The Effect of Carbon Content on Methanol Oxidation and Photo-Oxidation at Pt-TiO2-C Electrodes

 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
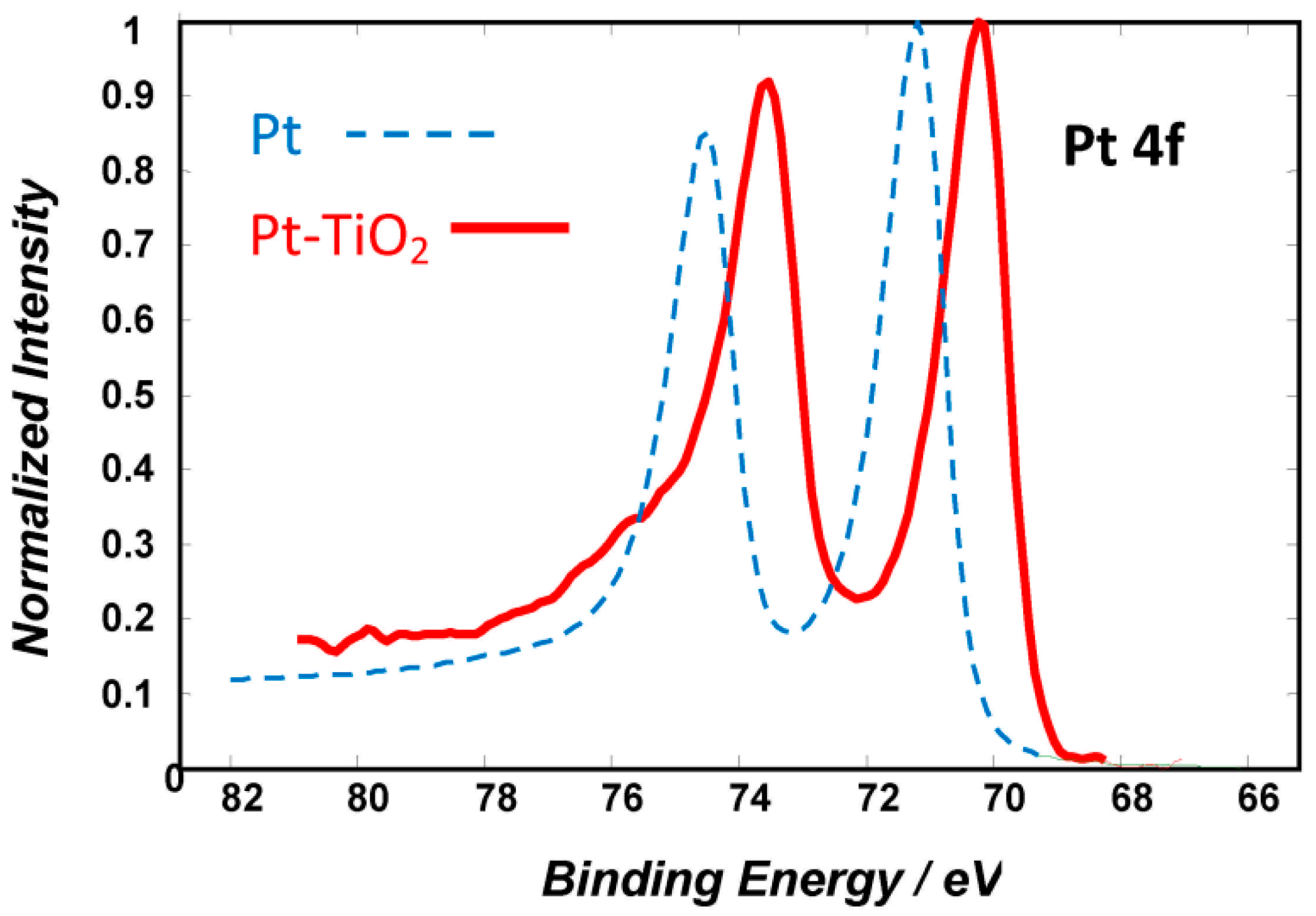
2.1. Microscopic (TEM) and Spectroscopic (EDS, XPS) Characterization of the Catalyst
