
Enhanced Catalytic Hydrogen Peroxide Production from Hydroxylamine Oxidation on Modified Activated Carbon Fibers: The Role of Surface Chemistry


Abstract
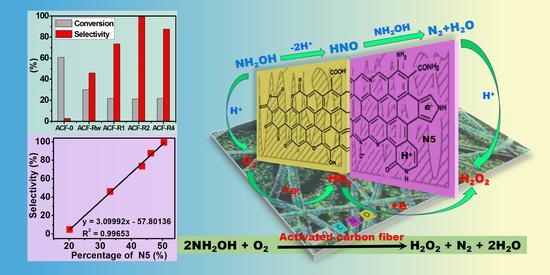
:
1. Introduction
2. Results and Discussion
2.1. Material Structure
2.2. Surface Properties
2.3. H2O2 Production
2.4. Effect of Surface Nitrogen- and Oxygen-Containing Groups
3. Materials and Methods
3.1. Surface Modification of ACF
3.2. Characterization of the ACF Catalysts
3.3. Catalyst Testing
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Campos-Martin, J.M.; Blanco-Brieva, G.; Fierro, J.L.G. Hydrogen peroxide synthesis: An out- look beyond the anthraquinone process. Angew. Chem. Int. Ed. 2006, 45, 6962–6984. [Google Scholar] [CrossRef]
- Xue, Y.; Wang, Y.; Pan, Z.; Sayama, K. Electrochemical and photoelectrochemical water oxidation for hydrogen peroxide production. Angew. Chem. Int. Ed. 2021, 60, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Goor, G.; Kunkel, W.; Weiberg, O. Hydrogen peroxide. In UllmannCs Encyclopedia of Industrial Chemistry, 5th ed.; Elvers, B., Hawkins, S., Ravenscroft, M., Schulz, G., Eds.; Wiley-VCH: New York, NY, USA; Basel, Switzerland; Cambridge, UK; Weinheim, Germany, 1989; Volume A13, pp. 443–466. [Google Scholar]
- Kholdeeva, O.; Maksimchuk, N. Metal-organic frameworks in oxidation catalysis with hydrogen peroxide. Catalysts 2021, 11, 283. [Google Scholar] [CrossRef]
- Puértolas, B.; Hillb, A.K.; García, T.; Solson, B.; Torrente-Murciano, L. In-situ synthesis of hydrogen peroxide in tandem with selective oxidation reactions: A mini-review. Catal. Today 2015, 248, 115–127. [Google Scholar] [CrossRef]
- Hu, X.; Zeng, X.; Liu, Y.; Lu, J.; Zhang, X. Carbon-based materials for photo- and electrocatalytic synthesis of hydrogen peroxide. Nanoscale 2020, 12, 16008–16027. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Beckman, E.J. One-pot green synthesis of propylene oxide using in situ generated hydrogen peroxide in carbon dioxide. Green Chem. 2008, 10, 934–938. [Google Scholar] [CrossRef]
- Miller, J.A.; Alexander, L.; Mori, D.I.; Ryabov, A.D.; Collins, T.J. In situ enzymatic generation of H2O2 from O2 for use in oxidative bleaching and catalysis by TAML activators. New J. Chem. 2013, 37, 3488–3495. [Google Scholar] [CrossRef]
- Asghar, A.; Raman, A.A.A.; Daud, W.M.A.W. Recent advances, challenges and prospects of in situ production of hydrogen peroxide for textile waste water treatment in microbial fuel cells. J. Chem. Technol. Biotechnol. 2014, 89, 1466–1480. [Google Scholar] [CrossRef]
- Pan, Z.; Wang, K.; Wang, Y.; Tsiakaras, P.; Song, S. In-situ electrosynthesis of hydrogen peroxide and wastewater treatment application: A novel strategy for graphite felt activation. Appl. Catal. B Environ. 2018, 237, 392–400. [Google Scholar] [CrossRef]
