Oxygen Reduction Reaction Catalyzed by Pt3M (M = 3d Transition Metals) Supported on O-doped Graphene


Abstract
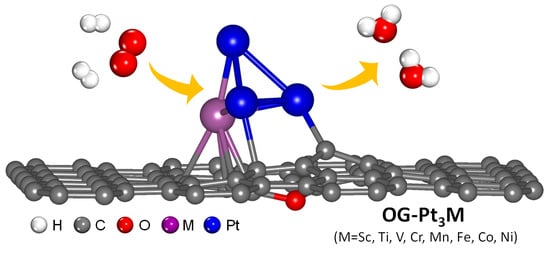
:
1. Introduction
2. Results and Discussion
2.1. Optimized Pt3M Structures on O-doped Graphene
2.2. Oxygen Adsorption and Reduction Mechanism
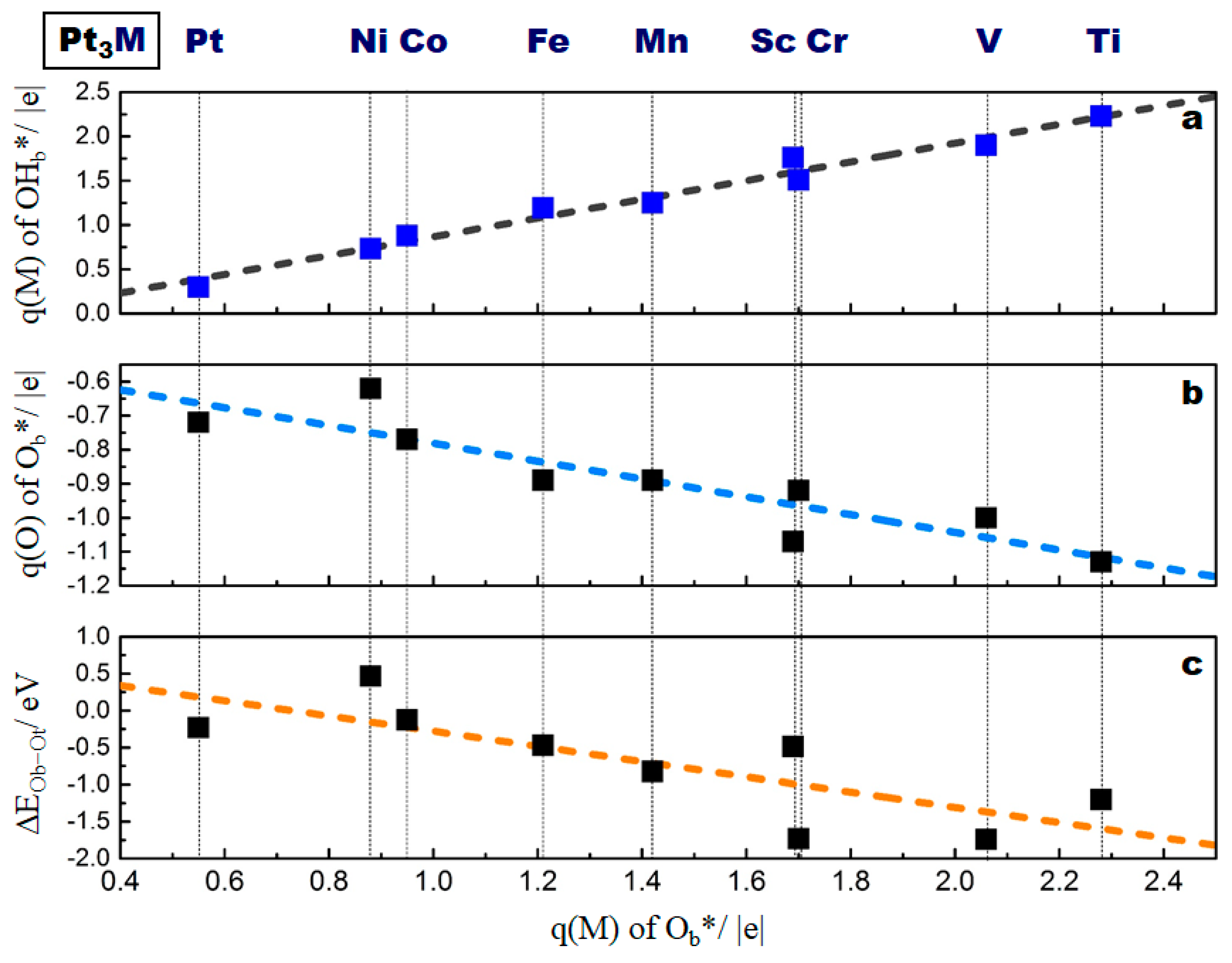
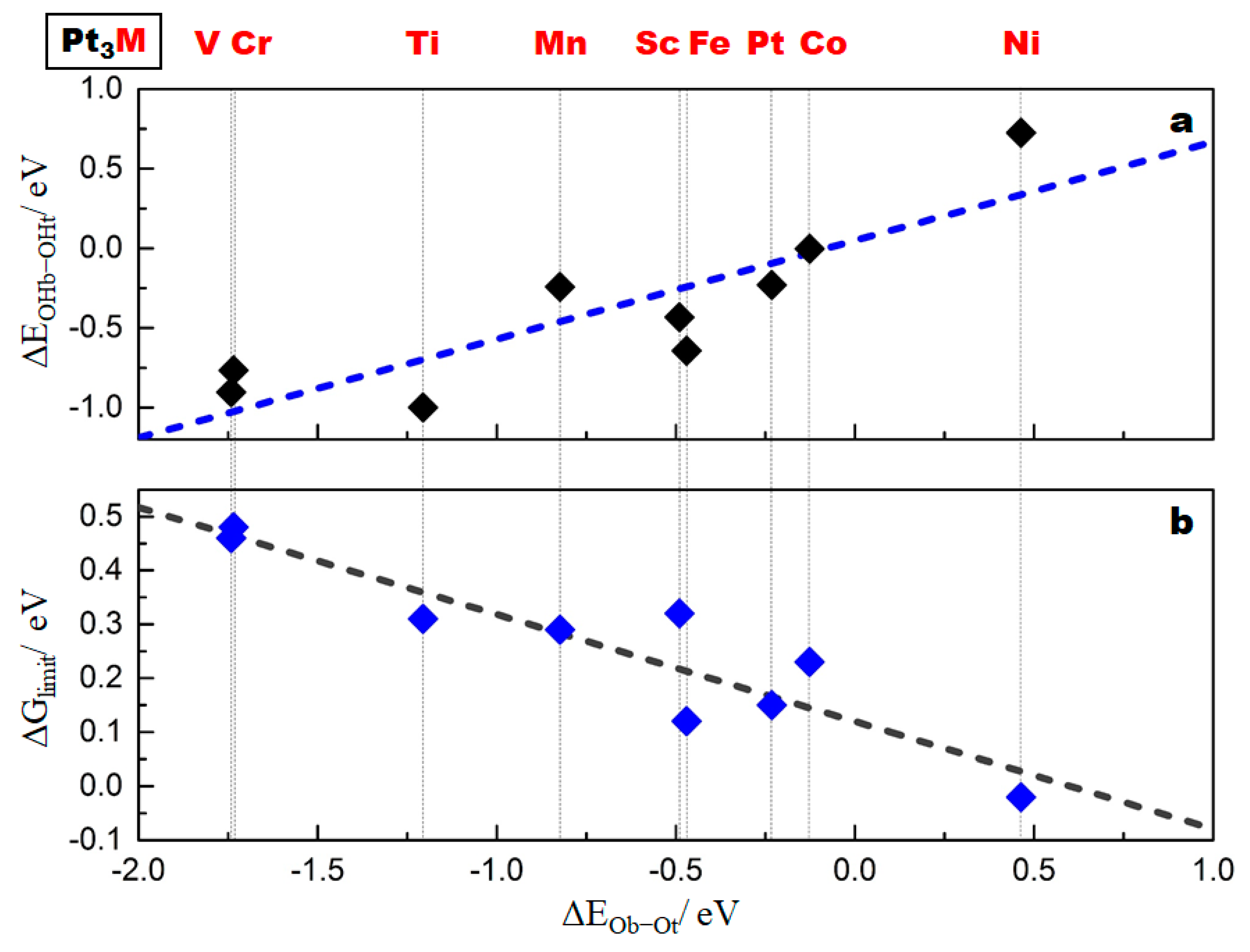
2.3. Correlation of the ORR Activity with the Oxygen Binding Energy Difference
3. Model and Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zagal, J.H.; Koper, M.T.M. Reactivity Descriptors for the Activity of Molecular MN4 Catalysts for the Oxygen Reduction Reaction. Angewandte Chemie Int. Ed. 2016, 55, 14510–14521. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Yu, H.; Wang, X.; Liu, L.; Jiang, Y.; Wang, L.; Zhuang, G.; Chu, Y.; Li, X.; Wang, J.-G. Pt@Au Nanorods Uniformly Decorated on Pyridyne Cycloaddition Graphene as a Highly Effective Electrocatalyst for Oxygen Reduction. ACS Appl. Mater. Interfaces 2014, 6, 13448–13454. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Zhang, Y.; Liu, W.; Sun, X. The chemistry, recent advancements and activity descriptors for macrocycles based electrocatalysts in oxygen reduction reaction. Coord. Chem. Rev. 2020, 402, 213047. [Google Scholar] [CrossRef]
- Sha, Y.; Yu, T.H.; Merinov, B.V.; Goddard, W.A., III. DFT Prediction of Oxygen Reduction Reaction on Palladium-Copper Alloy Surfaces. ACS Catal. 2014, 4, 1189–1197. [Google Scholar] [CrossRef]
- Antolini, E. Effect of Structural Characteristics of Binary Palladium-Cobalt Fuel Cell Catalysts on the Activity for Oxygen Reduction. ChemPlusChem 2014, 79, 765–775. [Google Scholar] [CrossRef]
- Wu, P.; Du, P.; Zhang, H.; Cai, C. Graphyne As a Promising Metal-Free Electrocatalyst for Oxygen Reduction Reactions in Acidic Fuel Cells: A DFT Study. J. Phys. Chem. C 2012, 116, 20472–20479. [Google Scholar] [CrossRef]
- Hyun, K.; Lee, J.H.; Yoon, C.W.; Kwon, Y. The Effect of Platinum Based Bimetallic Electrocatalysts on Oxygen Reduction Reaction of Proton Exchange Membrane Fuel Cells. Int. J. Electrochem. Sci. 2013, 8, 11752–11767. [Google Scholar]
- Zhang, B.-W.; Zhang, Z.-C.; Liao, H.-G.; Gong, Y.; Gu, L.; Qu, X.-M.; You, L.-X.; Liu, S.; Huang, L.; Tian, X.-C.; et al. Tuning Pt-skin to Ni-rich surface of Pt3Ni catalysts supported on porous carbon for enhanced oxygen reduction reaction and formic electro-oxidation. Nano Energy 2016, 19, 198–209. [Google Scholar] [CrossRef]