2.2. Electrochemical Characterization of the Catalyst (Cyclic Voltametry, CV)
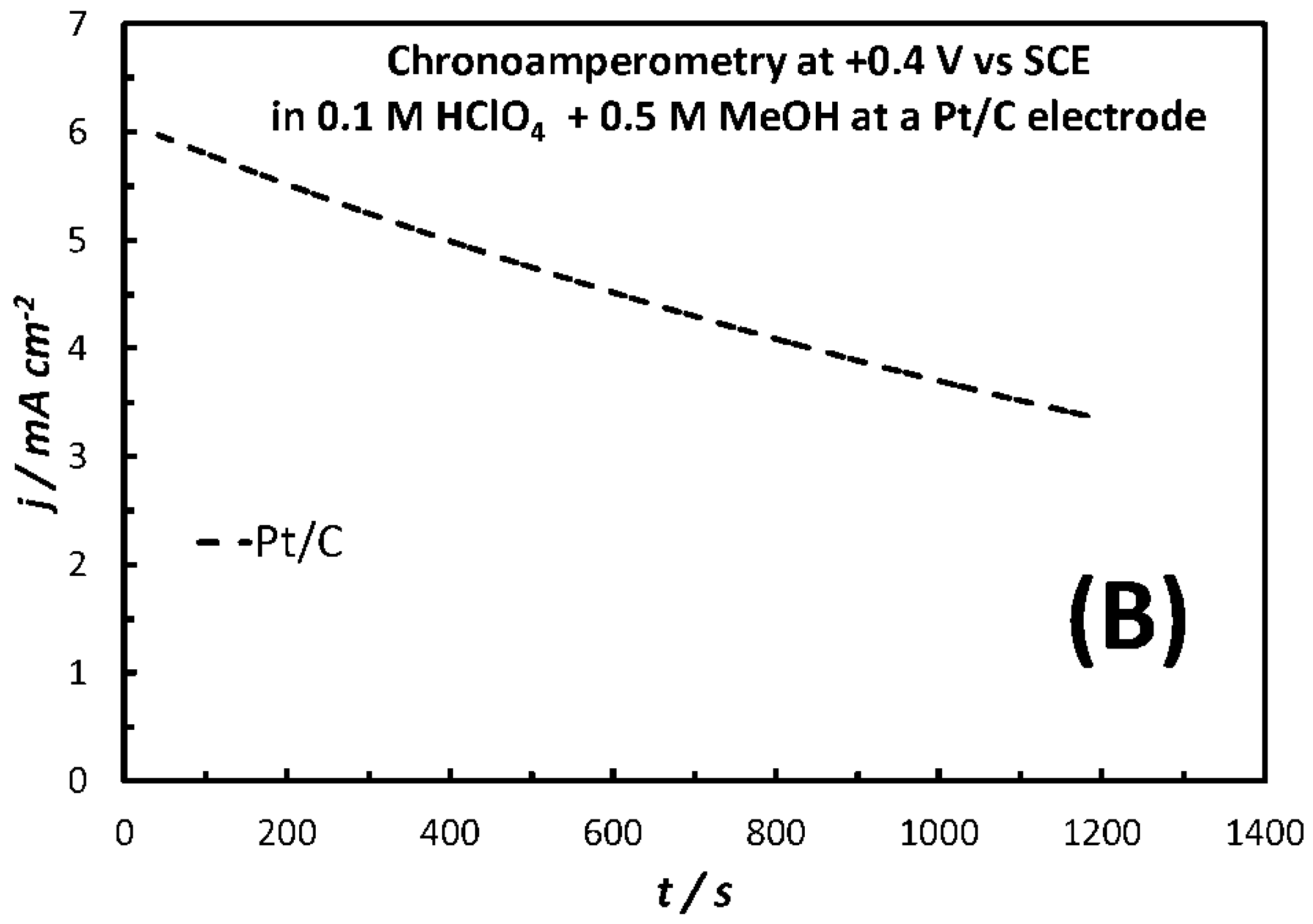
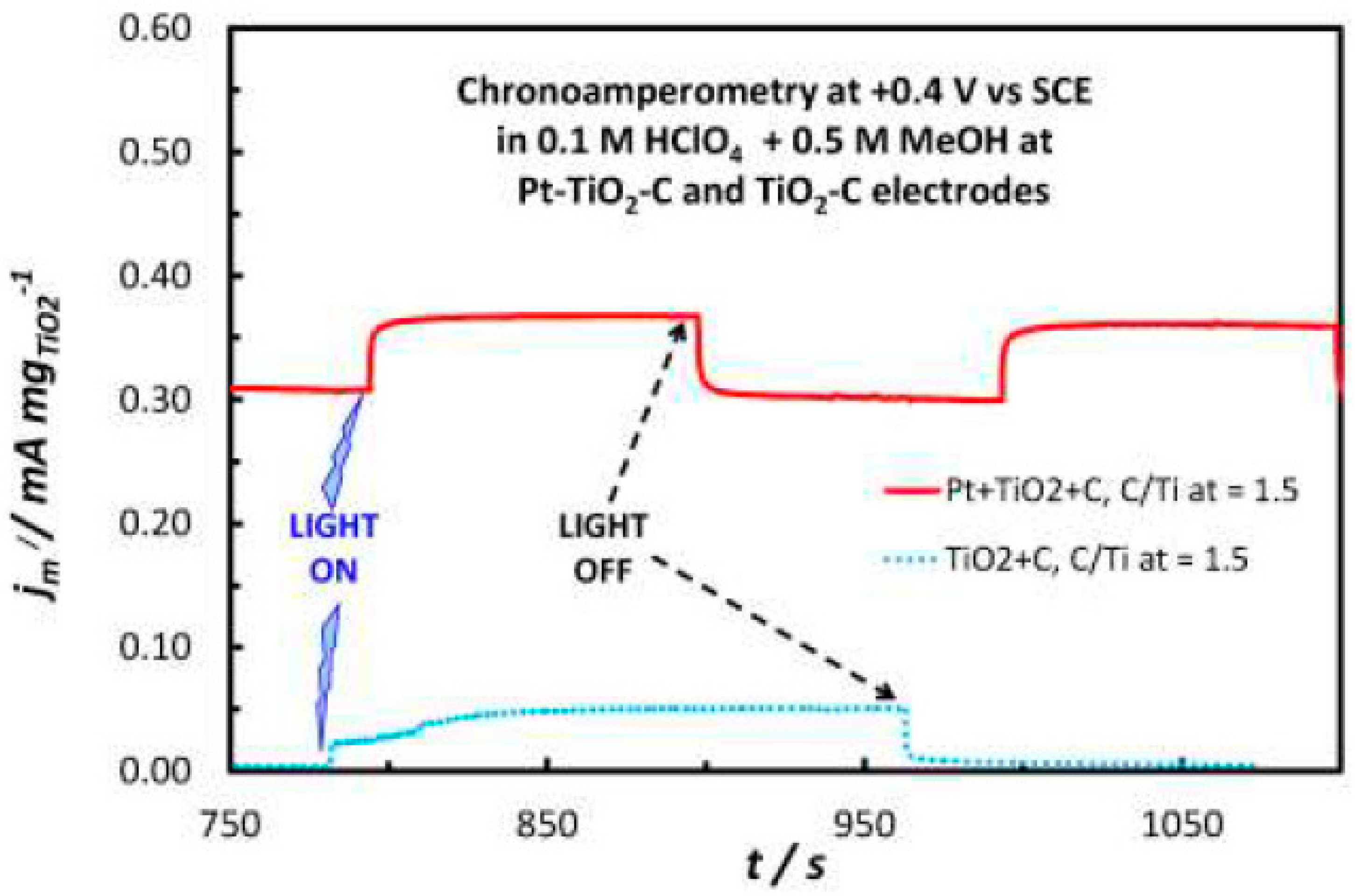
2.3. Methanol Oxidation Reaction (MOR) in the Dark and Under Illumination
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Electrode Preparation
3.3. Catalyst Characterization
3.4. Electrochemical/Photoelectrochemical Experiments
4. Conclusions
- Optimized Pt-TiO2-C catalytic electrodes prepared by photodeposition of Pt on TiO2 and subsequent mixing with C exhibited an intrinsic methanol oxidation activity that is higher than that of a commercial Pt/C catalyst.
