C–H Bond Activation of Silyl-Substituted Pyridines with Bis(Phenolate)Yttrium Catalysts as a Facile Tool towards Hydroxyl-Terminated Michael-Type Polymers


Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis of the Functionalized Pyridine 2
2.2. C–H Bond Activation of 2 Using 2-Methoxyethylamino-Bis(Phenolate)Yttrium Complex 3
2.3. Investigation on Catalytic Activity of Catalyst 4
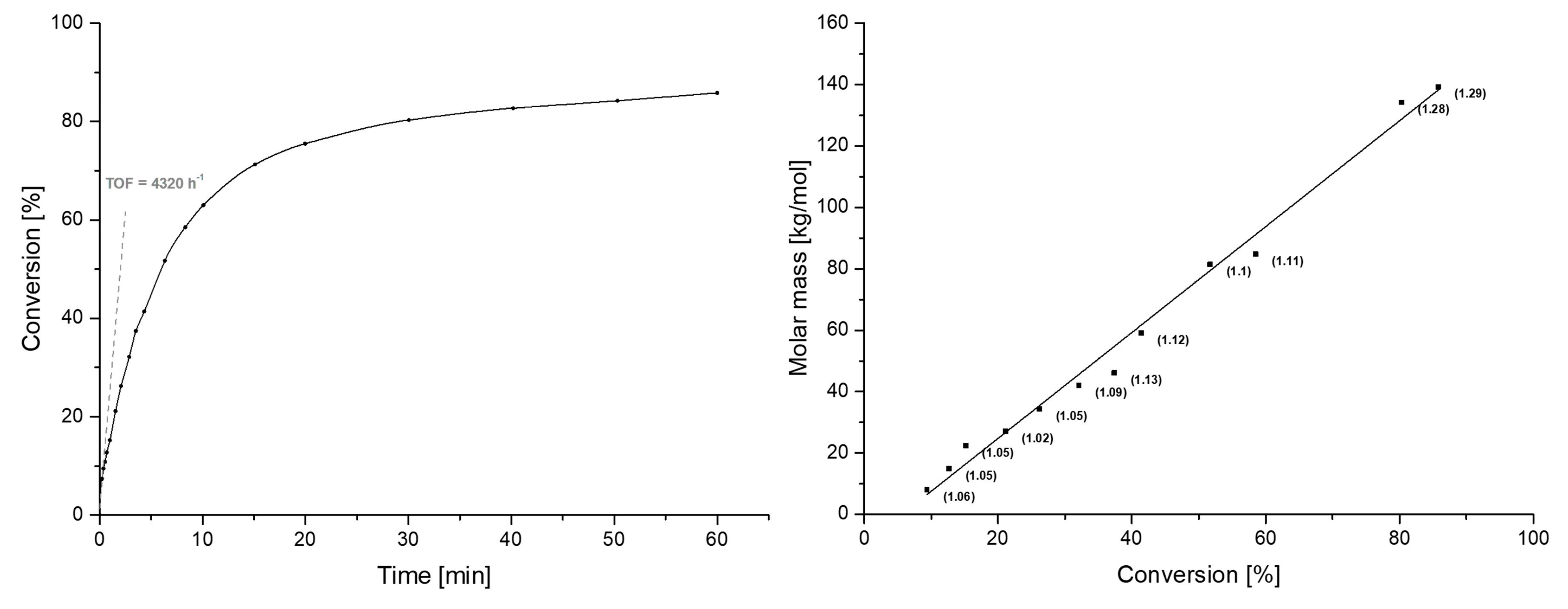
2.4. Polymerization Results
2.4.1. End-Group Analysis of PDEVP and P2VP Produced with Catalyst 4
2.4.2. Polymerization of DEVP and 2VP with Catalyst 4
2.5. Deprotection of PDEVP and P2VP
3. Materials and Methods
3.1. Materials
3.2. Instrumentalization
3.3. Synthesis of Catalyst 4 [(ONOO)tBuY(BenzPyOTBDMS)(thf)]
3.4. Polymerization Procedure
3.5. Activity Measurements
3.6. Polymer Deprotection
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Webster, O.W.; Hertler, W.R.; Sogah, D.Y.; Farnham, W.B.; RajanBabu, T.V. Group-transfer polymerization. 1. A new concept for addition polymerization with organosilicon initiators. J. Am. Chem. Soc. 1983, 105, 5706–5708. [Google Scholar] [CrossRef]
- Yasuda, H.; Yamamoto, H.; Yokota, K.; Miyake, S.; Nakamura, A. Synthesis of monodispersed high molecular weight polymers and isolation of an organolanthanide(III) intermediate coordinated by a penultimate poly(MMA) unit. J. Am. Chem. Soc. 1992, 114, 4908–4910. [Google Scholar] [CrossRef]
- Yasuda, H.; Ihara, E. Rare earth metal initiated polymerizations of polar and nonpolar monomers to give high molecular weight polymers with extremely narrow molecular weight distribution. Macromol. Chem. Phys. 1995, 196, 2417–2441. [Google Scholar] [CrossRef]
- Yasuda, H.; Ihara, E.; Nitto, Y.; Kakehi, T.; Morimoto, M.; Nodono, M. Organo Rare Earth Metal Initiated Living Polymerizations of Polar and Nonpolar Monomers. In Functional Polymers; Patil, A.O., Schulz, D.N., Novak, B.M., Eds.; American Chemical Society: Washington, DC, USA, 1998; Volume 704, pp. 149–162. [Google Scholar]
- Chen, E.Y.-X. Coordination polymerization of polar vinyl monomers by single-site metal catalysts. Chem. Rev. 2009, 109, 5157–5214. [Google Scholar] [CrossRef]
- Salzinger, S.; Rieger, B. Rare Earth metal-mediated group transfer polymerization of vinylphosphonates. Macromol. Rapid Commun. 2012, 33, 1327–1345. [Google Scholar] [CrossRef] [PubMed]
- Adams, F.; Pahl, P.; Rieger, B. Metal-Catalyzed Group-Transfer Polymerization: A Versatile Tool for Tailor-Made Functional (Co)Polymers. Chem. Eur. J. 2018, 24, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.; Ward, D.G. Group-transfer polymerization using cationic zirconocene compounds. J. Am. Chem. Soc. 1992, 114, 5460–5462. [Google Scholar] [CrossRef]
