Enzymatic Synthesis of Estolides from Castor Oil


Abstract
:1. Introduction
2. Results
2.1. Lipase Screening
2.1.1. Liquid Enzymes
2.1.2. Immobilized Enzymes
2.2. Optimizing the CALA Reaction
2.2.1. Temperature
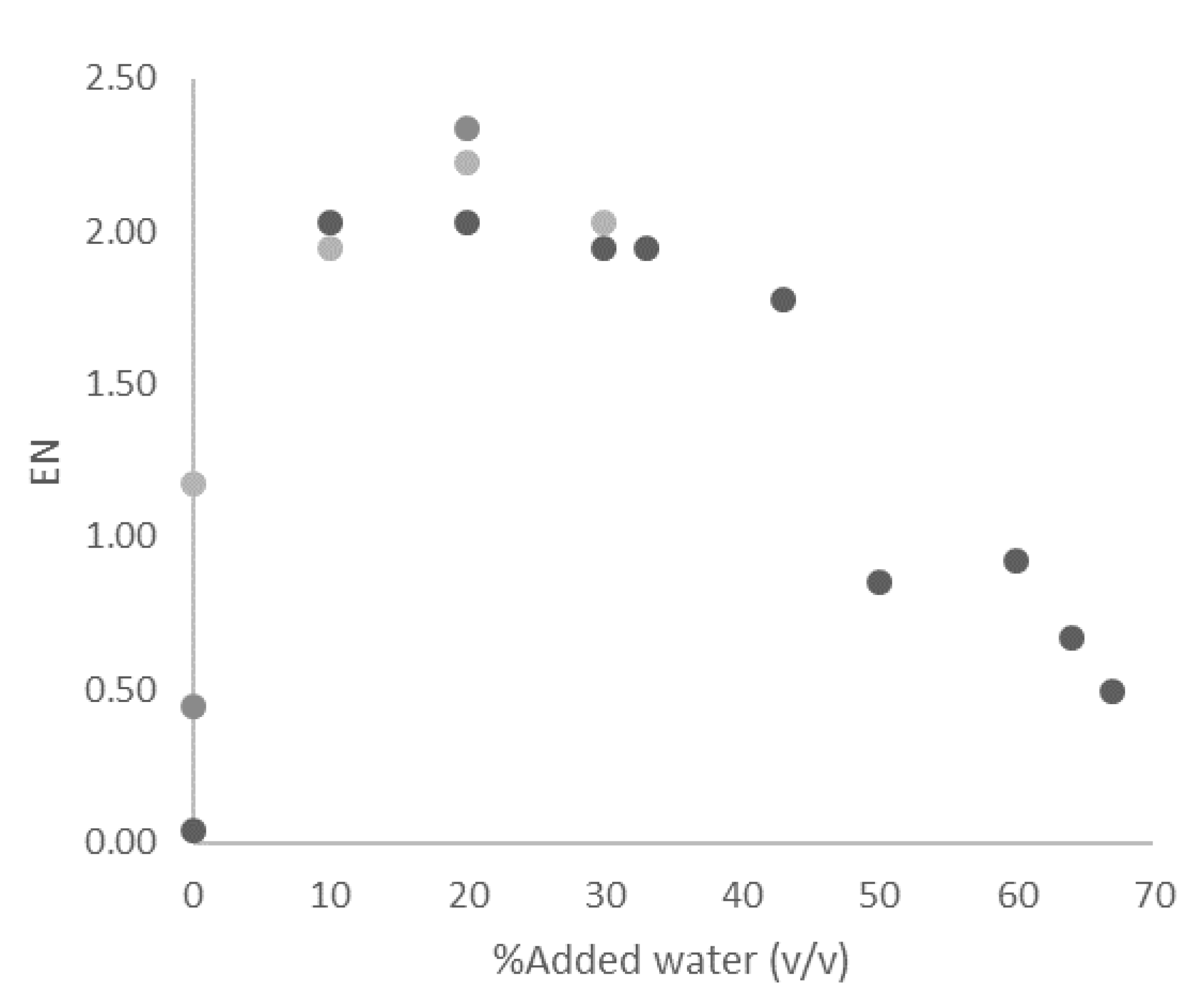
2.2.2. Water Content
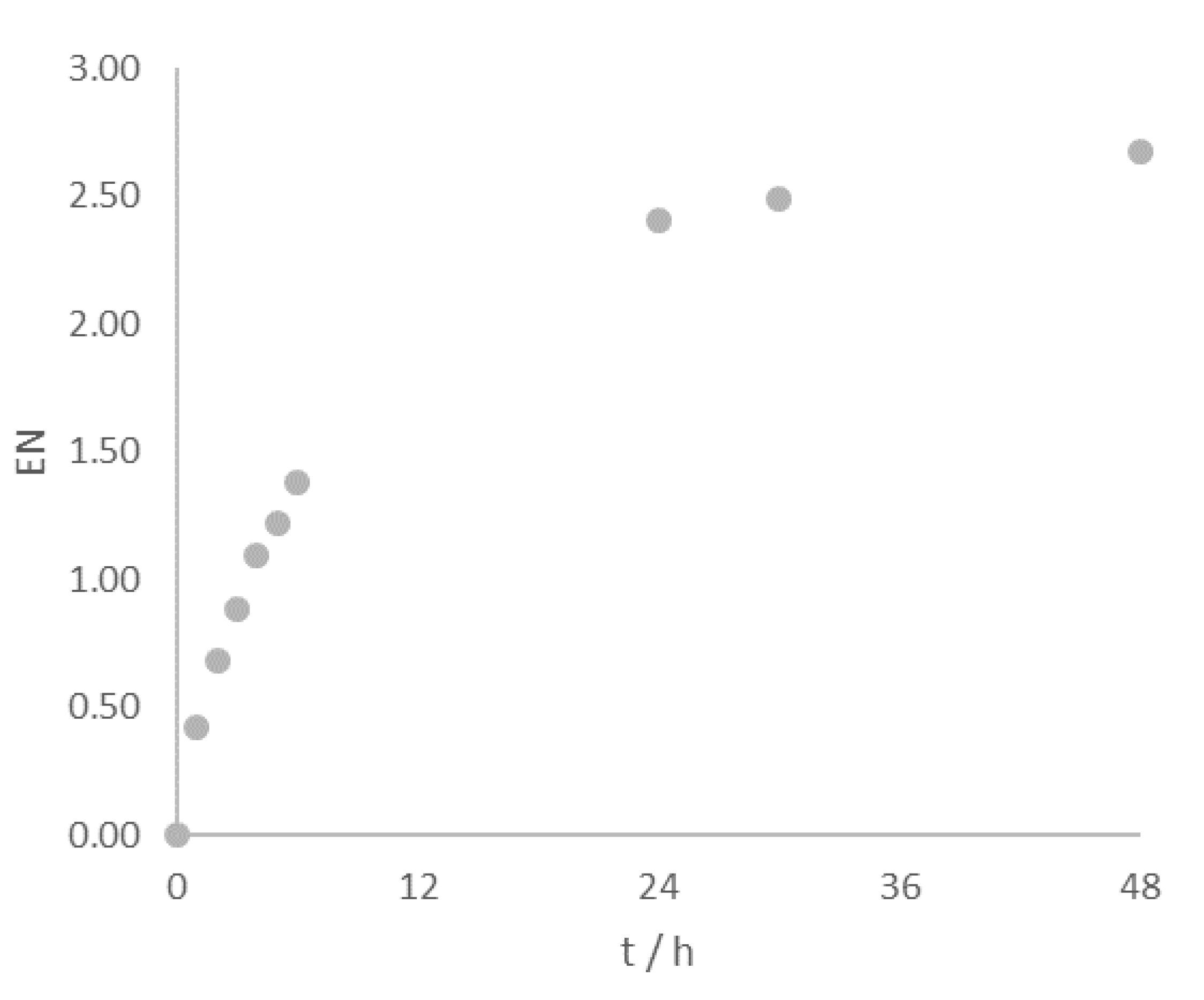
2.2.3. Time Course
2.2.4. pH-Effect
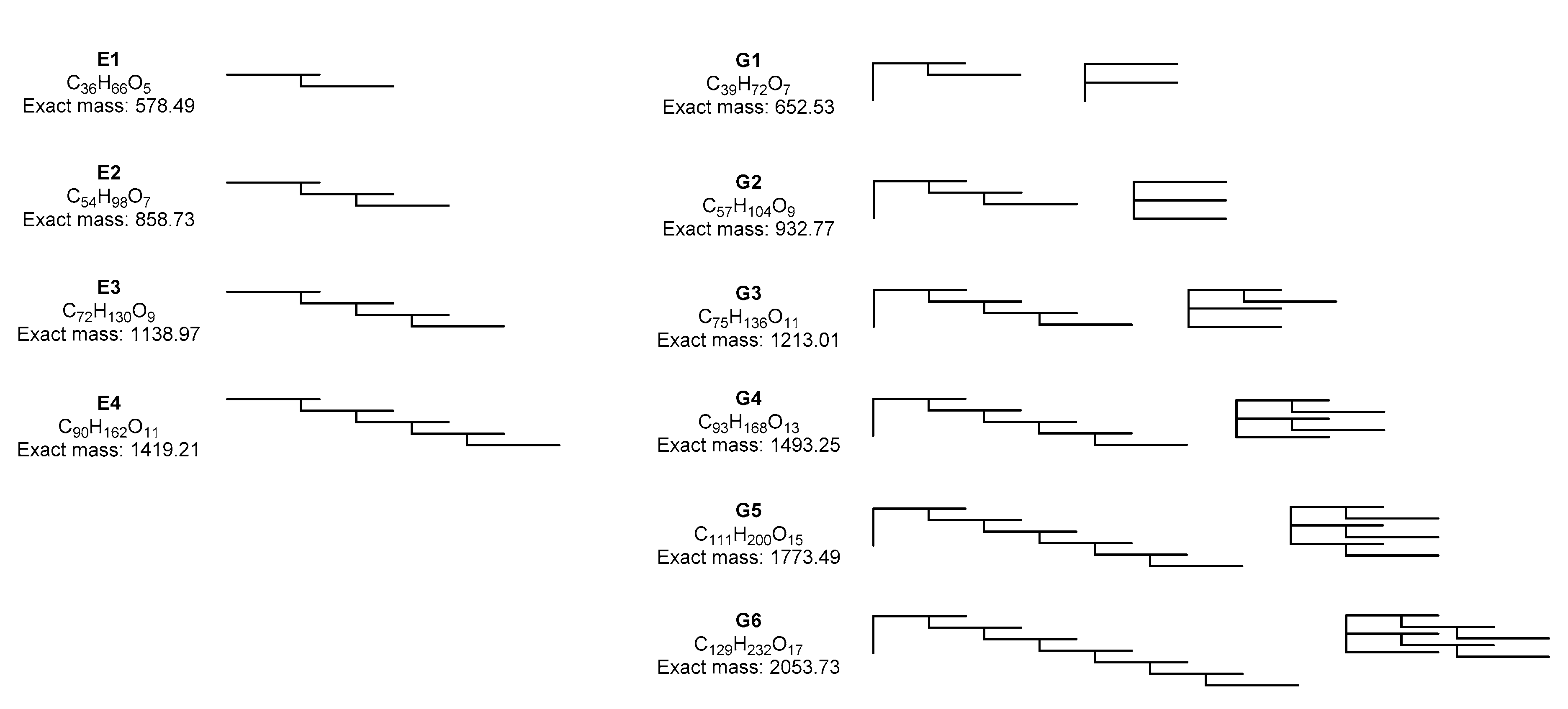
2.3. Two-Enzyme Process
2.3.1. Reaction Sequence
2.3.2. Scale up
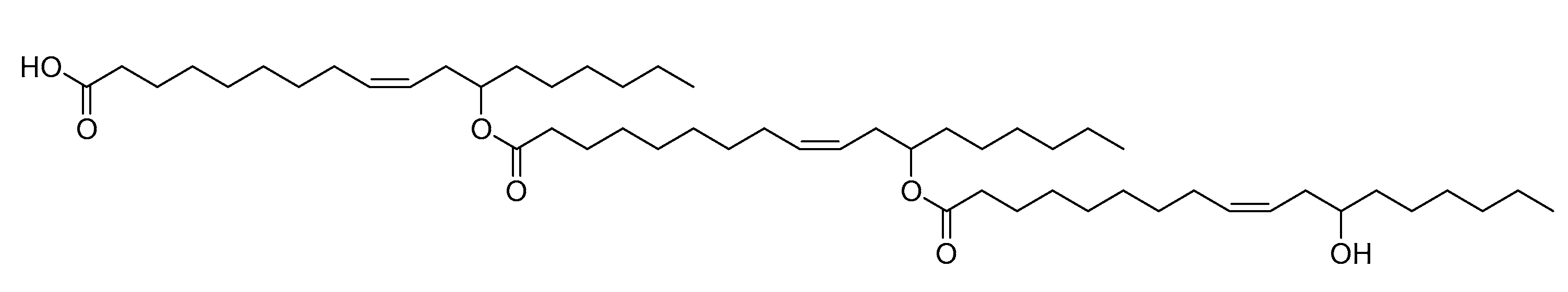
2.4. Three-Enzyme Process
3. Discussion
4. Materials and Methods
4.1. General Procedures
4.2. Analytical Procedures
4.3. Multi-Enzyme Processes
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kolar, M.J.; Nelson, A.T.; Chang, T.N.; Ertunc, M.E.; Christy, M.P.; Ohlsson, L.; Harrod, M.; Kahn, B.B.; Siegel, D.; Saghatelian, A. Faster protocol for endogenous fatty acid esters of hydroxy fatty acid (FAHFA) measurements. Anal. Chem. 2018, 90, 5358–5365. [Google Scholar] [CrossRef] [PubMed]
- Greco-Duarte, J.; Collaco, A.C.A.; Costa, A.M.M.; Silva, L.O.; Da Silva, J.A.C.; Torres, A.G.; Fernandez-Lafuente, R.; Freire, D.M.G. Understanding the degree of estolide enzymatic polymerization and the effects on its lubricant properties. Fuel 2019, 245, 286–293. [Google Scholar] [CrossRef]