- C plays a significant role, not only in increasing catalyst connectivity/conductivity, but also in increasing the catalytic activity towards methanol oxidation.
- The Pt mass-specific activity of the Pt-TiO2-C catalytic electrodes is currently lower than that of commercial Pt/C catalyst, due to limited catalyst connectivity/conductivity and it should be improved either by increasing the amount of Pt deposited (optimization of the photodeposition preparation route) or by increasing TiO2 conductivity (e.g., by doping or chemical treatment).
- UV illumination of the Pt-TiO2-C catalytic electrodes improved the stability of the methanol oxidation current in short term test and it should also be tested in long term experiments.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sharma, S.; Pollet, B.G. Support materials for PEMFC and DMFC electrocatalysts-A review. J. Power Sources 2012, 208, 96–119. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, J.; Gu, J.; Su, L.; Cheng, L. An overview of metal oxide materials as electrocatalysts and supports for polymer electrolyte fuel cells. Energy Environ. Sci. 2014, 7, 2535–2558. [Google Scholar] [CrossRef]
- Jung, N.; Chung, D.Y.; Ryu, J.; Yoo, S.J.; Sung, Y.-E. Pt-based nanoarchitecture and catalyst design for fuel cell applications. Nano Today 2014, 9, 433–456. [Google Scholar] [CrossRef]
- Cao, M.; Wu, D.; Cao, R. Recent advances in the stabilization of platinum electrocatalysts for fuel-cell reactions. ChemCatChem 2014, 6, 26–45. [Google Scholar] [CrossRef]
- Ali, I.; Suhail, M.; Alothman, Z.A.; Alwarthan, A. Recent advances in syntheses, properties and applications of TiO2 nanostructures. RSC Adv. 2018, 8, 30125–30147. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.; Chen, J.; Wang, M.; Sheng, X.; Chen, X.; Feng, X.; Mao, S.S. Titanium dioxide nanostructures for photoelectrochemical applications. Prog. Mater. Sci. 2018, 98, 299–385. [Google Scholar] [CrossRef]
- Abdullah, N.; Kamarudin, S.K. Titanium dioxide in fuel cell technology: An overview. J. Power Sources 2015, 278, 109–118. [Google Scholar] [CrossRef]
- Hayden, B.E.; Malevich, D.V.; Pletcher, D. Platinum catalysed nanoporous titanium dioxide electrodes in H2SO4 solutions. Electrochem. Commun. 2001, 3, 395–399. [Google Scholar] [CrossRef]
- Siracusano, S.; Stassi, A.; Modica, E.; Baglio, V.; Aricò, A.S. Preparation and characterisation of Ti oxide based catalyst supports for low temperature fuel cells. Int. J. Hydrog. Energy 2013, 38, 11600–11608. [Google Scholar] [CrossRef]
- Polo, A.S.; Santos, M.C.; De Souza, R.F.B.; Alves, W.A. Pt-Ru-TiO2 photoelectrocatalysts for methanol oxidation. J. Power Sources 2011, 196, 872–876. [Google Scholar] [CrossRef]
- Zheng, Y.; Chen, H.; Dai, Y.; Zhang, N.; Zhao, W.; Wang, S.; Lou, Y.; Li, Y.; Sun, Y. Preparation and characterization of Pt/TiO2 nanofibers catalysts for methanol electro-oxidation. Electrochim. Acta 2015, 178, 74–79. [Google Scholar] [CrossRef]
- Selvarani, G.; Maheswari, S.; Sridhar, P.; Pitchumani, S.; Shukla, A.K. Carbon-supported Pt-TiO2 as a methanol-tolerant oxygen-reduction catalyst for DMFCs. J. Electrochem. Soc. 2009, 156, B1354–B1360. [Google Scholar] [CrossRef]
