Co-immobilization of an Enzyme System on a Metal-Organic Framework to Produce a More Effective Biocatalyst



Abstract
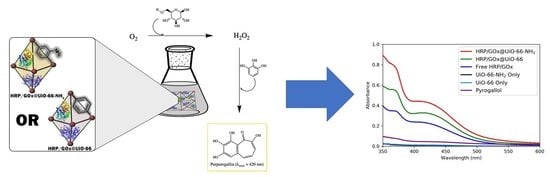
:
1. Introduction
2. Results and Discussion
2.1. The Biocatalytic Enzyme System
2.2. Zeta Potential Characterization
2.3. FT-IR Characterization
2.4. SEM of Enzyme/MOF Composites
2.5. Purpurogallin Synthesis
2.6. Enzyme/MOF Immobilization and Leaching
3. Materials and Methods
3.1. Materials
3.2. Methods
3.3. Synthesis of UiO-66 and UiO-66-NH2
3.4. Enzyme Immobilization and Characterization
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhou, H.-C. Enzyme–MOF (metal–organic framework) composites. Royal Society of Chemistry 2017. [Google Scholar]
- Rehm, B.H.A. Enzyme Engineering for in Situ Immobilization. Molecules 2016, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, Y.H.; Lo, S.H.; Yang, N.S.; Singco, B.; Cheng, Y.J.; Wu, C.Y.; Chang, I.H.; Huang, H.Y.; Lin, C.H. Trypsin-Immobilized Metal–Organic Framework as a Biocatalyst in Proteomics Analysis. ChemPlusChem 2012, 77, 982–986. [Google Scholar] [CrossRef]
- Ahmed, E.; Ismail, C.Z.D. Industrial Applications of Enzymes: Recent Advances, Techniques, and Outlooks. Catalysts 2018, 8, 238. [Google Scholar]
- Rodrigues, R.C.; Ortiz, C.; Berenguer-Murcia, Á.; Torres, R.; Fernández-Lafuente, R. Modifying enzyme activity and selectivity by immobilization. Chem. Soc. Rev. 2013, 42, 6290–6307. [Google Scholar] [CrossRef]
- Ki-Hyun Kim, A.D. Recent advances in enzyme immobilization techniques: Metal-organic frameworks as novel substrates. Elev. Coord. Chem. Rev. 2016, 322, 30–40. [Google Scholar]
- Sheldon, R.A. Role of Biocatalysis in Sustainable Chemistry. ACS Chem. Rev. 2017, 118, 801–838. [Google Scholar] [CrossRef]
- Gkaniatsou, E.; Sicard, C.; Ricoux, R.; Mahy, J.P.; Steunou, N.; Serre, C. Metal–organic frameworks: A novel host platform for enzymatic catalysis and detection. Mater. Horizons 2017, 5, 55–63. [Google Scholar] [CrossRef]
- Ren, S.; Li, C.; Jiao, X.; Jia, S.; Jiang, Y.; Bilal, M.; Cui, J. Recent progress in multienzymes co-immobilization and multienzyme system applications. Chem. Eng. J. 2019, 373, 1254–1278. [Google Scholar] [CrossRef]
- Homaei, A.A.; Sariri, R.; Vianello, F.; Stevanato, R. Enzyme immobilization: An update. J. Chem. Biol. 2013, 6, 185–205. [Google Scholar] [CrossRef] [Green Version]
- Jesionowski, T.; Zdarta, J.; Krajewska, B. Enzyme immobilization by adsorption: A review. Adsorption 2014, 20, 801–821. [Google Scholar] [CrossRef] [Green Version]
- Betancor, L.; Luckarift, H.R. Co-immobilized coupled enzyme systems in biotechnology. Biotechnol. Genet. Eng. Rev. 2013, 27, 95–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, S.L.; Yue, D.M.; Li, X.H.; Smith, T.J.; Li, N.; Zong, M.H.; Wu, H.; Ma, Y.Z.; Lou, W.Y. Novel Nano-/Micro-Biocatalyst: Soybean Epoxide Hydrolase Immobilized on UiO-66-NH2 MOF for Efficient Biosynthesis of Enantiopure (R)-1, 2-Octanediol in Deep Eutectic Solvents. ACS Sustain. Chem. Eng. 2016, 4, 3586–3595. [Google Scholar] [CrossRef]