- Greco-Duarte, J.; Cavalcanti-Oliveira, E.D.; Da Silva, J.A.C.; Fernandez-Lafuente, R.; Freire, D.M.G. Two-step enzymatic production of environmentally friendly biolubricants using castor oil: Enzyme selection and product characterization. Fuel 2017, 202, 196–205. [Google Scholar] [CrossRef]
- McNutt, J.; He, Q. Development of biolubricants from vegetable oils via chemical modification. J. Ind. Eng. Chem. 2016, 36, 1–12. [Google Scholar] [CrossRef]
- Stolp, L.J.; Joseph, E.; Kodali, D.R. Synthesis and evaluation of soy fatty acid ester estolides as bioplasticizers in poly (vinyl chloride). J. Am. Oil Chem. Soc. 2019, 96, 1291–1302. [Google Scholar] [CrossRef]
- Isbell, T.A. Chemistry and physical properties of estolides. Grasas Aceites 2011, 62, 8–20. [Google Scholar] [CrossRef] [Green Version]
- Cermak, S.C.; Isbell, T.A. Synthesis of estolides from oleic and saturated fatty acids. J. Am. Oil Chem. Soc. 2001, 78, 557–565. [Google Scholar] [CrossRef]
- Isbell, T.A.; Kleiman, R.; Plattner, B.A. Acid-catalyzed condensation of oleic-acid into estolides and polyestolides. J. Am. Oil Chem. Soc. 1994, 71, 169–174. [Google Scholar] [CrossRef]