- Altenbuchner, P.T.; Adams, F.; Kronast, A.; Herdtweck, E.; Pöthig, A.; Rieger, B. Stereospecific catalytic precision polymerization of 2-vinylpyridine via rare earth metal-mediated group transfer polymerization with 2-methoxyethylamino-bis(phenolate)-yttrium complexes. Polym. Chem. 2015, 6, 6796–6801. [Google Scholar] [CrossRef]
- Xu, T.-Q.; Yang, G.-W.; Lu, X.-B. Highly Isotactic and High-Molecular-Weight Poly(2-vinylpyridine) by Coordination Polymerization with Yttrium Bis(phenolate) Ether Catalysts. ACS Catal. 2016, 6, 4907–4913. [Google Scholar] [CrossRef]
- Fuchise, K.; Chen, Y.; Satoh, T.; Kakuchi, T. Recent progress in organocatalytic group transfer polymerization. Polym. Chem. 2013, 4, 4278. [Google Scholar] [CrossRef]
- Knaus, M.G.M.; Giuman, M.M.; Pöthig, A.; Rieger, B. End of Frustration: Catalytic Precision Polymerization with Highly Interacting Lewis Pairs. J. Am. Chem. Soc. 2016, 138, 7776–7781. [Google Scholar] [CrossRef]
- Zhang, Y.; Miyake, G.M.; Chen, E.Y.-X. Alane-based classical and frustrated Lewis pairs in polymer synthesis: Rapid polymerization of MMA and naturally renewable methylene butyrolactones into high-molecular-weight polymers. Angew. Chem. Int. Ed. 2010, 49, 10158–10162. [Google Scholar] [CrossRef]
- Salzinger, S.; Seemann, U.B.; Plikhta, A.; Rieger, B. Poly(vinylphosphonate)s Synthesized by Trivalent Cyclopentadienyl Lanthanide-Induced Group Transfer Polymerization. Macromolecules 2011, 44, 5920–5927. [Google Scholar] [CrossRef]
- Soller, B.S.; Salzinger, S.; Rieger, B. Rare Earth Metal-Mediated Precision Polymerization of Vinylphosphonates and Conjugated Nitrogen-Containing Vinyl Monomers. Chem. Rev. 2016, 116, 1993–2022. [Google Scholar] [CrossRef]
- Chen, X.; Caporaso, L.; Cavallo, L.; Chen, E.Y.-X. Stereoselectivity in metallocene-catalyzed coordination polymerization of renewable methylene butyrolactones: From stereo-random to stereo-perfect polymers. J. Am. Chem. Soc. 2012, 134, 7278–7281. [Google Scholar] [CrossRef]
- Salzinger, S.; Soller, B.S.; Plikhta, A.; Seemann, U.B.; Herdtweck, E.; Rieger, B. Mechanistic studies on initiation and propagation of rare earth metal-mediated group-transfer polymerization of vinylphosphonates. J. Am. Chem. Soc. 2013, 35, 13030–13040. [Google Scholar] [CrossRef]
- Soller, B.S.; Salzinger, S.; Jandl, C.; Pöthig, A.; Rieger, B. C–H Bond Activation by σ-Bond Metathesis as a Versatile Route toward Highly Efficient Initiators for the Catalytic Precision Polymerization of Polar Monomers. Organometallics 2014, 34, 2703–2706. [Google Scholar] [CrossRef]