- Giorgianni, G.; Abate, S.; Centi, G.; Perathoner, S. Direct synthesis of H2O2 on Pd based catalysts: Modelling the particle size effects and the promoting role of polyvinyl alcohol. ChemCatChem 2019, 11, 550–559. [Google Scholar] [CrossRef]
- Freakley, S.J.; Kochius, S.; van Marwijk, J.; Fenner, C.; Lewis, R.J.; Baldenius, K.; Marais, S.S.; Susan, D.J.O.; Harrison, T.L.; Alcalde, M.; et al. A chemo-enzymatic oxidation cascade to activate C-H bonds with in situ generated H2O2. Nat. Commun. 2019, 10, 4178. [Google Scholar] [CrossRef]
- Van Schie, M.M.C.H.; Kaczmarek, A.T.; Tieves, F.; de Santos, P.G.; Paul, C.E.; Arends, I.W.C.E.; Alcalde, M.; Schwarz, G.; Hollmann, F. Selective oxyfunctionalisation reactions driven by sulfite oxidase-catalysed in situ generation of H2O2. ChemCatChem 2020, 12, 3186–3189. [Google Scholar] [CrossRef]
- Lyu, J.; Niu, L.; Shen, F.; Wei, J.; Xiang, Y.; Yu, Z.; Zhang, G.; Ding, C.; Huang, Y.; Li, X. In situ hydrogen peroxide production for selective oxidation of benzyl alcohol over a Pd@hierarchical titanium silicalite catalyst. ACS Omega 2020, 5, 16865–16874. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, Y.; Wang, J. Fenton/Fenton-like processes with in-situ production of hydrogen peroxide/hydroxyl radical for degradation of emerging contaminants: Advances and prospects. J. Hazard. Mater. 2021, 404, 124191–124210. [Google Scholar] [CrossRef]
- Samanta, C. Direct synthesis of hydrogen peroxide from hydrogen and oxygen: An overview of recent developments in the process. Appl. Catal. A Gen. 2008, 350, 133–149. [Google Scholar] [CrossRef]
- Edwards, J.K.; Freakley, S.J.; Carley, A.F.; Kiely, C.J.; Hutchings, G.J. Strategies for designing supported gold-palladium bimetallic catalysts for the direct synthesis of hydrogen peroxide. Acc. Chem. Res. 2014, 47, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.K.; Freakley, S.J.; Lewis, R.J.; Pritchard, J.C.; Hutchings, G.J. Advances in the direct synthesis of hydrogen peroxide from hydrogen and oxygen. Catal. Today 2015, 248, 3–9. [Google Scholar] [CrossRef]
- Yi, Y.; Wang, L.; Li, G.; Guo, H. A review on research progress in the direct synthesis of hydrogen peroxide from hydrogen and oxygen: Noble-metal catalytic method, fuel-cell method and plasma method. Catal. Sci. Technol. 2016, 6, 1593–1610. [Google Scholar] [CrossRef]
- Gao, G.; Tian, Y.; Gong, X.; Pan, Z.; Yang, K.; Zong, B. Advances in the production technology of hydrogen peroxide. Chin. J. Catal. 2020, 41, 1039–1047. [Google Scholar] [CrossRef]
- Ntainjua, N.E.; Piccinini, M.; Pritchard, J.C.; Edwards, J.K.; Carley, A.F.; Moulijn, J.A.; Hutchings, G.J. Effect of halide and acid additives on the direct synthesis of hydrogen peroxide using supported gold–palladium catalysts. ChemSusChem 2009, 2, 575–580. [Google Scholar] [CrossRef]