- Shieh, F.K.; Wang, S.C.; Yen, C.I.; Wu, C.C.; Dutta, S.; Chou, L.Y.; Morabito, J.V.; Hu, P.; Hsu, M.H.; Wu, K.C.; et al. Imparting Functionality to Biocatalysts via Embedding Enzymes into Nanoporous Materials by a de Novo Approach: Size-Selective Sheltering of Catalase in Metal−Organic Framework Microcrystals. J. Am. Chem. Soc. 2015, 137, 4276–4279. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.L.; Yang, N.S.; Chen, Y.T.; Lirio, S.; Wu, C.Y.; Lin, C.H.; Huang, H.Y. Lipase-Supported Metal–Organic Framework Bioreactor Catalyzes Warfarin Synthesis. Chem. Eur. J. 2015, 21, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Liang, B.; Xiao, X.; Bai, L.; Tang, X.; Lojou, E.; Cosnier, S.; Liu, A. Controllable Display of Sequential Enzymes on Yeast Surface with Enhanced Biocatalytic Activity toward Efficient Enzymatic Biofuel Cells. J. Am. Chem. Soc. 2020, 4, 3222–3230. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.L.; Wu, C.Y.; Chen, C.Y.; Singco, B.; Lin, C.H.; Huang, H.Y. Fast Multipoint Immobilized MOF Bioreactor. Chem. Eur. J. Biocatal. 2014, 20, 8923–8928. [Google Scholar] [CrossRef]
- Jung, S.; Kim, Y.; Kim, S.J.; Kwon, T.H.; Huh, S.; Park, S. Bio-functionalization of metal–organic frameworks by covalent protein conjugation. ChemComm 2011, 47, 2904–2906. [Google Scholar] [CrossRef]
- Zhou, H.-C. Coupling two enzymes into a tandem nanoreactor utilizing a hierarchically structured MOF. RCS Chem. Sci. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Patra, S.; Crespo, T.H.; Permyakova, A.; Sicard, C.; Serre, C.; Chaussé, A.; Steunou, N.; Legrand, L. Design of metal organic framework–enzyme based bioelectrodes as a novel and highly sensitive biosensing platform. J. Mater. Chem. B 2015, 3. [Google Scholar] [CrossRef]
- Majewski, M.B.; Howarth, A.J.; Li, P.; Wasielewski, M.R.; Hupp, J.T.; Farha, O.K. Enzyme encapsulation in metal–organic frameworks for applications in catalysis. RCS CrystEngComm 2017. [Google Scholar] [CrossRef]
- Distefano, M.D. Site-Specific, Covalent Attachment of Proteins to a Solid Surface. Bioconjugate Chem. 2006, 17, 967–974. [Google Scholar] [CrossRef]
- Morten Meldal, S.S. Recent advances in covalent, site-specific protein immobilization. F1000Research 2016, 5, 1–11. [Google Scholar]
- Wang, X.; Lu, X.; Wu, L.; Chen, J. 3D metal-organic framework as highly efficient biosensing platform for ultrasensitive and rapid detection of bisphenol A. Biosens. Bioelectron. 2015, 65, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Ashlee, J.; Howarth, Y.L. Chemical, thermal and mechanical stabilities of metal–organic frameworks. Nat. Rev. Mater. 2016. [Google Scholar] [CrossRef]
- Liang, K. Biomimetic mineralization of metal-organic frameworks as protective coatings for biomacromolecules. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, J.P. Direct Observation of Amorphous Precursor Phases in the Nucleation of Protein−Metal−Organic Frameworks. J. Am. Chem. Soc. 2020, 1433–1442. [Google Scholar] [CrossRef]
- Kenneth, J.; Balkus, J. Hybrid materials for immobilization of MP-11 catalyst. Top. Catal. 2006, 36. [Google Scholar] [CrossRef]
- Feng, D.; Liu, T.F.; Su, J.; Bosch, M.; Wei, Z.; Wan, W.; Yuan, D.; Chen, Y.P.; Wang, X.; Wang, K.; et al. Stable metal-organic frameworks containing single-molecule traps for enzyme encapsulation. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Yang, R.; Zhai, J.; Tian, C. Bienzymatic glucose biosensor based on co-immobilization of peroxidase and glucose oxidase on a carbon nanotubes electrode. Biosens. Bioelectron. 2007, 23, 528–535. [Google Scholar] [CrossRef]