- Altenbuchner, P.T.; Soller, B.S.; Kissling, S.; Bachmann, T.; Kronast, A.; Vagin, S.I.; Rieger, B. Versatile 2-Methoxyethylaminobis(phenolate)yttrium Catalysts: Catalytic Precision Polymerization of Polar Monomers via Rare Earth Metal-Mediated Group Transfer Polymerization. Macromolecules 2014, 47, 7742–7749. [Google Scholar] [CrossRef]
- Mariott, W.R.; Chen, E.Y.-X. Stereospecific, Coordination Polymerization of Acrylamides by Chiral ansa -Metallocenium Alkyl and Ester Enolate Cations. Macromolecules 2004, 37, 4741–4743. [Google Scholar] [CrossRef]
- Rodriguez-Delgado, A.; Mariott, W.R.; Chen, E.Y.-X. Living and Syndioselective Polymerization of Methacrylates by Constrained Geometry Titanium Alkyl and Enolate Complexes. Macromolecules 2004, 37, 3092–3100. [Google Scholar] [CrossRef]
- Weger, M.; Grötsch, R.K.; Knaus, M.G.; Giuman, M.M.; Mayer, D.C.; Altmann, P.J.; Mossou, E.; Dittrich, B.; Pöthig, A.; Rieger, B. Non-Innocent Methylene Linker in Bridged Lewis Pair Initiators. Angew. Chem. Int. Ed. 2019, 58, 9797–9801. [Google Scholar] [CrossRef] [PubMed]
- Weger, M.; Giuman, M.M.; Knaus, M.G.; Ackermann, M.; Drees, M.; Hornung, J.; Altmann, P.J.; Fischer, R.A.; Rieger, B. Single-Site, Organometallic Aluminum Catalysts for the Precise Group Transfer Polymerization of Michael-Type Monomers. Chem. Eur. J. 2018, 24, 14950–14957. [Google Scholar] [CrossRef] [PubMed]
- Weger, M.; Pahl, P.; Schmidt, F.; Soller, B.S.; Altmann, P.J.; Pöthig, A.; Gemmecker, G.; Eisenreich, W.; Rieger, B. Isospecific Group-Transfer Polymerization of Diethyl Vinylphosphonate and Multidimensional NMR Analysis of the Polymer Microstructure. Macromolecules 2019, 52, 7073–7080. [Google Scholar] [CrossRef]
- Zhang, N.; Salzinger, S.; Soller, B.S.; Rieger, B. Rare earth metal-mediated group-transfer polymerization: From defined polymer microstructures to high-precision nano-scaled objects. J. Am. Chem. Soc. 2013, 135, 8810–8813. [Google Scholar] [CrossRef]
- Miyake, G.M.; Chen, E.Y.-X. Metallocene-Mediated Asymmetric Coordination Polymerization of Polar Vinyl Monomers to Optically Active, Stereoregular Polymers. Macromolecules 2008, 41, 3405–3416. [Google Scholar] [CrossRef]
- Kaneko, H.; Nagae, H.; Tsurugi, H.; Mashima, K. End-functionalized polymerization of 2-vinylpyridine through initial C–H bond activation of N-heteroaromatics and internal alkynes by yttrium ene-diamido complexes. J. Am. Chem. Soc. 2011, 133, 19626–19629. [Google Scholar] [CrossRef]
