Fine-Tuning of Sequence Specificity by Near Attack Conformations in Enzyme-Catalyzed Peptide Hydrolysis


Abstract
:1. Introduction
2. Results
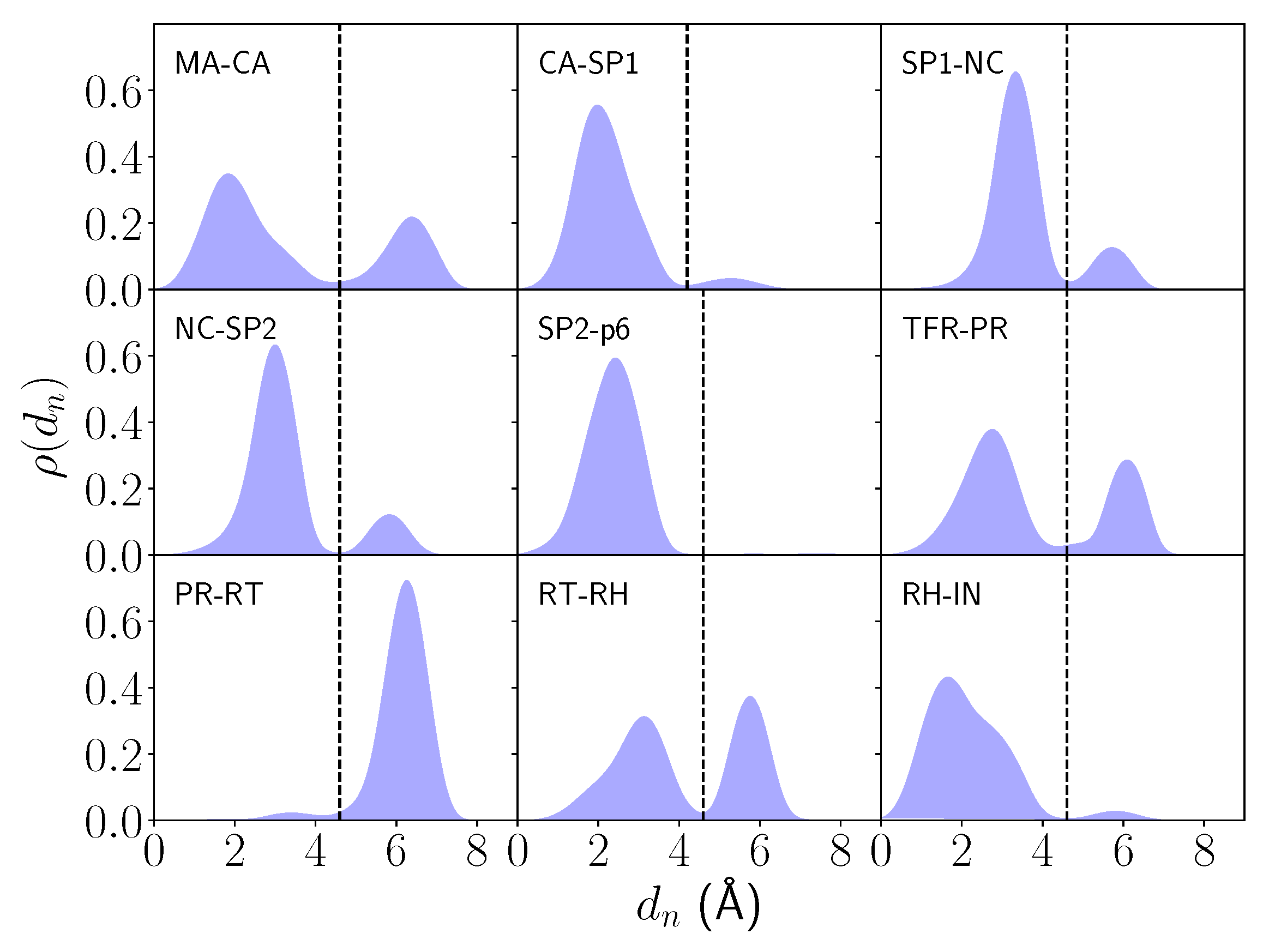
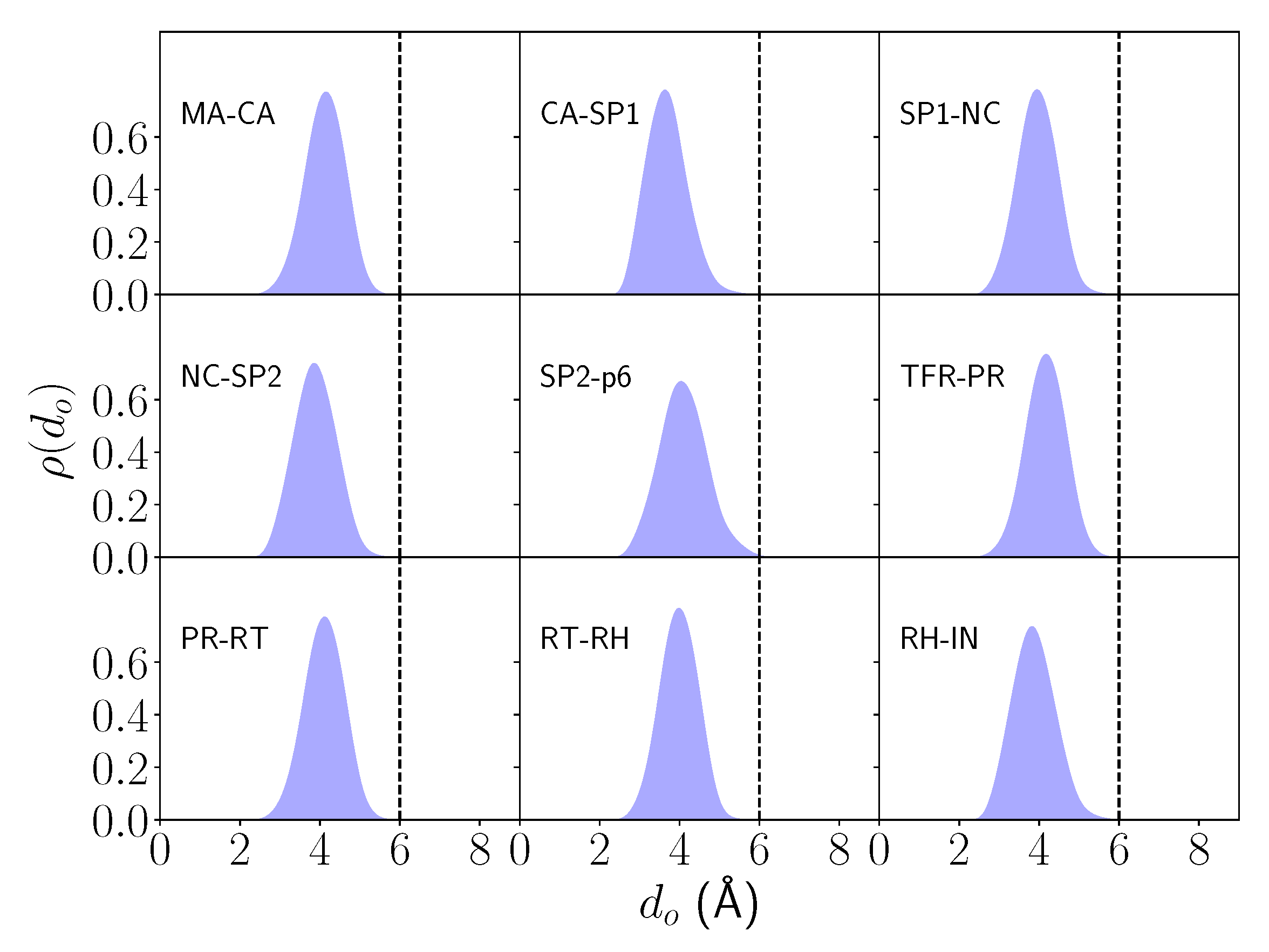
2.1. Differential Nucleophilic Water Binding
2.2. Analysis of the Hydrogen Bond Network
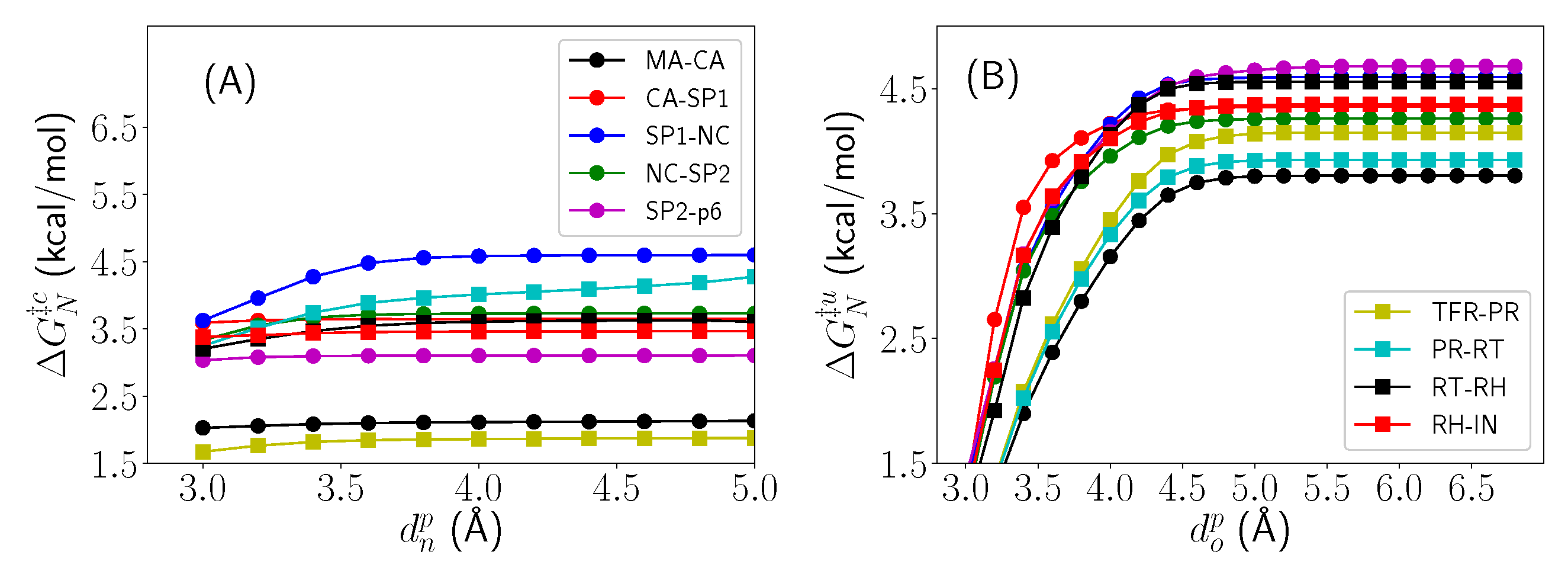
2.3. Thermodynamic Decomposition of Activation Free Energy Contributions
3. Discussion
4. Materials and Methods
4.1. Initial Preparation
4.2. Molecular Dynamics Equilibration and Production Simulation Protocol
4.3. Analysis
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Pauling, L. Molecular architecture and biological reactions. Chem. Eng. News 1946, 24, 1375–1377. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Viloca, M.; Gao, J.; Karplus, M.; Truhlar, D. How enzymes work: Analysis by modern rate theory and computer simulations. Science 2004, 303, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Eyring, H.; Stearn, A. The application of the theory of absolute reaction rates to proteins. Chem. Rev. 1939, 24, 253–270. [Google Scholar] [CrossRef]
- Cui, Q.; Karplus, M. Quantum mechanics/molecular mechanics studies of triosephosphate isomerase-catalyzed reactions: Effect of geometry and tunneling on proton-transfer rate constants. J. Am. Chem. Soc. 2002, 124, 3093–3124. [Google Scholar] [CrossRef]
- Masgrau, L.; Roujeinikova, A.; Johannissen, L.; Hothi, P.; Basran, J.; Ranaghan, K.; Mulholland, A.; Sutcliffe, M.; Scrutton, N.; Leys, D. Atomic description of an enzyme reaction dominated by proton tunneling. Science 2006, 312, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Truhlar, D. Quantum mechanical methods for enzyme kinetics. Ann. Rev. Phys. Chem. 2002, 53, 467–505. [Google Scholar] [CrossRef] [Green Version]
- Careri, G.; Fasella, P.; Gratton, E.; Jencks, W. Statistical time events in enzymes: A physical assessment. Crit. Rev. Biochem. Mol. Biol. 1975, 3, 141–164. [Google Scholar] [CrossRef]
- Welch, G.; Somogyi, B.; Damjanovich, S. The role of protein fluctuations in enzyme action: A review. Progr. Biophys. Mol. Biol. 1982, 39, 109. [Google Scholar] [CrossRef]