- Wang, G.S.; Sun, S.D. Synthesis of ricinoleic acid estolides by the esterification of ricinoleic acids using functional acid ionic liquids as catalysts. J. Oleo Sci. 2017, 66, ess17031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodalo-Santoyo, A.; Bastida-Rodriguez, J.; Maximo-Martin, M.F.; Montiel-Morte, M.C.; Murcia-Almagro, M.D. Enzymatic biosynthesis of ricinoleic acid estolides. Biochem. Eng. J. 2005, 26, 155–158. [Google Scholar] [CrossRef]
- Hayes, D.G.; Kleiman, R. Lipase-catalyzed synthesis and properties of estolides and their esters. J. Am. Oil Chem. Soc. 1995, 72, 1309–1316. [Google Scholar] [CrossRef]
- Martin-Arjol, I.; Isbell, T.A.; Manresa, A. Mono-estolide synthesis from trans-8-hydroxy-fatty acids by lipases in solvent-free media and their physical properties. J. Am. Oil Chem. Soc. 2015, 92, 1125–1141. [Google Scholar] [CrossRef]
- Todea, A.; Otten, L.G.; Frissen, A.E.; Arends, I.; Peter, F.; Boeriu, C.G. Selectivity of lipases for estolides synthesis. Pure Appl. Chem. 2015, 87, 51–58. [Google Scholar] [CrossRef]
- Bodalo, A.; Bastida, J.; Maximo, M.F.; Montiel, M.C.; Gomez, A.; Murcia, M.D. Production of ricinoleic acid estolide with free and immobilized lipase from Candida rugosa. Biochem. Eng. J. 2008, 39, 450–456. [Google Scholar] [CrossRef]