- Adams, F.; Machat, M.R.; Altenbuchner, P.T.; Ehrmaier, J.; Pöthig, A.; Karsili, T.N.V.; Rieger, B. Toolbox of Nonmetallocene Lanthanides: Multifunctional Catalysts in Group-Transfer Polymerization. Inorg. Chem. 2017, 56, 9754–9764. [Google Scholar] [CrossRef]
- Adams, F.; Altenbuchner, P.T.; Werz, P.D.L.; Rieger, B. Multiresponsive micellar block copolymers from 2-vinylpyridine and dialkylvinylphosphonates with a tunable lower critical solution temperature. RSC Adv. 2016, 6, 78750–78754. [Google Scholar] [CrossRef]
- Adams, F.; Pschenitza, M.; Rieger, B. Yttrium-Catalyzed Synthesis of Bipyridine-Functionalized AB-Block Copolymers: Micellar Support for Photocatalytic Active Rhenium-Complexes. ChemCatChem 2018, 10, 4309–4316. [Google Scholar] [CrossRef]
- Schwarzenböck, C.; Schaffer, A.; Nößner, E.; Nelson, P.J.; Huss, R.; Rieger, B. Fluorescent Polyvinylphosphonate Bioconjugates for Selective Cellular Delivery. Chem. Eur. J. 2018, 24, 2584–2587. [Google Scholar] [CrossRef]
- Schwarzenböck, C.; Schaffer, A.; Pahl, P.; Nelson, P.J.; Huss, R.; Rieger, B. Precise synthesis of thermoresponsive polyvinylphosphonate-biomolecule conjugates via thiol–ene click chemistry. Polym. Chem. 2018, 9, 284–290. [Google Scholar] [CrossRef]
- Altenbuchner, P.T.; Werz, P.D.L.; Schöppner, P.; Adams, F.; Kronast, A.; Schwarzenböck, C.; Pöthig, A.; Jandl, C.; Haslbeck, M.; Rieger, B. Next Generation Multiresponsive Nanocarriers for Targeted Drug Delivery to Cancer Cells. Chem. Eur. J. 2016, 22, 14576–14584. [Google Scholar] [CrossRef] [PubMed]
- Pahl, P.; Schwarzenböck, C.; Herz, F.A.D.; Soller, B.S.; Jandl, C.; Rieger, B. Core-First Synthesis of Three-Armed Star-Shaped Polymers by Rare Earth Metal-Mediated Group Transfer Polymerization. Macromolecules 2017, 50, 6569–6576. [Google Scholar] [CrossRef]
- Arndtsen, B.A.; Bergman, R.G.; Mobley, T.A.; Peterson, T.H. Selective Intermolecular Carbon-Hydrogen Bond Activation by Synthetic Metal Complexes in Homogeneous Solution. Acc. Chem. Res. 2002, 28, 154–162. [Google Scholar] [CrossRef]
- Labinger, J.A.; Bercaw, J.E. Understanding and exploiting C–H bond activation. Nature 2002, 417, 507–514. [Google Scholar] [CrossRef]
- Goldberg, K.I.; Goldman, A.S. (Eds.) Activation and Functionalization of C—H Bonds; American Chemical Society: Washington, DC, USA, 2004. [Google Scholar]
- Adams, F.; Rieger, B. From Michael-Type Systems to Biobased Lactones: Designing Novel Polymer Microstructures with Modified Bis(phenolate)lanthanides. Ph.D. Thesis, Technical University Munich, Munich, Germany, 2019. Available online: http://nbn-resolving.de/urn/resolver.pl?urn:nbn:de:bvb:91-diss-20190123-1464556-1-9 (accessed on 1 April 2020).