- Pritchard, J.C.; He, Q.; Ntainjua, E.N.; Piccinini, M.; Edwards, J.K.; Herzing, A.A.; Carley, A.F.; Moulijn, J.A.; Kiely, C.J.; Hutchings, G.J. The effect of catalyst preparation method on the performance of supported Au–Pd catalysts for the direct synthesis of hydrogen peroxide. Green Chem. 2010, 12, 915–921. [Google Scholar] [CrossRef]
- Pritchard, J.; Kesavan, L.; Piccinini, M.; He, Q.; Tiruvalam, R.; Dimitratos, N.; Lopez-Sanchez, J.A.; Carley, A.F.; Edwards, J.K.; Kiely, C.J.; et al. Direct synthesis of hydrogen peroxide and benzyl alcohol oxidation using Au−Pd catalysts prepared by sol immobilization. Langmuir 2010, 26, 16568–16577. [Google Scholar] [CrossRef] [PubMed]
- Tiruvalam, R.C.; Pritchard, J.C.; Dimitratos, N.; Lopez-Sanchez, J.A.; Edwards, J.K.; Carley, A.F.; Hutchings, G.J.; Kiely, C.J. Aberration corrected analytical electron microscopy studies of sol-immobilized Au + Pd, Au {Pd} and Pd {Au} catalysts used for benzyl alcohol oxidation and hydrogen peroxide production. Faraday Discuss. 2011, 152, 63–86. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.; Piccinini, M.; Tiruvalam, R.; He, Q.; Dimitratos, N.; Lopez-Sanchez, J.A.; Morgan, D.J.; Carley, A.F.; Edwards, J.K.; Kiely, C.J.; et al. Effect of heat treatment on Au–Pd catalysts synthesized by sol immobilisation for the direct synthesis of hydrogen peroxide and benzyl alcohol oxidation. Catal. Sci. Technol. 2013, 3, 308–317. [Google Scholar] [CrossRef]
- Hou, H.; Zeng, X.; Zhang, X. Production of hydrogen peroxide by photocatalytic processes. Angew. Chem. Int. Ed. 2020, 59, 17356–17376. [Google Scholar] [CrossRef]
- Song, H.; Wei, L.; Chen, L.; Zhang, H.; Su, J. Photocatalytic production of hydrogen peroxide over modifed semiconductor materials: A minireview. Top. Catal. 2020, 63, 895–912. [Google Scholar] [CrossRef]
- Hughes, M.N.; Nicklin, H.G. Autoxidation of hydroxylamine in alkaline solutions. J. Chem. Soc. A 1971, 1, 164–168. [Google Scholar] [CrossRef]
- Sheriff, T.S. Production of hydrogen peroxide from dioxygen and hydroxylamine or hydrazine catalysed by manganese complexes. J. Chem. Soc. Dalton Trans. 1992, 6, 1051–1058. [Google Scholar] [CrossRef]
- Sheriff, T.S.; Carr, P.; Piggott, B. Manganese catalysed reduction of dioxygen to hydrogen peroxide: Structural studies on a manganese (III)–catecholate complex. Inorg. Chim. Acta 2003, 348, 115–122. [Google Scholar] [CrossRef]
- Sheriff, T.S.; Carr, P.; Coles, S.J.; Hursthouse, M.B.; Lesin, J.; Light, M.E. Structural studies on manganese (III) and manganese (IV) complexes of tetrachlorocatechol and the catalytic reduction of dioxygen to hydrogen peroxide. Inorg. Chim. Acta 2004, 357, 2494–2502. [Google Scholar] [CrossRef]
- Choudhary, V.R.; Jana, P.; Bhargava, S.K. Reduction of oxygen by hydroxylammonium salt or hydroxylamine over supported Au nanoparticles for in situ generation of hydrogen peroxide in aqueous or non-aqueous medium. Catal. Commun. 2007, 8, 811–816. [Google Scholar] [CrossRef]