- Stephens, I.E.L.; Bondarenko, A.S.; Perez-Alonso, F.J.; Calle-Vallejo, F.; Bech, L.; Johansson, T.P.; Jepsen, A.K.; Frydendal, R.; Knudsen, B.P.; Rossmeisl, J.; et al. Tuning the Activity of Pt (111) for Oxygen Electroreduction by Subsurface Alloying. J. Am. Chem. Soc. 2011, 133, 5485–5491. [Google Scholar] [CrossRef]
- Deng, K.; Xu, Y.; Li, Y.; Dai, Z.; Wang, Z.; Li, X.; Wang, H.; Wang, L. Integration mesoporous surface and hollow cavity into PtPdRh nano-octahedra for enhanced oxygen reduction electrocatalysis. Nanotechnology 2020, 31, 025401. [Google Scholar] [CrossRef]
- Zhao, Z.; Chen, C.; Liu, Z.; Huang, J.; Wu, M.; Liu, H.; Li, Y.; Huang, Y. Pt-Based Nanocrystal for Electrocatalytic Oxygen Reduction. Adv. Mater. 2019, 31, 1808115. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Dang, D.; Tian, X. Designing Robust Support for Pt Alloy Nanoframes with Durable Oxygen Reduction Reaction Activity. ACS Appl. Mater. Interfaces 2019, 11, 9117–9124. [Google Scholar] [CrossRef] [PubMed]
- Stamenkovic, V.R.; Mun, B.S.; Arenz, M.; Mayrhofer, K.J.J.; Lucas, C.A.; Wang, G.; Ross, P.N.; Markovic, N.M. Trends in electrocatalysis on extended and nanoscale Pt-bimetallic alloy surfaces. Nat. Mater. 2007, 6, 241–247. [Google Scholar] [CrossRef]
- Cheng, D.; Qiu, X.; Yu, H. Enhancing oxygen reduction reaction activity of Pt-shelled catalysts via subsurface alloying. Phys. Chem. Chem. Phys. 2014, 16, 20377–20381. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Wakabayashi, R.H.; Yang, M.; Abruna, H.D.; DiSalvo, F.J. Synthesis of carbon supported ordered tetragonal pseudo-ternary Pt2M′M′′ (M = Fe, Co, Ni) nanoparticles and their activity for oxygen reduction reaction. J. Power Sources 2015, 280, 459–466. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Tao, H.; Li, Z.; Wang, G. Effect of iron precursor on the activity and stability of PtFe/C catalyst for oxygen reduction reaction. J. Alloys Compd. 2020, 814, 152212. [Google Scholar] [CrossRef]
- Lin, S.-P.; Wang, K.-W.; Liu, C.-W.; Chen, H.-S.; Wang, J.-H. Trends of Oxygen Reduction Reaction on Platinum Alloys: A Computational and Experimental Study. J. Phys. Chem. C 2015, 119, 15224–15231. [Google Scholar] [CrossRef]