- Bodalo, A.; Bastida, J.; Maximo, M.F.; Montiel, M.C.; Murcia, M.D.; Ortega, S. Influence of the operating conditions on lipase-catalysed synthesis of ricinoleic acid estolides in solvent-free systems. Biochem. Eng. J. 2009, 44, 214–219. [Google Scholar] [CrossRef]
- Zerkowski, J.A.; Nunez, A.; Solaiman, D.K.Y. Structured estolides: Control of length and sequence. J. Am. Oil Chem. Soc. 2008, 85, 277–284. [Google Scholar] [CrossRef]
- Naik, S.; Basu, A.; Saikia, R.; Madan, B.; Paul, P.; Chaterjee, R.; Brask, J.; Svendsen, A. Lipases for use in industrial biocatalysis: Specificity of selected structural groups of lipases. J. Mol. Catal. B Enzym. 2010, 65, 18–23. [Google Scholar] [CrossRef]
- Bantchev, G.B.; Cermak, S.C.; Durham, A.L.; Price, N.P.J. Estolide molecular weight distribution via gel permeation chromatography. J. Am. Oil Chem. Soc. 2019, 96, 365–380. [Google Scholar] [CrossRef]
- Isbell, T.A.; Kleiman, R. Characterization of estolides produced from the acid-catalyzed condensation of oleic-acid. J. Am. Oil Chem. Soc. 1994, 71, 379–383. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipase | EN | AV |
|---|---|---|
| Blank | 0.0 | 2 |
| CALA | 2.45 | 25 |
| CALB | 0.02 | 25 |
| TLL | 0.05 | 79 |
| RML | 0.09 | 110 |
| GCL | 0.52 | 43 |
| HIC | 0.45 | 88 |
| Lipase | R | E1 | E2 | E3 | E4 | G1 | G2 | G3 | G4 | G5 | G6 |
| CALA | + | + | + | + | + | + | + | + | + | + | + |