- Watson, P.L. Facile C–H activation by lutetium–methyl and lutetium–hydride complexes. J. Chem. Soc. Chem. Commun. 1983, 276–277. [Google Scholar] [CrossRef]
- Watson, P.L. Methane exchange reactions of lanthanide and early-transition-metal methyl complexes. J. Am. Chem. Soc. 1983, 105, 6491–6493. [Google Scholar] [CrossRef]
- Schaffer, A.; Kränzlein, M.; Rieger, B. Synthesis and Application of Functional Group-Bearing Pyridyl-based Initiators to Rare-Earth Metal-Mediated Group-Transfer Polymerization. Macromolecules. under review.
- Zhang, N.; Salzinger, S.; Deubel, F.; Jordan, R.; Rieger, B. Surface-initiated group transfer polymerization mediated by rare earth metal catalysts. J. Am. Chem. Soc. 2012, 134, 7333–7336. [Google Scholar] [CrossRef]
- Ajellal, N.; Carpentier, J.-F.; Guillaume, C.; Guillaume, S.M.; Helou, M.; Poirier, V.; Sarazin, Y.; Trifonov, A.A. Bridging the gap in catalysis via multidisciplinary approaches. Dalton Trans. 2010, 39, 8354. [Google Scholar]
- Anastasaki, A.; Nikolaou, V.; Nurumbetov, G.; Wilson, P.; Kempe, K.; Quinn, J.F.; Davis, T.P.; Whittaker, M.R.; Haddleton, D.M. Cu(0)-Mediated Living Radical Polymerization: A Versatile Tool for Materials Synthesis. Chem. Rev. 2016, 116, 835–877. [Google Scholar] [CrossRef]
- Matyjaszewski, K. Advanced Materials by Atom Transfer Radical Polymerization. Adv. Mater. 2018, 30, e1706441. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, M.; Hong, C.R.; Wong, W.W.; Liew, L.P.; Shome, A.; Wang, J.; Gu, Y.; Stevenson, R.J.; Qi, W.; Anderson, R.F.; et al. Next-Generation Hypoxic Cell Radiosensitizers: Nitroimidazole Alkylsulfonamides. J. Med. Chem. 2018, 61, 1241–1254. [Google Scholar] [CrossRef] [PubMed]
- Doundoulakis, T.; Xiang, A.X.; Lira, R.; Agrios, K.A.; Webber, S.E.; Sisson, W.; Aust, R.M.; Shah, A.M.; Showalter, R.E.; Appleman, J.R.; et al. Myxopyronin B analogs as inhibitors of RNA polymerase, synthesis and biological evaluation. Bioorg. Med. Chem. Lett. 2004, 14, 5667–5672. [Google Scholar] [CrossRef] [PubMed]
- Greene, T.W.; Wuts, P.G.M. Protective Groups in Organic Synthesis, 3rd ed.; Wiley-Interscience: Hoboken, NJ, USA, 2002; pp. 201–270. [Google Scholar]
- Nelson, T.D.; Crouch, R.D. Selective Deprotection of Silyl Ethers. Synthesis 1996, 1996, 1031–1069. [Google Scholar] [CrossRef]
- Seemann, U.B. Polyvinylphosphonate und Deren Copolymere Durch Seltenerdmetall Initiierte Gruppen-Transfer-Polymerisation. Ph.D. Thesis, Technical University Munich, Munich, Germany, 2010. Available online: http://nbn-resolving.de/urn/resolver.pl?urn:nbn:de:bvb:91-diss-20101013-992998-1-7 (accessed on 1 April 2020).