- Choudhary, V.R.; Jana, P. In situ generation of hydrogen peroxide from reaction of O2 with hydroxylamine from hydroxylammonium salt in neutral aqueous or non-aqueous medium using reusable Pd/Al2O3 catalyst. Catal. Commun. 2007, 8, 1578–1582. [Google Scholar] [CrossRef]
- Choudhary, V.R.; Jana, P. Factors influencing the in situ generation of hydrogen peroxide from the reduction of oxygen by hydroxylamine from hydroxylammonium sulfate over Pd/alumina. Appl. Catal. A Gen. 2008, 335, 95–102. [Google Scholar] [CrossRef]
- Song, W.; Li, J.; Liu, J.; Shen, W. Production of hydrogen peroxide by the reaction of hydroxylamine and molecular oxygen over activated carbons. Catal. Commun. 2008, 9, 831–836. [Google Scholar] [CrossRef]
- Song, W.; Yu, L.; Xie, X.; Hao, Z.; Sun, M.; Wen, H.; Li, Y. Effect of textual features and surface properties of activated carbon on the production of hydrogen peroxide from hydroxylamine oxidation. RSC Adv. 2017, 7, 25305–25313. [Google Scholar] [CrossRef] [Green Version]
- Melchionna, M.; Fornasiero, P.; Prato, M. The rise of hydrogen peroxide as the main product by metal-free catalysis in oxygen reductions. Adv. Mater. 2019, 31, 1802920–1802924. [Google Scholar] [CrossRef]
- Sun, Y.; Sinev, I.; Ju, W.; Bergmann, A.; Dresp, S.; Kühl, S.; Spöri, C.; Schmies, H.; Wang, H.; Bernsmeier, D.; et al. Efficient electrochemical hydrogen peroxide production from molecular oxygen on nitrogen-doped mesoporous carbon catalysts. ACS Catal. 2018, 8, 2844–2856. [Google Scholar] [CrossRef]
- Raymundo-Piñero, E.; Cazorla-Amorós, D.; Linares-Solano, A. Temperature programmed desorption study on the mechanism of SO2 oxidation by activated carbon and activated carbon fibres. Carbon 2001, 39, 231–242. [Google Scholar] [CrossRef]
- Boehm, H.P. Catalytic properties of nitrogen-containing carbons. In Carbon Materials for Catalysis, 1st ed.; Serp, P., Figueiredo, J.L., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009; Chapter 7; pp. 219–238. [Google Scholar]
- De la Puente, G.; Pis, J.J.; Menéndez, J.A.; Grange, P. Thermal stability of oxygenated functions in activated carbons. J. Anal. Appl. Pyrolysis 1997, 43, 125–138. [Google Scholar] [CrossRef]
- Prahas, D.; Kartika, Y.; Indraswati, N.; Ismadji, S. Activated carbon from jackfruit peel waste by H3PO4 chemical activation: Pore structure and surface chemistry characterization. Chem. Eng. J. 2008, 140, 32–42. [Google Scholar] [CrossRef]
- Yang, S.; Li, L.; Xiao, T.; Zheng, D.; Zhang, Y. Role of surface chemistry in modified ACF (activated carbon fiber)-catalyzed peroxymonosulfate oxidation. Appl. Surf. Sci. 2016, 383, 142–150. [Google Scholar] [CrossRef]
- Macías-García, A.; Díaz-Díez, M.A.; Cuerda-Correa, E.M.; Olivares-Marín, M.; Gañan-Gómez, J. Study of the pore size distribution and fractal dimension of HNO3-treated activated carbons. Appl. Surf. Sci. 2006, 252, 5972–5975. [Google Scholar] [CrossRef]