- Olsson, M.; Parson, W.; Warshel, A. Dynamical contributions to enzyme catalysis: Critical tests of a popular hypothesis. Chem. Rev. 2006, 106, 1737–1756. [Google Scholar] [CrossRef]
- Eisenmesser, E.; Millet, O.; Labeikovsky, W.; Korzhnev, D.; Wolf-Watz, M.; Bosco, D.; Skalicky, J.; Kay, L.; Kern, D. Intrinsic dynamics of an enzyme underlies catalysis. Nature 2005, 438, 117–121. [Google Scholar] [CrossRef]
- Boehr, D.; McElheny, D.; Dyson, H.; Wright, P. The dynamic energy landscape of dihydrofolate reductase catalysis. Science 2006, 313, 1638–1642. [Google Scholar] [CrossRef]
- Henzler-Wildman, K.A.; Thai, V.; Lei, M.; Ott, M.; Wolf-Watz, M.; Fenn, T.; Pozharski, E.; Wilson, M.A.; Petsko, G.A.; Karplus, M.; et al. Intrinsic motions along an enzymatic reaction trajectory. Nature 2007, 450, 838–844. [Google Scholar] [CrossRef]
- Bruice, T. A view at the millennium: The efficiency of enzymatic catalysis. Accounts. Chem. Res. 2002, 35, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Falzone, C.; Wright, P.; Benkovic, S. Dynamics of a flexible loop in dihydrofolate reductase from Escherichia coli and its implication for catalysis. Biochemistry 1994, 33, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Osborne, M.; Schnell, J.; Benkovic, S.; Dyson, H.; Wright, P. Backbone dynamics in dihydrofolate reductase complexes: Role of loop flexibility in the catalytic mechanism. Biochemistry 2001, 40, 9846–9859. [Google Scholar] [CrossRef] [PubMed]
- Villà, J.; Warshel, A. Energetics and dynamics of enzymatic reactions. J. Phys. Chem. B 2001, 105, 7887–7907. [Google Scholar] [CrossRef]
- Warshel, A. Electrostatic origin of the catalytic power of enzymes and the role of preorganized active sites. J. Biol. Chem. 1998, 273, 27035–27038. [Google Scholar] [CrossRef] [Green Version]
- Cui, Q.; Elstner, M.; Karplus, M. A theoretical analysis of the proton and hydride transfer in liver alcohol dehydrogenase (LADH). J. Phys. Chem. B 2002, 106, 2721–2740. [Google Scholar] [CrossRef]
- Hansson, T.; Nordlund, P.; Åqvist, J. Energetics of nucleophile activation in a protein tyrosine phosphatase. J. Mol. Biol. 1997, 265, 118–127. [Google Scholar] [CrossRef]