- Zhang, L.; Fischer, J.M.T.A.; Jia, Y.; Yan, X.; Xu, W.; Wang, X.; Chen, J.; Yang, D.; Liu, H.; Zhuang, L.; et al. Coordination of Atomic Co-Pt Coupling Species at Carbon Defects as Active Sites for Oxygen Reduction Reaction. J. Am. Chem. Soc. 2018, 140, 10757–10763. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Zhao, X.; Su, Y.-Q.; Wang, L.; Wang, H.; Dang, D.; Chi, B.; Liu, H.; Hensen, E.J.M.; Lou, X.W.; et al. Engineering bunched Pt-Ni alloy nanocages for efficient oxygen reduction in practical fuel cells. Science 2019, 366, 850–856. [Google Scholar] [CrossRef]
- Ou, L. The origin of enhanced electrocatalytic activity of Pt-M (M = Fe, Co, Ni, Cu, and W) alloys in PEM fuel cell cathodes: A DFT computational study. Comput. Theor. Chem. 2014, 1048, 69–76. [Google Scholar] [CrossRef]
- Gatalo, M.; Moriau, L.; Petek, U.; Ruiz-Zepeda, F.; Sala, M.; Grom, M.; Galun, T.; Jovanovic, P.; Pavlisic, A.; Bele, M.; et al. CO-assisted ex-situ chemical activation of Pt-Cu/C oxygen reduction reaction electrocatalyst. Electrochim. Acta 2019, 306, 377–386. [Google Scholar] [CrossRef]
- Zhu, C.; Dong, S. Recent progress in graphene-based nanomaterials as advanced electrocatalysts towards oxygen reduction reaction. Nanoscale 2013, 5, 1753–1767. [Google Scholar] [CrossRef] [PubMed]
- Higgins, D.; Zamani, P.; Yu, A.; Chen, Z. The application of graphene and its composites in oxygen reduction electrocatalysis: A perspective and review of recent progress. Energy Environ. Sci. 2016, 9, 357–390. [Google Scholar] [CrossRef]
- Chen, H.S.; Liang, Y.T.; Chen, T.Y.; Tseng, Y.C.; Liu, C.W.; Chung, S.R.; Hsieh, C.T.; Lee, C.E.; Wang, K.W. Graphene-supported Pt and PtPd nanorods with enhanced electrocatalytic performance for the oxygen reduction reaction. Chem. Commun. 2014, 50, 11165–11168. [Google Scholar] [CrossRef]
- Yan, Z.H.; Wang, M.; Huang, B.X.; Liu, R.M.; Zhao, J.S. Graphene Supported Pt-Co Alloy Nanoparticles as Cathode Catalyst for Microbial Fuel Cells. Int. J. Electrochem. Sci. 2013, 8, 149–158. [Google Scholar]
- Boone, C.V.; Maia, G. Lowering metal loadings onto Pt-Pd-Cu/graphene nanoribbon nanocomposites affects electrode collection efficiency and oxygen reduction reaction performance. Electrochim. Acta 2019, 303, 192–203. [Google Scholar] [CrossRef]