- Garcia-Galan, C.; Berenguer-Murcia, Á.; Fernandez-Lafuente, R.; Rodrigues, R.C. Potential of Different Enzyme Immobilization Strategies to Improve Enzyme Performance. Adv. Synth. Catal. 2011, 353, 2885–2904. [Google Scholar] [CrossRef]
- Pitzalis, F.; Monduzzi, M.; Salis, A. A bienzymatic biocatalyst constituted by glucose oxidase and Horseradish peroxidase immobilized on ordered mesoporous silica. Microporous Mesoporous Mater. 2017, 241, 145–154. [Google Scholar] [CrossRef]
- Barbosa, O.; Torres, R.; Ortiz, C.; Berenguer-Murcia, A.; Rodrigues, R.C.; Fernandez-Lafuente, R. Rodrigues, Roberto Fernandez-Lafuentee. Heterofunctional Supports in Enzyme Immobilization: From Traditional Immobilization Protocols to Opportunities in Tunning Enzyme Properties. Biomacromolecules 2013, 8, 2433–2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Don, A.; Cowana, R.F.-L. Enhancing the functional properties of thermophilic enzymes by chemical modification and immobilization. Enzyme Microb. Technol. 2011, 49, 326–346. [Google Scholar]
- Rios, N.S.; Arana-Peña, S.; Mendez-Sanchez, C.; Ortiz, C.; Gonçalves, L.R.; Fernandez-Lafuente, R. Gonçalves, and Roberto Fernandez-Lafuente Reuse of Lipase from Pseudomonas fluorescens via Its Step-by-Step Coimmobilization on Glyoxyl-Octyl Agarose Beads with Least Stable Lipases. Catalysts 2019, 9, 487. [Google Scholar] [CrossRef] [Green Version]
- Marshall, R.J.; Forgan, R.S. Postsynthetic Modification of Zirconium Metal-Organic Frameworks. Eur. J. Inorg. Chem. 2016, 4310–4331. [Google Scholar] [CrossRef]
- Smith, M.R.; Gao, H.; Prabhu, P.; Bugada, L.F.; Roth, C.; Mutukuri, D.; Yee, C.M.; Lee, L.; Ziff, R.M.; Lee, J.K.; et al. Elucidating structure–performance relationships in whole-cell cooperative enzyme catalysis. Nat. Catal. 2019, 2, 809–819. [Google Scholar] [CrossRef]
- Bugada, L.F.; Smith, M.R.; Wen, F. Engineering Spatially Organized Multienzyme Assemblies for Complex Chemical Transformation. ACS Catal. 2018, 8, 7898–7906. [Google Scholar] [CrossRef] [Green Version]
- Ananthanarayan, L. Glucose oxidase—An overview. Biotechnol. Adv. 2009, 27, 489–501. [Google Scholar] [CrossRef]
- Veitch, N.C. Horseradish peroxidase: A modern view of a classic enzyme. Phytochemistry 2004, 65, 249–259. [Google Scholar] [CrossRef]
- Gao, F.; Guo, Y.; Fan, X.; Hu, M.; Li, S.; Zhai, Q.; Jiang, Y.; Wang, X. Enhancing the catalytic performance of chloroperoxidase by coimmobilization with glucose oxidase on magnetic graphene oxide. Biochem. Eng. J. 2018, 143, 101–109. [Google Scholar] [CrossRef]
- Liu, Y. Hemin@metal–organic framework with peroxidaselike activity and its application to glucose detection. RCS Catal. Sci. Technol. 2013, 3, 2761–2768. [Google Scholar] [CrossRef]
- Jia, F.; Zhang, Y.; Narasimhan, B.; Mallapragada, S.K. Block Copolymer-Quantum Dot Micelles for Multienzyme Colocalization. ACS Langmuir 2012, 28, 17389–17395. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, H.; Küchler, A.; Holmberg, K.; Walde, P. Co-immobilization of enzymes with the help of a dendronized polymer and mesoporous silica nanoparticles. RCS Mater. Chem. B 2012, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.L. Combined cross-linked enzyme aggregates of horseradish peroxidase and glucose oxidase for catalyzing cascade chemical reactions. Enzyme Microbial Technol. 2017, 100, 52–59. [Google Scholar] [CrossRef]