- Warshel, A.; Sharma, P.; Kato, M.; Xiang, Y.; Liu, H.; Olsson, M. Electrostatic basis for enzyme catalysis. Chem. Rev. 2006, 106, 3210–3235. [Google Scholar] [CrossRef]
- Kienhöfer, A.; Kast, P.; Hilvert, D. Selective stabilization of the chorismate mutase transition state by a positively charged hydrogen bond donor. J. Am. Chem. Soc. 2003, 125, 3206–3207. [Google Scholar] [CrossRef] [PubMed]
- Lyne, P.; Mulholland, A.; Richards, W. Insights into chorismate mutase catalysis from a combined QM/MM simulation of the enzyme reaction. J. Am. Chem. Soc. 1995, 117, 11345–11350. [Google Scholar] [CrossRef]
- Shurki, A.; Štrajbl, M.; Villà, J.; Warshel, A. How much do enzymes really gain by restraining their reacting fragments? J. Am. Chem. Soc. 2002, 124, 4097–4107. [Google Scholar] [CrossRef] [PubMed]
- Štrajbl, M.; Shurki, A.; Kato, M.; Warshel, A. Apparent NAC effect in chorismate mutase reflects electrostatic transition state stabilization. J. Am. Chem. Soc. 2003, 125, 10228–10237. [Google Scholar] [CrossRef]
- Bruice, T.C.; Lightstone, F. Ground state and transition state contributions to the rates of intramolecular and enzymatic reactions. Acc. Chem. Res. 1999, 32, 127–136. [Google Scholar] [CrossRef]
- Bruice, T.C.; Benkovic, S.J. Chemical basis for enzyme catalysis. Biochemistry 2000, 39, 6267–6274. [Google Scholar] [CrossRef]
- Schowen, R. How an enzyme surmounts the activation energy barrier. Proc. Natl. Acad. Sci. USA 2003, 100, 11931–11932. [Google Scholar] [CrossRef] [Green Version]
- Hur, S.; Bruice, T.C. The near attack conformation approach to the study of the chorismate to prephenate reaction. Proc. Natl. Acad. Sci. USA 2003, 100, 12015–12020. [Google Scholar] [CrossRef] [Green Version]
- Hur, S.; Bruice, T.C. The mechanism of catalysis of the chorismate to prephenate reaction by the Escherichia coli mutase enzyme. Proc. Natl. Acad. Sci. USA 2002, 99, 1176–1181. [Google Scholar] [CrossRef] [Green Version]
- Hur, S.; Bruice, T.C. Comparison of formation of reactive conformers (NACs) for the Claisen rearrangement of chorismate to prephenate in water and in the E. coli mutase: The efficiency of the enzyme catalysis. J. Am. Chem. Soc. 2003, 125, 5964–5972. [Google Scholar] [CrossRef]