- Fazio, G.; Ferrighi, L.; Di Valentin, C. Boron-doped graphene as active electrocatalyst for oxygen reduction reaction at a fuel-cell cathode. J. Catal. 2014, 318, 203–210. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.Y.; Zhang, L.P.; Xia, Z.H.; Roy, A.; Chang, D.W.; Baek, J.B.; Dai, L.M. BCN Graphene as Efficient Metal-Free Electrocatalyst for the Oxygen Reduction Reaction. Angewandte Chemie Int. Ed. 2012, 51, 4209–4212. [Google Scholar] [CrossRef]
- Zhang, L.P.; Xia, Z.H. Mechanisms of Oxygen Reduction Reaction on Nitrogen-Doped Graphene for Fuel Cells. J. Phys. Chem. C 2011, 115, 11170–11176. [Google Scholar] [CrossRef]
- del Cueto, M.; Ocon, P.; Poyato, J.M.L. Comparative Study of Oxygen Reduction Reaction Mechanism on Nitrogen-, Phosphorus-, and Boron-Doped Graphene Surfaces for Fuel Cell Applications. J. Phys. Chem. C 2015, 119, 2004–2009. [Google Scholar] [CrossRef]
- Shi, X.; Zhang, J.; Huang, T. Significant advantages of sulfur-doped graphene in neutral media as electrocatalyst for oxygen reduction comparing with Pt/C. IOP Conf. Ser. Earth Environ. Sci. 2018, 121, 022011. [Google Scholar] [CrossRef]
- Cui, H.; Zhou, Z.; Jia, D. Heteroatom-Doped Graphene as Electrocatalysts for Air Cathodes. Mater. Horiz. 2017, 4, 7–19. [Google Scholar] [CrossRef]
- Xu, J.; Wang, Y.; Hu, S. Nanocomposites of graphene and graphene oxides: Synthesis, molecular functionalization and application in electrochemical sensors and biosensors. A review. Microchim. Acta 2017, 184, 1–44. [Google Scholar] [CrossRef]
- Su, C.; Loh, K.P. Carbocatalysts: Graphene Oxide and Its Derivatives. Acc. Chem. Res. 2013, 46, 2275–2285. [Google Scholar] [CrossRef]
- Tang, Y.; Chen, T.; Guo, W. Surfactant-Free Synthesis of Reduced Graphene Oxide Supported Well-Defined Polyhedral Pd-Pt Nanocrystals for Oxygen Reduction Reaction. Catalysts 2019, 9, 756. [Google Scholar] [CrossRef] [Green Version]
- Jin, N.; Han, J.; Wang, H.; Zhu, X.; Ge, Q. A DFT study of oxygen reduction reaction mechanism over O-doped graphene-supported Pt-4, Pt3Fe and Pt3V alloy catalysts. Int. J. Hydrogen Energy 2015, 40, 5126–5134. [Google Scholar] [CrossRef]
- Stamenkovic, V.; Mun, B.S.; Mayrhofer, K.J.J.; Ross, P.N.; Markovic, N.M.; Rossmeisl, J.; Greeley, J.; Norskov, J.K. Changing the activity of electrocatalysts for oxygen reduction by tuning the surface electronic structure. Angewandte Chemie Int. Ed. 2006, 45, 2897–2901. [Google Scholar] [CrossRef]