- Memon, A.H.; Ding, R.; Yuan, Q.; Liang, H.; Wei, Y. Coordination of GMP ligand with Cu to enhance the multiple enzymes stability and substrate specificity by co-immobilization process. Biochem. Eng. J. 2018, 136, 102–108. [Google Scholar] [CrossRef]
- Wu, X.; Yang, C.; Ge, J. Green synthesis of enzyme/ metal-organic framework composites with high stability in protein denaturing solvents. Bioresour. Bioprocess. 2017, 4. [Google Scholar] [CrossRef]
- Chen, S.; Wen, L.; Svec, F.; Tan, T.; Lv, Y. Magnetic metal–organic frameworks as scaffolds for spatial co-location and positional assembly of multi-enzyme systems enabling enhanced cascade biocatalysis. RCS Adv. 2017, 7, 21205–21213. [Google Scholar] [CrossRef] [Green Version]
- Vriezema, D.M.; Garcia, P.M.; Sancho Oltra, N.; Hatzakis, N.S.; Kuiper, S.M.; Nolte, R.J.; Rowan, A.E.; van Hest, J.C. Positional Assembly of Enzymes in Polymersome Nanoreactors for Cascade Reactions. Angew. Chem. Commun. Nanoreactors 2007, 46, 7378–7382. [Google Scholar] [CrossRef]
- Kazenwadel, F. Synthetic enzyme supercomplexes: Coimmobilization of enzyme cascades. RSC Anal. Methods 2015, 7, 4030–4037. [Google Scholar] [CrossRef] [Green Version]
- van de Velde, F.; Lourenço, N.D.; Bakker, M.; van Rantwijk, F.; Sheldon, R.A. Sheldon. Improved Operational Stability of Peroxidases by Coimmobilization with Glucose Oxidase. Biotechnol. Bioeng. 2000, 69, 286–291. [Google Scholar] [CrossRef]
- Jia, F.; Narasimhan, B.; Mallapragada, S.K. Biomimetic Multienzyme Complexes Based on Nanoscale Platforms. AlChE 2012, 59. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Y.; Su, Y.; Ouyang, P.; Ge, J.; Liu, Z. Spatial co-localization of multi-enzymes by inorganic nanocrystal–protein complexes. RCS ChemComm 2014, 50, 12465–12468. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Niemeyer, C.M. DNA-directed assembly of artificial multienzyme complexes. Biochem. Biophys. Res. Commun. 2008, 377, 62–67. [Google Scholar] [CrossRef]
- Park, H.Y. Purpurogallin exerts anti-inflammatory effects in lipopolysaccharide-stimulated BV2 microglial cells through the inactivation of the NF-κB and MAPK signaling pathways. Int. J. Mol. Med. 2013, 32, 1171–1178. [Google Scholar] [CrossRef] [Green Version]
- Bilal, M.; Mehmood, S.; Rasheed, T.; Iqbal, H. Bio-Catalysis and Biomedical Perspectives of Magnetic Nanoparticles as Versatile Carriers. Magnetochemistry 2019, 5. [Google Scholar] [CrossRef] [Green Version]
- Bosio, V.E.; Islan, G.A.; Martínez, Y.N.; Durán, N.; Castro, G.R. Nanodevices for the immobilization of therapeutic enzymes. Crit. Rev. Biotechnol. 2016, 36, 447–464. [Google Scholar] [CrossRef]
- Wang, W.; Wang, L.; Li, Y.; Liu, S.; Xie, Z.; Jing, X. Nanoscale Polymer Metal–Organic Framework Hybrids for Effective Photothermal Therapy of Colon Cancers. Adv. Mater. 2016, 28, 9320–9325. [Google Scholar] [CrossRef]
- Ramasarma, T. New insights of superoxide dismutase inhibition of pyrogallol autoxidation. Mol. Cell. Biochem. 2014, 400, 277–285. [Google Scholar] [CrossRef]