- Kima, M.J.; Song, E.J.; Kim, K.H.; Choi, S.S.; Lee, Y.S. The textural and chemical changes in ACFs with e-beam and their influence on the detection of nerve agent simulant gases. J. Ind. Eng. Chem. 2019, 79, 465–472. [Google Scholar] [CrossRef]
- Shi, M.; Bao, D.; Li, S.; Wulan, B.; Yan, J.; Jiang, Q. Anchoring PdCu amorphous nanocluster on graphene for electrochemical reduction of N2 to NH3 under ambient conditions in aqueous solution. Adv. Energy Mater. 2018, 8, 1800124–1800129. [Google Scholar] [CrossRef]
- Terzyk, A.P. The influence of activated carbon surface chemical composition on the adsorption of acetaminophen (paracetamol) in vitro: Part II. TG, FTIR, and XPS analysis of carbons and the temperature dependence of adsorption kinetics at the neutral pH. Colloids Surf. A 2001, 177, 23–45. [Google Scholar] [CrossRef]
- Swiatkowski, A.; Pakula, M.; Biniak, S.; Walczyk, M. Influence of the surface chemistry of modified activated carbon on its electrochemical behaviour in the presence of lead(II) ions. Carbon 2004, 42, 3057–3069. [Google Scholar] [CrossRef]
- Brazhnyk, D.V.; Zaitsev, Y.P.; Bacherikova, I.V.; Zazhigalov, V.A.; Stoch, J.; Kowal, A. Oxidation of H2S on activated carbon KAU and influence of the surface state. Appl. Catal. B Environ. 2007, 70, 557–566. [Google Scholar] [CrossRef]
- Fels, J.R.; Kapteijn, F.; Moulijn, J.A.; Zhu, Q.; Thomas, K.M. Evolution of nitrogen functionalities in carbonaceous materials during pyrolysis. Carbon 1995, 33, 1641–1653. [Google Scholar]
- Pietrzak, R. XPS study and physico-chemical properties of nitrogen-enriched microporous activated carbon from high volatile bituminous coal. Fuel 2009, 88, 1871–1877. [Google Scholar] [CrossRef]
- Kundu, S.; Xia, W.; Busser, W.; Kundu, S.; Xia, W.; Busser, W.; Becker, M.; Schmidt, D.A.; Havenith, M.; Muhle, M. The formation of nitrogen-containing functional groups on carbon nanotube surfaces: A quantitative XPS and TPD study. Phys. Chem. Chem. Phys. 2010, 12, 4351–4359. [Google Scholar] [CrossRef]
- Yang, G.; Chen, H.; Qin, H.; Yang, G.; Chen, H.; Qin, H.; Feng, Y. Amination of activated carbon for enhancing phenol adsorption: Effect of nitrogen-containing functional groups. Appl. Surf. Sci. 2014, 293, 299–305. [Google Scholar] [CrossRef]
- Sun, H.; Kwan, C.; Wang, S.; Sun, H.; Kwan, C.K.; Suvorova, A.; Ang, H.M.; Tadé, M.O.; Wang, S. Catalytic oxidation of organic pollutants on pristine and surface nitrogen-modifified carbon nanotubes with sulfate radicals. Appl. Catal. B Environ. 2014, 154–155, 134–141. [Google Scholar] [CrossRef]
- Boehm, H.P. Surface oxides on carbon and their analys is: A critical assessment. Carbon 2002, 40, 145–149. [Google Scholar] [CrossRef]
- Zielke, U.; Hüttinger, K.J.; Hoffman, W.P. Surface-oxidized carbon fibers: I. Surface structure and chemistry. Carbon 1996, 34, 983–998. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, H.; Cheng, M.; Lu, Q. Tailoring the electrochemical production of H2O2: Strategies for the rational design of high-performance electrocatalysts. Small 2020, 16, 1902845–1902861. [Google Scholar] [CrossRef]