- de Tacconi, N.R.; Chenthamarakshan, C.R.; Rajeshwar, K.; Lin, W.-Y.; Carlson, T.F.; Nikiel, L.; Wampler, W.A.; Sambandam, S.; Ramanic, V. Photocatalytically Generated Pt/C–TiO2 Electrocatalysts with Enhanced Catalyst Dispersion for Improved Membrane Durability in Polymer Electrolyte Fuel Cells. J. Electrochem. Soc. 2008, 155, B1102–B1109. [Google Scholar] [CrossRef]
- Sambandam, S.; Valluri, V.; Chanmanee, W.; De Tacconi, N.R.; Wampler, W.A.; Lin, W.-Y.; Carlson, T.F.; Ramani, V.; Rajeshwar, K. Platinum-carbon black-titanium dioxide nanocomposite electrocatalysts for fuel cell applications. J. Chem. Sci. 2009, 121, 655–664. [Google Scholar] [CrossRef]
- Georgieva, J.; Valova, E.; Mintsouli, I.; Sotiropoulos, S.; Tatchev, D.; Armyanov, S.; Hubin, A.; Dille, J.; Hoell, A.; Raghuwanshi, V.; et al. Pt(Ni) electrocatalysts for methanol oxidation prepared by galvanic replacement on TiO2 and TiO2–C powder supports. J. Electroanal. Chem. 2015, 754, 65–74. [Google Scholar] [CrossRef]
- Dimitrova, N.; Georgieva, J.; Sotiropoulos, S.; Boiadjieva-Scherzer, T.Z.; Valova, E.; Armyanov, S.; Steenhaut, O.; Hubin, A.; Karashanova, D. Pt(Cu) catalyst on TiO2 powder support prepared by photodeposition-galvanic replacement method. J. Electroanal. Chem. 2018, 823, 624–632. [Google Scholar] [CrossRef]
- Qin, Y.-H.; Li, Y.; Lv, R.-L.; Wang, T.-L.; Wang, W.-G.; Wang, C.-W. Enhanced methanol oxidation activity and stability of Pt particles anchored on carbon-doped TiO2 nanocoating support. J. Power Sources 2015, 278, 639–644. [Google Scholar] [CrossRef]
- Lee, J.-M.; Han, S.-B.; Kim, J.-Y.; Lee, Y.-W.; Ko, A.-R.; Roh, B.; Hwang, I.; Park, K.-W. TiO2@carbon core-shell nanostructure supports for platinum and their use for methanol electrooxidation. Carbon 2010, 48, 2290–2296. [Google Scholar] [CrossRef]
- Liao, S.-Y.; Yang, Y.-C.; Huang, S.-H.; Gan, J.-Y. Synthesis of Pt@TiO2@CNTs hierarchical structure catalyst by atomic layer deposition and their photocatalytic and photoelectrochemical activity. Nanomaterials 2017, 7, 97. [Google Scholar] [CrossRef]
- Song, H.; Xiao, P.; Qiu, X.; Zhu, W. Design and preparation of highly active carbon nanotube-supported sulfated TiO2 and platinum catalysts for methanol electrooxidation. J. Power Sources 2010, 195, 1610–1614. [Google Scholar] [CrossRef]
- Wang, M.; Wang, Z.; Wei, L.; Li, J.; Zhao, X. Catalytic performance and synthesis of a Pt/graphene-TiO2 catalyst using an environmentally friendly microwave-assisted solvothermal method. Cuihua Xuebao Chin. J. Catal. 2017, 38, 1680–1687. [Google Scholar] [CrossRef]
- Winter, M.; Brodd, R.J. What Are Batteries, Fuel Cells, and Supercapacitors? Chem. Rev. 2004, 104, 4245–4269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maillant, F.; Pronkin, S.; Savinova, E.R. Influence of size on the electrocatalyticactivities of supported metal nanoparticles in fuel cell-related reactions. In Handbook of Fuel Cells–Fundamentals, Technology and Applications, Volume 5: Advances in Electocatalysis, Materials, Diagnostics and Durability; Vielstich, W., Gasteiger, H.A., Yokokawa, H., Eds.; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Yang, L.; Ge, J.; Liu, C.; Wang, G.; Xing, W. Approaches to improve the performance of anode methanol oxidation reaction—A short review. Curr. Opin. Electrochem. 2017, 4, 83–88. [Google Scholar] [CrossRef]