- Sadiq, S.K.; Coveney, P.V. Computing the role of near attack conformations in an enzyme-catalyzed nucleophilic bimolecular reaction. J. Chem. Theor. Comput. 2015, 11, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Suh, J.; Lee, S. Ab initio studies on the catalytic mechanism of aspartic proteinases: Nucleophilic versus general acid/general base mechanism. J. Am. Chem. Soc. 2000, 122, 3901–3908. [Google Scholar] [CrossRef]
- Piana, S.; Bucher, D.; Carloni, P.; Rothlisberger, U. Reaction mechanism of HIV-1 protease by hybrid Car-Parrinello/classical MD simulations. J. Phys. Chem. B 2004, 108, 11139–11149. [Google Scholar] [CrossRef]
- Bürgi, H.B.; Dunitz, J.D.; Shefter, E. Geometrical reaction coordinates. II. Nucleophilic addition to a carbonyl group. J. Am. Chem. Soc. 1973, 95, 5065–5067. [Google Scholar] [CrossRef]
- Bürgi, H.B.; Lehn, J.M.; Wipff, G. Ab initio study of nucleophilic addition to a carbonyl group. J. Am. Chem. Soc. 1974, 96, 1956–1957. [Google Scholar] [CrossRef]
- Dunitz, J.D.; Lehn, J.M.; Wipff, G. Stereochemistry of reaction paths at carbonyl centres. Tetrahedron 1974, 30, 1563–1572. [Google Scholar]
- Bürgi, H.B.; Dunitz, J.D.; Shefter, E. Chemical reaction paths. IV. Aspects of O⋯C=O interactions in crystals. Acta. Crystall. Sect. B 1974, 30, 1517–1527. [Google Scholar] [CrossRef]
- Shehu-Xhilaga, M.; Crowe, S.M.; Mak, J. Maintenance of the Gag/Gag-Pol ratio is important for human immunodeficiency virus type 1 RNA dimerization and viral infectivity. J. Virol. 2001, 75, 1834–1841. [Google Scholar] [CrossRef] [Green Version]
- Maschera, B.; Darby, G.; Palu, G.; Wright, L.L.; Tisdale, M.; Myers, R.; Blair, E.D.; Furfine, E.S. Human immunodeficiency virus mutations in the viral protease that confer resistance to saquinavir increase the dissociation rate constant of the protease-saquinavir complex. J. Biol. Chem. 1996, 271, 33231–33235. [Google Scholar] [CrossRef] [Green Version]
- Tözsér, J.; Bláha, I.; Copeland, T.D.; Wondrak, E.M.; Oroszlan, S. Comparison of the HIV-1 and HIV-2 proteinases using oligopeptide substrates representing cleavage sites in Gag and Gag-Pol polyproteins. FEBS Lett. 1991, 281, 77–80. [Google Scholar] [CrossRef] [Green Version]