- Xu, L.; Ge, Q. Effect of defects and dopants in graphene on hydrogen interaction in graphene-supported NaAlH4. Int. J. Hydrogen Energy 2013, 38, 3670–3680. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Z.; Zhu, Z.P. A Density Functional Theory Study on Mechanism of Electrochemical Oxygen Reduction on FeN3-Graphene. J. Electrochem. Soc. 2015, 162, F1262–F1267. [Google Scholar] [CrossRef]
- Qaseem, A.; Chen, F.Y.; Wu, X.Q.; Johnston, R.L. Pt-free silver nanoalloy electrocatalysts for oxygen reduction reaction in alkaline media. Catal. Sci. Technol. 2016, 6, 3317–3340. [Google Scholar] [CrossRef] [Green Version]
- Roudgar, A.; Eikerling, M.; van Santen, R. Ab initio study of oxygen reduction mechanism at Pt-4 cluster. Phys. Chem. Chem. Phys. 2010, 12, 614–620. [Google Scholar] [CrossRef]
- Fu, C.; Liu, C.; Li, T.; Zhang, X.; Wang, F.; Yang, J.; Jiang, Y.; Cui, P.; Li, H. DFT calculations: A powerful tool for better understanding of electrocatalytic oxygen reduction reactions on Pt-based metallic catalysts. Comput. Mater. Sci. 2019, 170, 109202. [Google Scholar] [CrossRef]
- Lim, D.-H.; Wilcox, J. Mechanisms of the oxygen reduction reaction on defective graphene-supported Pt nanoparticles from first-principles. J. Phys. Chem. C 2012, 116, 3653–3660. [Google Scholar] [CrossRef] [Green Version]
- Greeley, J.; Stephens, I.E.L.; Bondarenko, A.S.; Johansson, T.P.; Hansen, H.A.; Jaramillo, T.F.; Rossmeisl, J.; Chorkendorff, I.; Norskov, J.K. Alloys of platinum and early transition metals as oxygen reduction electrocatalysts. Nat. Chem. 2009, 1, 552–556. [Google Scholar] [CrossRef]
- Liu, J.; Liu, H.; Chen, H.; Du, X.; Zhang, B.; Hong, Z.; Sun, S.; Wang, W. Progress and Challenges Toward the Rational Design of Oxygen Electrocatalysts Based on a Descriptor Approach. Adv. Sci. 2019. [Google Scholar] [CrossRef]
- Pasti, I.A.; Gavrilov, N.M.; Mentus, S.V. DFT study of chlorine adsorption on bimetallic surfaces—Case study of Pd3M and Pt3M alloy surfaces. Electrochim. Acta 2014, 130, 453–463. [Google Scholar] [CrossRef]
- Hwang, S.J.; Kim, S.K.; Lee, J.G.; Lee, S.C.; Jang, J.H.; Kim, P.; Lim, T.H.; Sung, Y.E.; Yoo, S.J. Role of Electronic Perturbation in Stability and Activity of Pt-Based Alloy Nanocatalysts for Oxygen Reduction. J. Am. Chem. Soc. 2012, 134, 19508–19511. [Google Scholar] [CrossRef]
- Karlberg, G.S.; Rossmeisl, J.; Norskov, J.K. Estimations of electric field effects on the oxygen reduction reaction based on the density functional theory. Phys. Chem. Chem. Phys. 2007, 9, 5158–5161. [Google Scholar] [CrossRef] [Green Version]