- Jiang, Y.; Ni, P.; Chen, C.; Lu, Y.; Yang, P.; Kong, B.; Fisher, A.; Wang, X. Selective electrochemical H2O2 production through two-electron oxygen electrochemistry. Adv. Energy Mater. 2018, 8, 1801909–1801933. [Google Scholar] [CrossRef]
- Figueiredo, J.L.; Pereira, M.F.R. Carbon as Catalyst. In Carbon Materials for Catalysis, 1st ed.; Serp, P., Figueiredo, J.L., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009; Chapter 6; pp. 196–198. [Google Scholar]
- Song, W.; Li, Y.; Guo, X.; Li, J.; Huang, X.; Shen, W. Selective surface modifification of activated carbon for enhancing the catalytic performance in hydrogen peroxide production by hydroxylamine oxidation. J. Mol. Catal. Chem. 2010, 328, 53–59. [Google Scholar] [CrossRef]
- Figueiredo, J.L.; Pereira, M.F.R.; Freitas, M.M.A.; Órfão, J.J.M. Modification of the surface chemistry of activated carbons. Carbon 1999, 37, 1379–1389. [Google Scholar] [CrossRef]
- Clapp, P.A.; Evans, D.F.; Sheriff, T.S. Spectrophotometric determination of hydrogen peroxide after extraction with ethyl acetate. Anal. Chim. Acta 1989, 218, 331–334. [Google Scholar] [CrossRef]
- Yang, M. Hydroxylamine hydrochloride was determined by indirect spectrophotometry. Chem. Ind. Eng. (China) 1999, 16, 233–235. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | SBET[a] (m2/g) | Smic.[b] (m2/g) | Smes.[b] (m2/g) | Vmic.[b] (cm3/g) | VTotal[c] (cm3/g) | dpore[d] (nm) |
|---|---|---|---|---|---|---|
| ACF-0 | 944 | 909 | 35 | 0.374 | 0.411 | 0.87 |
| ACF-Rw | 934 | 895 | 39 | 0.361 | 0.395 | 0.85 |
| ACF-R1 | 686 | 601 | 85 | 0.261 | 0.323 | 0.94 |
| ACF-R2 | 481 | 356 | 124 | 0.146 | 0.229 | 0.95 |
| ACF-R4 | 368 | 322 | 46 | 0.132 | 0.160 | 0.95 |
| Catalyst | Functional Groups/Binding Energy (eV) | ||||
|---|---|---|---|---|---|
| Peak I C–graphite 284.7–284.8 | Peak II C–O 286.0–286.3 | Peak III C=O 287.7–288.1 | Peak IV –COO– 289.2–289.6 | Peak V π–π* 291.2 | |
| ACF-0 | 72.5 | 18.8 | 5.7 | 2.0 | 1.0 |
| ACF-Rw | 67.9 | 19.9 | 6.4 | 4.5 | 1.3 |
| ACF-R1 | 63.4 | 23.4 | 7.8 | 5.4 | ― |
| ACF-R2 | 57.4 | 24.8 | 9.5 | 7.9 | 1.9 |
| ACF-R4 | 56.5 | 23.7 | 10.3 | 8.7 | 0.8 |
| Catalyst | Functional Groups/Binding Energy (eV) | O/C (%) | |||
|---|---|---|---|---|---|
| Peak I C=O 531.6–531.8 | Peak II C–O 532.9–533.0 | Peak III H2Oads, O2ads 534.3–534.7 | Peak IV CO2ads, COads 536.9–537.0 | ||
| ACF-0 | 23.2 | 51.0 | 23.3 | 2.5 | 12.0 |
| ACF-Rw | 25.5 | 54.2 | 20.1 | 0.7 | 21.3 |
| ACF-R1 | 27.6 | 56.3 | 15.3 | 0.9 | 24.9 |
| ACF-R2 | 30.7 | 56.0 | 12.9 | 0.5 | 32.2 |
| ACF-R4 | 30.5 | 55.8 | 13.5 | 0.2 | 32.9 |
| Catalyst | Functional Groups/Binding Energy (eV) | N/C (%) | ||||
|---|---|---|---|---|---|---|
| N6 Pyridine 398.7 | N5 Pyrrolic/Pyridone 400.1–400.2 | NQ Quaternary N 401.2–401.3 | N4 Pyridine-N-oxide 402.6 | NX –NO2 406.0 | ||