- Fehér, A.; Weber, I.T.; Bagossi, P.; Boross, P.; Mahalingam, B.; Louis, J.M.; Copeland, T.D.; Torshin, I.Y.; Harrison, R.W.; Tözsér, J. Effect of sequence polymorphism and drug resistance on two HIV-1 Gag processing sites. Eur. J. Biochem. 2002, 269, 4114–4120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radzicka, A.; Wolfenden, R. Rates of uncatalyzed peptide bond hydrolysis in neutral solution and the transition state affinities of proteases. J. Am. Chem. Soc. 1996, 118, 6105–6109. [Google Scholar] [CrossRef]
- Sadiq, S.K.; Könnyü, B.; Müller, V.; Coveney, P.V. Reaction kinetics of catalyzed competitive heteropolymer cleavage. J. Phys. Chem. B 2011, 115, 11017–11027. [Google Scholar] [CrossRef]
- Könnyü, B.; Sadiq, S.K.; Turányi, T.; Hírmondó, R.; Müller, B.; Kräusslich, H.G.; Coveney, P.V.; Müller, V. Gag-Pol processing during HIV-1 virion maturation: A systems biology approach. PLoS Comput. Biol. 2013, 9, e1003103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirambeau, G.; Lyonnais, S.; Coulaud, D.; Hameau, L.; Lafosse, S.; Jeusset, J.; Borde, I.; Reboud-Ravaux, M.; Restle, T.; Gorelick, R.J.; et al. HIV-1 protease and reverse transcriptase control the architecture of their nucleocapsid partner. PLoS ONE 2007, 2, e669. [Google Scholar] [CrossRef]
- Wlodawer, A.; Miller, M.; Jaskólski, M.; Sathyanarayana, B.K.; Baldwin, E.; Weber, I.T.; Selk, L.M.; Clawson, L.; Schneider, J.; Kent, S.B.H. Conserved folding in retroviral proteases: Crystal structure of a synthetic HIV-1 protease. Science 1989, 245, 616–621. [Google Scholar] [CrossRef]
- Sadiq, S.K.; Noé, F.; De Fabritiis, G. Kinetic characterization of the critical step in HIV-1 protease maturation. Proc. Natl. Acad. Sci. USA 2012, 109, 20449–20454. [Google Scholar] [CrossRef] [Green Version]
- Nijhuis, M.; Schuurman, R.; de Jong, D.; Erickson, J.; Gustchina, E.; Albert, J.; Schipper, P.; Gulnik, S.; Boucher, C.A.B. Increased fitness of drug resistant HIV-1 protease as a result of acquisition of compensatory mutations during suboptimal therapy. AIDS 1999, 13, 2349–2359. [Google Scholar] [CrossRef]