- Tao, H.; Liu, S.; Luo, J.-L.; Choi, P.; Liu, Q.; Xu, Z. Descriptor of catalytic activity of metal sulfides for oxygen reduction reaction: A potential indicator for mineral flotation. J. Mater. Chem. A 2018, 6, 9650–9656. [Google Scholar] [CrossRef]
- Xin, H.; Vojvodic, A.; Voss, J.; Norskov, J.K.; Abild-Pedersen, F. Effects of d-band shape on the surface reactivity of transition-metal alloys. Phys. Rev. B 2014, 89, 115114. [Google Scholar] [CrossRef]
- Xie, Y.; Wang, Z.-W.; Zhu, T.-Y.; Shu, D.-J.; Hou, Z.-F.; Terakura, K. Breaking the scaling relations for oxygen reduction reaction on nitrogen-doped graphene by tensile strain. Carbon 2018, 139, 129–136. [Google Scholar] [CrossRef]
- Yu, T.H.; Hofmann, T.; Sha, Y.; Merinov, B.V.; Myers, D.J.; Heske, C.; Goddard, W.A. Finding Correlations of the Oxygen Reduction Reaction Activity of Transition Metal Catalysts with Parameters Obtained from Quantum Mechanics. J. Phys. Chem. C 2013, 117, 26598–26607. [Google Scholar] [CrossRef] [Green Version]
- Norskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jonsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Hansen, H.A.; Viswanathan, V.; Norskov, J.K. Unifying Kinetic and Thermodynamic Analysis of 2 e(−) and 4 e(−) Reduction of Oxygen on Metal Surfaces. J. Phys. Chem. C 2014, 118, 6706–6718. [Google Scholar] [CrossRef]
- Seitz, L.C.; Dickens, C.F.; Nishio, K.; Hikita, Y.; Montoya, J.; Doyle, A.; Kirk, C.; Vojvodic, A.; Hwang, H.Y.; Norskov, J.K.; et al. A highly active and stable IrOx/SrIrO3 catalyst for the oxygen evolution reaction. Science 2016, 353, 1011–1014. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Blöchl, P.E.; Jepsen, O.; Andersen, O.K. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B 1994, 49, 16223. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Rossmeisl, J.; Karlberg, G.S.; Jaramillo, T.; Norskov, J.K. Steady state oxygen reduction and cyclic voltammetry. Faraday Discuss. 2008, 140, 337–346. [Google Scholar] [CrossRef]
- Tripković, V.; Skúlason, E.; Siahrostami, S.; Nørskov, J.K.; Rossmeisl, J. The oxygen reduction reaction mechanism on Pt (111) from density functional theory calculations. Electrochim. Acta 2010, 55, 7975–7981. [Google Scholar] [CrossRef]
- Johnson, R.D., III. Computational Chemistry Comparison and Benchmark Database; NIST Standard Reference Database Number 101; U.S. Secretary of Commerce; NIST: Gaithersburg, MD, USA, 2010; Volume 219, p. 611. [Google Scholar]