- Lamy, C.; Lima, A.; LeRhun, V.; Delime, F.; Coutencau, C.; Léger, J.-M. Recent advances in the development of direct alcohol fuel cells (DAFC). J. Power Sources 2002, 105, 283. [Google Scholar] [CrossRef]
- Antolini, E.; Salgado, J.R.C.; Gonzalez, E.R. The methanol oxidation reaction on platinum alloys with the first row transition metals. The case of Pt–Co and –Ni alloy electrocatalysts for DMFCs: A short review. Appl. Catal. B Environ. 2006, 63, 137–149. [Google Scholar] [CrossRef]
- Antolini, E. Platinum-based ternary catalysts for low temperature fuel cells. Part II. Electrochemical properties. Appl. Catal. B Environ. 2007, 74, 337–350. [Google Scholar] [CrossRef]
- Antolini, E.; Lopes, T.; Gonzalez, E.R. An overview of platinum-based catalysts as methanol-resistant oxygen reduction materials for direct methanol fuel cells. J. Alloys Compd. 2008, 461, 253–262. [Google Scholar] [CrossRef]
- Kulesza, P.J.; Pieta, I.S.; Rutkowska, I.A.; Wadas, A.; Marks, D.; Klak, K.; Stobinski, L.; Cox, J.A. Electrocatalytic oxidation of small organic molecules in acid medium: Enhancement of activity of noble metal nanoparticles and their alloys by supporting or modifying them with metal oxides. Electrochim. Acta 2013, 110, 474–483. [Google Scholar] [CrossRef] [Green Version]
- Antolini, E. Photo-assisted methanol oxidation on Pt-TiO2 catalysts for direct methanol fuel cells: A short review. Appl. Catal. B Environ. 2018, 237, 491–503. [Google Scholar] [CrossRef]
- Camacho, R.G.G.; Huerta, M.A.; Valenzuela, N. Alonso-Vante, Preparation and Characterization of Pt/C and Pt/TiO2 Electrocatalysts by Liquid Phase Photodeposition. Top. Catal. 2011, 54, 512–518. [Google Scholar] [CrossRef]
- Geboes, B.; Mintsouli, I.; Wouters, B.; Georgieva, J.; Kakaroglou, A.; Sotiropoulos, S.; Valova, E.; Armyanov, S.; Hubin, A.; Breugelmans, T. Surface and electrochemical characterisation of a Pt-Cu/C nano-structured electrocatalyst, prepared by galvanic displacement. Appl. Catal. B Environ. 2014, 150–151, 249–256. [Google Scholar] [CrossRef]
- Huang, H.; Leung, D.Y.C. Complete elimination of indoor formaldehyde over supported Pt catalysts with extremely low Pt content at ambient temperature. J. Catal. 2011, 280, 60–67. [Google Scholar] [CrossRef]
- Nie, L.; Yu, J.; Li, X.; Cheng, B.; Liu, G.; Jaroniec, M. Enhanced performance of NaOH-modified Pt/TiO2 toward Room temperature selective oxidation of formaldehyde. Environ. Sci. Technol. 2013, 47, 2777–2783. [Google Scholar] [CrossRef] [PubMed]
- Cui, E.; Hou, G.; Shao, R.; Guan, R. Facet-Dependent Activity of Pt Nanoparticles as Cocatalyst on TiO2 Photocatalyst for Dye-Sensitized Visible-Light Hydrogen Generation. J. Nanomater. 2016, 3469393. [Google Scholar] [CrossRef]
- Spichiger-Ulmann, M.; Monnier, A.; Koudelka, M.; Augustynski, J. Electrochemical study of the state of platinum in various SMSI and non-SMSI Pt/TiO2 catalysts. Prepr. Symp. 1985, 30, 182. [Google Scholar]
- Spichiger-Ulmann, M.; Monnier, A.; Koudelka, M.; Augustynski, J. Pectroscopic and Electrochemical Study of the State of Pt in Pt-TiO2 Catalysts; ACS Symposium Series; ACS: Washington, DC, USA, 1986; pp. 212–227. [Google Scholar]