| ACF-0 | 15.6 | 20.2 | 46.8 | 17.5 | ― | 1.4 |
| ACF-Rw | 3.2 | 33.2 | 26.0 | 5.4 | 32.3 | 1.5 |
| ACF-R1 | 2.8 | 43.4 | 24.3 | 7.3 | 22.3 | 1.7 |
| ACF-R2 | 5.2 | 50.5 | 22.8 | 6.2 | 15.3 | 2.4 |
| ACF-R4 | 4.7 | 46.3 | 22.9 | 7.1 | 19.0 | 2.0 |
| Catalyst | CO2 Desorption (μmol/g) | |||
|---|---|---|---|---|
| Carboxyl [a] | Anhydride [b] | Lactone [c] | Total | |
| ACF-0 | 32 | 15 | 17 | 65 |
| ACF-Rw | 195 | 132 | 73 | 400 |
| ACF-R1 | 317 | 245 | 116 | 678 |
| ACF-R2 | 407 | 420 | 136 | 963 |
| ACF-R4 | 406 | 425 | 122 | 953 |
| Catalyst | CO Desorption (μmol/g) | |||
|---|---|---|---|---|
| Anhydride [d] | Phenol [e] | Carbonyl [f] | Total | |
| ACF-0 | 15 | 83 | 146 | 244 |
| ACF-Rw | 132 | 739 | 137 | 1008 |
| ACF-R1 | 245 | 823 | 139 | 1207 |
| ACF-R2 | 420 | 627 | 125 | 1172 |
| ACF-R4 | 425 | 551 | 67 | 1043 |
| Catalyst | [H2O2] (mmol/L) | Reaction Time (h) | Hydrogen Source | Conversion (%) | Selectivity (%) | Yield (%) | Reference |
|---|---|---|---|---|---|---|---|
| ACF-Rw | 60.8 | 11.0 | NH2OH | 72.9 | 33.3 | 24.3 | This work |
| ACF-R1 | 88.5 | 11.0 | NH2OH | 50.5 | 70.1 | 35.4 | This work |
| ACF-R4 | 112 | 11.0 | NH2OH | 47.4 | 94.5 | 44.8 | This work |
| ACF-R2 | 123 | 11.0 | NH2OH | 49.2 | 100 | 49.2 | This work |
| ACF-R2 a | 117 | 11.0 | NH2OH | 56.0 | 83.6 | 46.8 | This work |
| ACF-R2 | 53 | 3.0 | NH2OH | 21.3 | 99.3 | 21.2 | This work |
| ACH | 114 | 7.0 | NH2OH | - | - | 46.7 | [35] |
| ACP | ~50 | 3.0 | NH2OH | ~23 | ~87 | 20.0 | [36] |
| Mn2+-Tiron | ~225 | 6.0 | NH2OH | - | - | ~90 | [29] |
| Mn3+- Complex b | ~185 | 0.75 | NH2OH | - | - | ~74 | [30] |
| Au/MgO | 32.6 | 1.0 | NH2OH | - | - | 81.5 | [32] |
| Pd/Al2O3 | 37.6 | 1.0 | NH2OH | - | - | 94.0 | [33] |
| Au-Pd/C c | 69 | 0.5 | H2 | - | - | - | [21] |
| AuPd/C c | 62 | 0.5 | H2 | - | - | - | [22] |
| Au-Pd Catalyst c | 94 | 0.5 | H2 | - | - | - | [23] |
| Au + Pd/C c | 79 | 0.5 | H2 | - | - | - | [24] |
| Pd{Au}/C c | 77.5 | 0.5 | H2 | - | - | - | [25] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, W.; Zhao, R.; Yu, L.; Xie, X.; Sun, M.; Li, Y. Enhanced Catalytic Hydrogen Peroxide Production from Hydroxylamine Oxidation on Modified Activated Carbon Fibers: The Role of Surface Chemistry. Catalysts 2021, 11, 1515. https://doi.org/10.3390/catal11121515
Song W, Zhao R, Yu L, Xie X, Sun M, Li Y. Enhanced Catalytic Hydrogen Peroxide Production from Hydroxylamine Oxidation on Modified Activated Carbon Fibers: The Role of Surface Chemistry. Catalysts. 2021; 11(12):1515. https://doi.org/10.3390/catal11121515
Chicago/Turabian StyleSong, Wei, Ran Zhao, Lin Yu, Xiaowei Xie, Ming Sun, and Yongfeng Li. 2021. "Enhanced Catalytic Hydrogen Peroxide Production from Hydroxylamine Oxidation on Modified Activated Carbon Fibers: The Role of Surface Chemistry" Catalysts 11, no. 12: 1515. https://doi.org/10.3390/catal11121515