- Mammano, F.; Trouplin, V.; Zennou, V.; Clavel, F. Retracing the evolutionary pathways of human immunodeficiency virus type 1 resistance to protease inhibitors: Virus fitness in the absence and in the presence of drug. J. Virol. 2000, 74, 8524–8531. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Picado, J.; Savara, A.V.; Sutton, L.; D’Aquila, R.T. Replicative fitness of protease inhibitor-resistant mutants of human immunodeficiency virus type 1. J. Virol. 1999, 73, 3744–3752. [Google Scholar] [CrossRef] [Green Version]
- Prabu-Jeyabalan, M.; Nalivaika, E.; Schiffer, C.A. Substrate shape determines specificity of recognition for HIV-1 protease: Analysis of crystal structures of six substrate complexes. Structure 2002, 10, 369–381. [Google Scholar] [CrossRef]
- Prabu-Jeyabalan, M.; Nalivaika, E.; Schiffer, C.A. How does a symmetric dimer recognize an asymmetric substrate? A substrate complex of HIV-1 protease. J. Mol. Biol. 2000, 301, 1207–1220. [Google Scholar] [CrossRef] [PubMed]
- Prabu-Jeyabalan, M.; Nalivaika, E.; King, N.M.; Schiffer, C.A. Structural basis for coevolution of a human immunodeficiency virus type 1 nucleocapsid-p1 cleavage site with a V82A drug-resistant mutation in viral protease. J. Virol. 2004, 78, 12446–12454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittayanarakul, K.; Hannongbua, S.; Feig, M. Accurate prediction of protonation state as a prerequisite for reliable MM-PB (GB) SA binding free energy calculations of HIV-1 protease inhibitors. J. Comput. Chem. 2008, 29, 673–685. [Google Scholar] [CrossRef]
- Kovalskyy, D.; Dubyna, V.; Mark, A.E.; Korenelyuk, A. A molecular dynamics study of the structural stability of HIV-1 protease under physiological conditions: The role of Na+ ions in stabilizing the active site. Proteins Struct. Funct. Bioinf. 2005, 58, 450–458. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Harvey, M.J.; Giupponi, G.; Fabritiis, G.D. ACEMD: Accelerating biomolecular dynamics in the microsecond time scale. J. Chem. Theor. Comput. 2009, 5, 1632–1639. [Google Scholar] [CrossRef] [Green Version]
- Sadiq, S.K.; De Fabritiis, G. Explicit solvent dynamics and energetics of HIV-1 protease flap opening and closing. Proteins 2010, 78, 2873–2885. [Google Scholar] [CrossRef]