| CALB | + | - | - | - | - | - | + | - | - | - | - |
| TLL | + | - | - | - | - | + | + | - | - | - | - |
| RML | + | - | - | - | - | + | + | - | - | - | - |
| GCL | + | + | + | - | - | + | + | + | - | - | - |
| Step 1 | Step 2 | |
|---|---|---|
| (i) CALA, TLL | 2.70 | 2.70 |
| (ii) TLL, CALA | 0.05 | 0.56 |
| (iii) CALA+TLL | 0.61 | - |
| Process | R | E1 | E2 | E3 | E4 | G1 | G2 | G3 | G4 | G5 | G6 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| (i) step 1 | + | + | + | + | + | + | + | + | + | + | + |
| (i) step 2 | + | + | + | + | + | - | - | - | - | - | - |
| (ii) step 1 | + | - | - | - | - | + | + | - | - | - | - |
| (ii) step 2 | + | + | + | - | - | + | + | + | - | - | - |
| (iii) | + | + | + | - | - | + | - | - | - | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arslan, A.; Rancke-Madsen, A.; Brask, J. Enzymatic Synthesis of Estolides from Castor Oil. Catalysts 2020, 10, 835. https://doi.org/10.3390/catal10080835
Arslan A, Rancke-Madsen A, Brask J. Enzymatic Synthesis of Estolides from Castor Oil. Catalysts. 2020; 10(8):835. https://doi.org/10.3390/catal10080835
Chicago/Turabian StyleArslan, Amine, Anders Rancke-Madsen, and Jesper Brask. 2020. "Enzymatic Synthesis of Estolides from Castor Oil" Catalysts 10, no. 8: 835. https://doi.org/10.3390/catal10080835