- Nie, L.; Zhou, P.; Yu, J.; Jaroniec, M. Deactivation and regeneration of Pt/TiO2 nanosheet-type catalysts with exposed (0 0 1) facets for room temperature oxidation of formaldehyde. J. Mol. Catal. A Chem. 2014, 390, 7–13. [Google Scholar] [CrossRef]
- Lewera, A.; Timperman, L.; Roguska, A.; Alonso-Vante, N. Metal support interactions between nanosized Pt and metal oxides (WO3 and TiO2) studied using X-ray photoelectron spectroscopy. J. Phys. Chem. C 2011, 115, 20153–20159. [Google Scholar] [CrossRef]
- Bardeen, J. Surface states and rectification at a metal semi-conductor contact. Phys. Rev. 1947, 71, 717–727. [Google Scholar] [CrossRef]
- Kim, H.; Kim, D.-W.; Phark, S.-H. Transport behaviours and nanoscopic resistance profiles of electrically stressed Pt/TiO2/Ti planar junctions. J. Phys. D Appl. Phys. 2010, 43, 505305. [Google Scholar] [CrossRef]
- Braslau, N. Alloyed ohmic contacts to GaAs. J. Vac. Sci. Technol. 1981, 19, 803–807. [Google Scholar] [CrossRef]
- Poh, C.K.; Tian, Z.; Gao, J.; Liu, Z.; Lin, J.; Feng, Y.P.; Su, F. Nanostructured trimetallic Pt/FeRuC, Pt/NiRuC, and Pt/CoRuC catalysts for methanol electrooxidation. J. Mater. Chem. 2012, 22, 13643–13652. [Google Scholar] [CrossRef]
- Bligaard, T.; Nørskov, J.K. Ligand effects in heterogeneous catalysis and electrochemistr. Electrochim. Acta 2007, 52, 5512–5516. [Google Scholar] [CrossRef]
- Weinert, M.; Watson, R.E. Core-level shifts in bulk alloys and surface adlayers. Phys. Rev. B 1995, 51, 17168–17180. [Google Scholar] [CrossRef] [PubMed]
- Lindström, R.W.; Kortsdottir, K.; Wesselmark, M.; Oyarce, A.; Lagergren, C.; Lindbergh, G. Active area determination of porous Pt electrodes used in polymer electrolyte fuel cells: Temperature and humidity effects. J. Electrochem. Soc. 2010, 157, B1795–B1801. [Google Scholar] [CrossRef]
- Haghnegahdar, S.; Noroozifar, M. Deposition of PdPtAu Nanoparticles on Hollow Nanospheres of Fe3O4 as a New Catalyst for Methanol Electrooxidation: Application in Direct Methanol Fuel Cell. Electroanalysis 2017, 29, 2896–2905. [Google Scholar] [CrossRef]
- Momeni, M.M. Evaluation of the performance of Pt-MWCNTs nanocomposites electrodeposited on titanium for methanol electro-oxidation. Port. Electrochim. Acta 2015, 33, 331–341. [Google Scholar] [CrossRef]
- Zhang, Z.; Ye, M.; Harvey, E.J.; Merle, G. Methanol electrooxidation with platinum decorated hematene Nanosheet. J. Electrochem. Soc. 2019, 166, H135–H139. [Google Scholar] [CrossRef]
- Bagotzky, V.S.; Vassilyev, Y.B. Mechanism of electro-oxidation of methanol on the platinum electrode. Electrochim. Acta 1967, 12, 1323–1343. [Google Scholar] [CrossRef]
- Fan, Y.; Yang, Z.; Huang, P.; Zhang, X.; Liu, Y.-M. Pt/TiO2-C with hetero interfaces as enhanced catalyst for methanol electrooxidation. Electrochim. Acta 2013, 105, 157–161. [Google Scholar] [CrossRef]
- Hasa, B.; Kalamaras, E.; Papaioannou, E.I.; Sygellou, L.; Katsaounis, A. Electrochemical oxidation of alcohols on Pt-TiO2 binary electrodes. Int. J. Hydrog. Energy 2013, 38, 15395–15404. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, W.; Du, Y.; Yang, P.; Wang, C.; Xu, J. Enhanced electrocatalytic performance for. methanol oxidation on Pt-TiO2/ITO electrode under UV illuminatio. Int. J. Hydrog. Energy 2010, 35, 13290–13297. [Google Scholar] [CrossRef]