- Fernandez, M.; Frydman, R.B.; Hurst, J.; Buldain, G. Structure/activity relationships in porphobilinogen oxygenase and horseradish peroxidase an analysis using synthetic hemins. Eur. J. Biochem. 1993, 218, 251–259. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Elsevier Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Salopek, B.; Krasic, D.; Filipovic, S. Measurement and application of zeta-potential. Rudarsko-geolosko-Naftni Zbornik 1992, 4, 147–151. [Google Scholar]
- Guo, X. Facile synthesis of morphology- and size-controlled zirconium metal-organic framework UiO-66: The role of hydrofluoric acid in crystallization. CrystEngComm 2015, 17, 6434–6440. [Google Scholar] [CrossRef]
- Wu, L. Electronic effects of ligand substitution on metal–organic framework photocatalysts: The case study of UiO-66. PCCP 2015, 17, 117–121. [Google Scholar] [CrossRef]
- Wang, S. Amino-functionalized Zr-MOF nanoparticles for adsorption of CO2 and CH4. Int. J. Smart Nano Mater. 2013, 4, 72–82. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.J.; Wang, R.; Liu, X.L.; Peng, F.M.; Li, C.H.; Teng, F.; Yuan, Y.P. In situ growth of CdS nanoparticles on UiO-66 metal-organic framework octahedrons for enhanced photocatalytic hydrogen production under visible light irradiation. Appl. Surf. Sci. 2015, 346, 278–283. [Google Scholar] [CrossRef]
- Boudrant, J.; Woodley, J.M.; Fernandez-Lafuente, R. Parameters necessary to define an immobilized enzyme preparation. Process Biochem. 2019, 90, 66–80. [Google Scholar] [CrossRef]
- Lillerud, K.P. Tuned to Perfection: Ironing out the Defects in Metal-Organic Framework UiO-66. Chem. Mater. 2016, 14, 4068–4071. [Google Scholar] [CrossRef]
- Weltz, J.S.; Kienle, D.F.; Schwartz, D.K.; Kaar, J.L. Reduced Enzyme Dynamics upon Multipoint Covalent Immobilization Leads to Stability-Activity Trade-off. J. Am. Chem. Soc. 2020, 142, 3463–3471. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composite | Zeta Potential (mV) |
|---|---|
| HRP/GOx@UiO-66-NH2 | −3.13 (s = ±8.73) |
| HRP@UiO-66-NH2 | −8.46 (s = ±0.14) |
| GOx@UiO-66-NH2 | −10.00 (s = ±5.64) |
| UiO-66-NH2 | −26.67 (s = ±6.68) |
| HRP/GOx@UiO-66 | −10.80 (s = ±4.86) |
| HRP@UiO-66 | −7.97 (s = ±4.89) |
| GOx@UiO-66 UiO-66 | −3.38 (s = ±4.50) −18.00 (s = ±5.96) |
| Composite | % Immobilization a | % Leached from Composite b | Enzyme Activity (U/mg) d |
|---|---|---|---|
| HRP/GOx@UiO-66-NH2 HRP/GOx@UiO-66 | 9.91 (s = ±0.033) 3.91 (s = ±0.007) | 35.6 (s = ±0.025) 100 (s = ±0.045) | 189 143 |
| HRP/GOx | 0 | 6 c | 100 |
| UiO-66-NH2 UiO-66 | 0 0 | 0 0 | 0 0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, R.; Shanahan, J.; Rizaldo, S.; Kissel, D.S.; Stone, K.L. Co-immobilization of an Enzyme System on a Metal-Organic Framework to Produce a More Effective Biocatalyst. Catalysts 2020, 10, 499. https://doi.org/10.3390/catal10050499
Ahmad R, Shanahan J, Rizaldo S, Kissel DS, Stone KL. Co-immobilization of an Enzyme System on a Metal-Organic Framework to Produce a More Effective Biocatalyst. Catalysts. 2020; 10(5):499. https://doi.org/10.3390/catal10050499
Chicago/Turabian StyleAhmad, Raneem, Jordan Shanahan, Sydnie Rizaldo, Daniel S. Kissel, and Kari L. Stone. 2020. "Co-immobilization of an Enzyme System on a Metal-Organic Framework to Produce a More Effective Biocatalyst" Catalysts 10, no. 5: 499. https://doi.org/10.3390/catal10050499