- Tshuva, E.Y.; Groysman, S.; Goldberg, I.; Kol, M.; Goldschmidt, Z. [ONXO]-Type Amine Bis(phenolate) Zirconium and Hafnium Complexes as Extremely Active 1-Hexene Polymerization Catalysts. Organometallics 2002, 21, 662–670. [Google Scholar] [CrossRef]
- Hultzsch, K.C.; Voth, P.; Beckerle, K.; Spaniol, T.P.; Okuda, J. Single-Component Polymerization Catalysts for Ethylene and Styrene: Synthesis, Characterization, and Reactivity of Alkyl and Hydrido Yttrium Complexes Containing a Linked Amido−Cyclopentadienyl Ligand. Organometallics 2000, 19, 228–243. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | M | [M]/[4] a | Conv. b (%) | Mn,calc c (kg mol−1) | Mn,abs d (kg mol−1) | Đ d (-) | I* e (%) | TOF (h−1) | TOF* f (h−1) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | DEVP | 600/1 | 86 | 83.1 | 139 | 1.29 | 68 | 4320 | 6350 |
| 2 | 2VP | 400/1 | 99 | 40.8 | 55.7 | 1.02 | 67 | 500 | 750 |
| Entry | [DEVP]/[4] a | Solvent b | Conv. c (%) | Mn,calc d (kg mol−1) | Mn,abs e (kg mol−1) | Mn,NMR f (kg mol−1) | Đ e (-) | I g (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 200/1 | toluene | 99 | 31.8 | 34.6 | 42.3 | 1.07 | 92 |
| 2 | 200/1 | thf | 99 | 32.8 | 38.0 | 48.3 | 1.13 | 86 |
| 3 | 200/1 | dcm | 44 | 14.8 | 25.0 | 34.0 | 1.29 | 59 |
| 4 | 100/1 | toluene | 99 | 15.8 | 16.6 | 22.5 | 1.04 | 95 |
| 5 | 400/1 | toluene | 96 | 61.8 | 104 | 122.3 | 1.33 | 58 |
| Entry | [2VP]/[4] a | Solvent b | Conv. c (%) | Mn,calc d (kg mol−1) | Mn,abs e (kg mol−1) | Mn,NMR f (kg mol−1) | Đ e (-) | I g (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 200/1 | toluene | 99 | 22.1 | 25.0 | 27.2 | 1.04 | 89 |
| 2 | 200/1 | thf | 22 | 5.3 | 21.5 | 21.5 | 1.31 | 25 |
| 3 | 200/1 | dcm | 99 | 19.0 | 22.6 | 26.9 | 1.06 | 84 |
| 4 | 100/1 | toluene | 99 | 10.5 | 12.8 | 11.1 | 1.09 | 82 |
| 5 | 400/1 | toluene | 98 | 42.2 | 58.4 | 58.8 | 1.01 | 72 |
| Entry | Polymer | Deprotection Procedure | Before Deprotection | After Deprotection | ||
|---|---|---|---|---|---|---|
| Mn,abs a (kg mol−1) | Đ a (-) | Mn,abs a (kg mol−1) | Đ a (-) | |||
| 1 | PDEVP | A | 43.4 | 1.11 | 40.6 | 1.18 |
| 2 | P2VP | B | 25.2 | 1.05 | 29.0 | 1.05 |
| 3 | PDEVP | C | 38.0 | 1.13 | 34.3 | 1.11 |
| 4 | P2VP | C | 25.8 | 1.09 | 28.7 | 1.10 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pehl, T.M.; Kränzlein, M.; Adams, F.; Schaffer, A.; Rieger, B. C–H Bond Activation of Silyl-Substituted Pyridines with Bis(Phenolate)Yttrium Catalysts as a Facile Tool towards Hydroxyl-Terminated Michael-Type Polymers. Catalysts 2020, 10, 448. https://doi.org/10.3390/catal10040448
Pehl TM, Kränzlein M, Adams F, Schaffer A, Rieger B. C–H Bond Activation of Silyl-Substituted Pyridines with Bis(Phenolate)Yttrium Catalysts as a Facile Tool towards Hydroxyl-Terminated Michael-Type Polymers. Catalysts. 2020; 10(4):448. https://doi.org/10.3390/catal10040448
Chicago/Turabian StylePehl, Thomas M., Moritz Kränzlein, Friederike Adams, Andreas Schaffer, and Bernhard Rieger. 2020. "C–H Bond Activation of Silyl-Substituted Pyridines with Bis(Phenolate)Yttrium Catalysts as a Facile Tool towards Hydroxyl-Terminated Michael-Type Polymers" Catalysts 10, no. 4: 448. https://doi.org/10.3390/catal10040448