- Henkelman, G.; Arnaldsson, A.; Jonsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Bader, R.F. Atoms in Molecules: A Quantum Theory; Oxford University Press: New York, NY, USA, 1990. [Google Scholar]
- Seifitokaldani, A.; Savadogo, O.; Perrier, M. Density Functional Theory (DFT) Computation of the Oxygen Reduction Reaction (ORR) on Titanium Nitride (TiN) Surface. Electrochim. Acta 2014, 141, 25–32. [Google Scholar] [CrossRef]
- Skulason, E.; Tripkovic, V.; Bjorketun, M.E.; Gudmundsdottir, S.; Karlberg, G.; Rossmeisl, J.; Bligaard, T.; Jonsson, H.; Norskov, J.K. Modeling the Electrochemical Hydrogen Oxidation and Evolution Reactions on the Basis of Density Functional Theory Calculations. J. Phys. Chem. C 2010, 114, 18182–18197. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster | Pt4 | Pt3Sc | Pt3Ti | Pt3V | Pt3Cr | Pt3Mn | Pt3Fe | Pt3Co | Pt3Ni | |
|---|---|---|---|---|---|---|---|---|---|---|
| Bader Charge | C1 | +0.72 | +0.78 | +0.93 | +0.70 | +0.71 | +0.68 | +0.73 | +0.62 | +0.75 |
| C2 | +0.65 | +0.73 | +0.32 | +0.67 | +0.57 | +0.58 | +0.60 | −0.20 | −0.22 | |
| O | −1.61 | −1.63 | −1.42 | −1.65 | −1.64 | −1.64 | −1.65 | −1.64 | −1.61 | |
| Pt1 | −0.16 | −0.59 | −0.84 | −0.61 | −0.51 | −0.49 | −0.44 | −0.39 | −0.35 | |
| Pt2 | +0.25 | −0.1 | +0.04 | −0.13 | −0.02 | −0.03 | +0.01 | +0.06 | +0.09 | |
| Pt3 | +0.19 | −0.11 | −0.52 | −0.09 | −0.07 | −0.04 | +0.01 | +0.08 | +0.11 | |
| M(or Pt0) | −0.12 | +1.56 | +2.13 | +1.70 | +1.28 | +1.12 | +0.97 | +0.71 | +0.57 | |
| d(O-OG)/Å | 0.17 | 0.51 | 0.60 | 0.57 | 0.42 | 0.38 | 0.40 | 0.20 | 0.18 | |
 | ||||||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, C.; Sun, M.; Zhu, X.; Han, J.; Wang, H.; Ge, Q. Oxygen Reduction Reaction Catalyzed by Pt3M (M = 3d Transition Metals) Supported on O-doped Graphene. Catalysts 2020, 10, 156. https://doi.org/10.3390/catal10020156
Cui C, Sun M, Zhu X, Han J, Wang H, Ge Q. Oxygen Reduction Reaction Catalyzed by Pt3M (M = 3d Transition Metals) Supported on O-doped Graphene. Catalysts. 2020; 10(2):156. https://doi.org/10.3390/catal10020156
Chicago/Turabian StyleCui, Chaonan, Mengnan Sun, Xinli Zhu, Jinyu Han, Hua Wang, and Qingfeng Ge. 2020. "Oxygen Reduction Reaction Catalyzed by Pt3M (M = 3d Transition Metals) Supported on O-doped Graphene" Catalysts 10, no. 2: 156. https://doi.org/10.3390/catal10020156