- Chen, C.-S.; Pan, F.-M. Electrocatalytic activity of Pt nanoparticles deposited on porous TiO2 supports toward methanol oxidation. Appl. Catal. B Environ. 2009, 91, 663–669. [Google Scholar] [CrossRef]
- Song, Y.-Y.; Gao, Z.-D.; Schmuki, P. Highly uniform Pt nanoparticle decoration on TiO2 nanotube arrays: A refreshable platform for methanol electrooxidation. Electrochem. Commun. 2011, 13, 290–293. [Google Scholar] [CrossRef]
- Wu, J.W.; Madani, S.H.; Biggs, M.J.; Phillip, P.; Lei, C.; Hu, E.J. Characterizations of Activated Carbon-Methanol Adsorption Pair Including the Heat of Adsorptions. J. Chem. Eng. Data 2015, 60, 1727–1731. [Google Scholar] [CrossRef]
- Al-Dawery, S.K. Adsorption of methanol from methanol–water mixture by activated carbon and its. regeneration using photo-oxidation process. Desalin. Water Treat. 2016, 57, 3065–3073. [Google Scholar] [CrossRef]
- Stevanović, S.; Panić, V.; Tripković, D.; Jovanović, V.M. Promoting effect of carbon functional groups in methanol oxidation on supported Pt catalyst. Electrochem. Commun. 2009, 11, 18–21. [Google Scholar] [CrossRef]
- Georgieva, J.; Armyanov, S.; Poulios, I.; Jannakoudakis, A.D.; Sotiropoulos, S. Gas phase photoelectrochemistry in a polymer electrolyte cell with a titanium dioxide/carbon/Nafion® photoanode. Electrochem. Solid State Lett. 2010, 13, P11–P13. [Google Scholar] [CrossRef]
- Svetlozar, I.; Ioanna, M.; Jenia, G.; Stephan, A.; Eugenia, V.; Georgios, K.; Ioannis, P.; Sotiris, S. Platinized titanium dioxide electrodes for methanol oxidation and photo-oxidation. J. Electrochem. Sci. Eng. 2012, 2, 155–236. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst Notation | C/Ti Atom Concentration Ratio | % w/w C in Catalyst | % w/w Pt in Catalyst | Pt Mass-Specific EASA/ m2 g−1 |
|---|---|---|---|---|
| Pt/C | n/a | 90 | 10 | 41 |
| Pt-TiO2-C, C/Ti at = 3 | 3 | 29 | 5.5 | 4.3 |
| Pt-TiO2-C, C/Ti at = 1.5 | 1.5 | 17 | 6.3 | 3.3 |
| Pt-TiO2-C, C/Ti at = 0.8 | 0.8 | 10 | 6.9 | 2.1 |
| Pt-TiO2-C, C/Ti at = 0.4 | 0.4 | 5.2 | 7.2 | 0.8 |
| TiO2-C, C/Ti at = 1.5 | 1.5 | 17 | n/a | n/a |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papaderakis, A.; Spyridou, O.; Karanasios, N.; Touni, A.; Banti, A.; Dimitrova, N.; Armyanov, S.; Valova, E.; Georgieva, J.; Sotiropoulos, S. The Effect of Carbon Content on Methanol Oxidation and Photo-Oxidation at Pt-TiO2-C Electrodes. Catalysts 2020, 10, 248. https://doi.org/10.3390/catal10020248
Papaderakis A, Spyridou O, Karanasios N, Touni A, Banti A, Dimitrova N, Armyanov S, Valova E, Georgieva J, Sotiropoulos S. The Effect of Carbon Content on Methanol Oxidation and Photo-Oxidation at Pt-TiO2-C Electrodes. Catalysts. 2020; 10(2):248. https://doi.org/10.3390/catal10020248
Chicago/Turabian StylePapaderakis, Athanasios, Olga Spyridou, Nikolaos Karanasios, Aikaterini Touni, Angeliki Banti, Nina Dimitrova, Stephan Armyanov, Eugenia Valova, Jenia Georgieva, and Sotiris Sotiropoulos. 2020. "The Effect of Carbon Content on Methanol Oxidation and Photo-Oxidation at Pt-TiO2-C Electrodes" Catalysts 10, no. 2: 248. https://doi.org/10.3390/catal10020248