, A.; Meunier, F.C.; Tibiletti, D.; Breen, J.P.; Burch, R. Spectrokinetic investigation of reverse water-gas-shift reaction intermediates over a Pt/CeO2 catalyst. J. Phys. Chem. B 2004, 108, 20240–20246. [Google Scholar] [CrossRef] [Green Version]
- Bobadilla, L.F.; Santos, J.L.; Ivanova, S.; Odriozola, J.A.; Urakawa, A. Unravelling the Role of Oxygen Vacancies in the Mechanism of the Reverse Water–Gas Shift Reaction by Operando DRIFTS and Ultraviolet–Visible Spectroscopy. ACS Catal. 2018, 8, 7455–7467. [Google Scholar] [CrossRef]
- Álvarez, A.; Borges, M.; Corral-Pérez, J.J.; Olcina, J.G.; Hu, L.; Cornu, D.; Huang, R.; Stoian, D.; Urakawa, A. CO2 Activation over Catalytic Surfaces. ChemPhysChem 2017, 18, 3135–3141. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Goddard, W.A.; Cheng, T.; Liu, Y. Cu metal embedded in oxidized matrix catalyst to promote CO2 activation and CO dimerization for electrochemical reduction of CO2. Proc. Natl. Acad. Sci. USA 2017, 114, 6685–6688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daza, Y.A.; Kuhn, J.N. CO2 conversion by reverse water gas shift catalysis: Comparison of catalysts, mechanisms and their consequences for CO2 conversion to liquid fuels. RSC Adv. 2016, 6, 49675–49691. [Google Scholar] [CrossRef]
- Oshima, K.; Shinagawa, T.; Nogami, Y.; Manabe, R.; Ogo, S.; Sekine, Y. Low temperature catalytic reverse water gas shift reaction assisted by an electric field. Catal. Today 2014, 232, 27–32. [Google Scholar] [CrossRef]
- Lambeets, S.V.; Barroo, C.; Owczarek, S.; Jacobs, L.; Genty, E.; Gilis, N.; Kruse, N.; Visart de Bocarmé, T. Adsorption and Hydrogenation of CO2 on Rh Nanosized Crystals: Demonstration of the Role of Interfacet Oxygen Spillover and Comparative Studies with O2, N2O, and CO. J. Phys. Chem. C 2017, 121, 16238–16249. [Google Scholar] [CrossRef]
- Calvaresi, M.; Martinez, R.V.; Losilla, N.S.; Martinez, J.; Garcia, R.; Zerbetto, F. Splitting CO2 with Electric Fields: A Computational Investigation. J. Phys. Chem. Lett. 2010, 1, 3256–3260. [Google Scholar] [CrossRef]
- Sterrer, M.; Freund, H.-J. Properties of Oxide Surfaces. Surf. Interface Sci. 2013, 229–278. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter Mater. Phys. 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558. [Google Scholar] [CrossRef] [PubMed]
- Lejaeghere, K.; Bihlmayer, G.; Björkman, T.; Blaha, P.; Blügel, S.; Blum, V.; Caliste, D.; Castelli, I.E.; Clark, S.J.; Dal Corso, A.; et al. Reproducibility in density functional theory calculations of solids. Science 2016, 351, aad3000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 11–19. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Che, F.; Ha, S.; McEwen, J.S. Elucidating the role of the electric field at the Ni/YSZ electrode: A DFT study. J. Phys. Chem. C 2016, 120, 14608–14620. [Google Scholar] [CrossRef]
- Shishkin, M.; Ziegler, T. Oxidation of H2, CH4, and CO molecules at the interface between nickel and yttria-stabilized zirconia: A theoretical study based on DFT. J. Phys. Chem. C 2009, 113, 21667–21678. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Che, F.; Ha, S.; McEwen, J.S. Hydrogen Oxidation and Water Dissociation over an Oxygen-Enriched Ni/YSZ Electrode in the Presence of an Electric Field: A First-Principles-Based Microkinetic Model. Ind. Eng. Chem. Res. 2017, 56, 1201–1213. [Google Scholar] [CrossRef]
- Neugebauer, J.; Scheffler, M. Adsorbate-substrate and adsorbate-adsorbate interactions of Na and K adlayers on Al(111). Phys. Rev. B 1992, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feibelman, P.J. Surface-diffusion mechanism versus electric field: Pt/Pt(001). Phys. Rev. B Condens. Matter Mater. Phys. 2001, 64, 1–6. [Google Scholar] [CrossRef]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Wiberg, K.B.; Rablen, P.R. Comparison of atomic charges derived via different procedures. J. Comput. Chem. 1993, 14, 1504–1518. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Studt, F.; Abild-Pedersen, F.; Bligaard, T. Fundamental Concepts in Heterogeneous Catalysis; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; ISBN 9781118892114. [Google Scholar]
- Che, F.; Zhang, R.; Hensley, A.J.; Ha, S.; McEwen, J.-S. Density functional theory studies of methyl dissociation on a Ni(111) surface in the presence of an external electric field. Phys. Chem. Chem. Phys. 2014, 16, 2399–2410. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Applied Electric Field, V/Å | Bonding d-States (F), Electrons/vol | Antibonding d-States (U), Electrons/vol | U/(Nd) Ratio |
|---|---|---|---|
| 0 | 0.94 | 2.73 | 0.74 |
| 1 | 0.98 | 2.45 | 0.71 |
| −1 | 1.22 | 3.81 | 0.76 |
| Electric Field, V/Å | Bader Partial Charge = Valence − Bader Charge | ||
|---|---|---|---|
| Y | Oterminal | Osublattice | |
| 0 | 2.19 | −1.29 | −1.33 |
| 1 | 2.20 | −1.30 | −1.33 |
| −1 | 2.20 | −1.28 | −1.33 |
| Applied Electric Field, V/Å | Oterminal | Osublattice | ||
|---|---|---|---|---|
| U/Np Ratio (p-States) | Total Bonding States (s + p), Electrons/vol | U/Np Ratio (p-States) | Total Bonding States (s + p), Electrons/vol | |
| 0 | 0.14 | 4.36 | 0.08 | 4.74 |
| 1 | 0.10 | 3.28 | 0.06 | 5.24 |
| −1 | 0.11 | 4.50 | 0.06 | 5.31 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ulumuddin, N.; Che, F.; Yang, J.-I.; Ha, S.; McEwen, J.-S. Elucidating the Influence of Electric Fields toward CO2 Activation on YSZ (111). Catalysts 2021, 11, 271. https://doi.org/10.3390/catal11020271
Ulumuddin N, Che F, Yang J-I, Ha S, McEwen J-S. Elucidating the Influence of Electric Fields toward CO2 Activation on YSZ (111). Catalysts. 2021; 11(2):271. https://doi.org/10.3390/catal11020271
Chicago/Turabian StyleUlumuddin, Nisa, Fanglin Che, Jung-Il Yang, Su Ha, and Jean-Sabin McEwen. 2021. "Elucidating the Influence of Electric Fields toward CO2 Activation on YSZ (111)" Catalysts 11, no. 2: 271. https://doi.org/10.3390/catal11020271