- Pettit, S.C.; Henderson, G.J.; Schiffer, C.A.; Swanstrom, R. Replacement of the P1 amino acid of human immunodeficiency virus type 1 Gag processing sites can inhibit or enhance the rate of cleavage by the viral protease. J. Virol. 2002, 76, 10226–10233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundquist, W.I.; Kräusslich, H.G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Cleavage Sequence | (Å) | G |
|---|---|---|---|
| MA-CA | SGNY-PIVQ | 4.6 | −0.4 ± 0.1 |
| CA-SP1 | ARVL-AEAM | 4.2 | −1.9 ± 0.5 |
| SP1-NC | ATIM-MQRG | 4.6 | −1.0 ± 0.1 |
| NC-SP2 | RQAN-FLGK | 4.6 | −1.1 ± 0.3 |
| SP2-p6 | PYNF-LQSR | 4.6 | −3.7 ± 1.2 |
| TFR-PR | SFNF-PQIT | 4.6 | −0.4 ± 0.2 |
| PR-RT | TLNF-PISP | 4.6 | 2.0 ± 0.3 |
| RT-RH | AETP-YVDG | 4.6 | −0.1 ± 0.2 |
| RH-IN | RKIL-FLDG | 4.6 | −2.2 ± 0.7 |
| Uncatalyzed | Catalyzed | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| System | G | G | G | G | G | G | G | G | G |
| MA-CA | ∼30 | 3.8 ± 0.1 | 26.2 | 15.7 | 2.1 ± 0.1 | 13.6 | −14.3 | −1.7 ± 0.2 | −12.6 |
| CA-SP1 | 4.4 ± 0.1 | 25.6 | 17.0 | 3.7 ± 0.2 | 13.3 | −13.0 | −0.7 ± 0.3 | −12.3 | |
| SP1-NC | 4.6 ± 0.1 | 25.4 | 15.3 | 4.6 ± 0.2 | 10.7 | −14.7 | 0.0 ± 0.3 | −14.7 | |
| NC-SP2 | 4.3 ± 0.1 | 25.7 | 16.7 | 3.8 ± 0.2 | 12.9 | −13.3 | −0.5 ± 0.3 | −12.8 | |
| SP2-p6 | 4.8 ± 0.3 | 25.2 | 18.4 | 3.1 ± 0.1 | 15.3 | −11.6 | −1.7 ± 0.4 | −9.9 | |
| TFR-PR | 4.1 ± 0.1 | 25.9 | 15.3 | 1.9 ± 0.2 | 13.4 | −14.7 | −2.2 ± 0.3 | −12.5 | |
| PR-RT | 3.9 ± 0.1 | 26.1 | 15.8 | 4.2 ± 0.5 | 11.6 | −14.2 | 0.3 ± 0.6 | −14.5 | |
| RT-RH | 4.7 ± 0.4 | 25.3 | 17.2 | 3.7 ± 0.4 | 13.5 | −12.8 | −1.0 ± 0.8 | −11.8 | |
| RH-IN | 4.4 ± 0.1 | 25.6 | 16.6 | 3.5 ± 0.1 | 13.1 | −13.4 | −0.9 ± 0.2 | −12.5 | |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadiq, S.K. Fine-Tuning of Sequence Specificity by Near Attack Conformations in Enzyme-Catalyzed Peptide Hydrolysis. Catalysts 2020, 10, 684. https://doi.org/10.3390/catal10060684
Sadiq SK. Fine-Tuning of Sequence Specificity by Near Attack Conformations in Enzyme-Catalyzed Peptide Hydrolysis. Catalysts. 2020; 10(6):684. https://doi.org/10.3390/catal10060684
Chicago/Turabian StyleSadiq, S. Kashif. 2020. "Fine-Tuning of Sequence Specificity by Near Attack Conformations in Enzyme-Catalyzed Peptide Hydrolysis" Catalysts 10, no. 6: 684. https://doi.org/10.3390/catal10060684