Understanding Hydrodechlorination of Chloromethanes. Past and Future of the Technology

 ,
,  ,
,
Abstract
:
1. Introduction
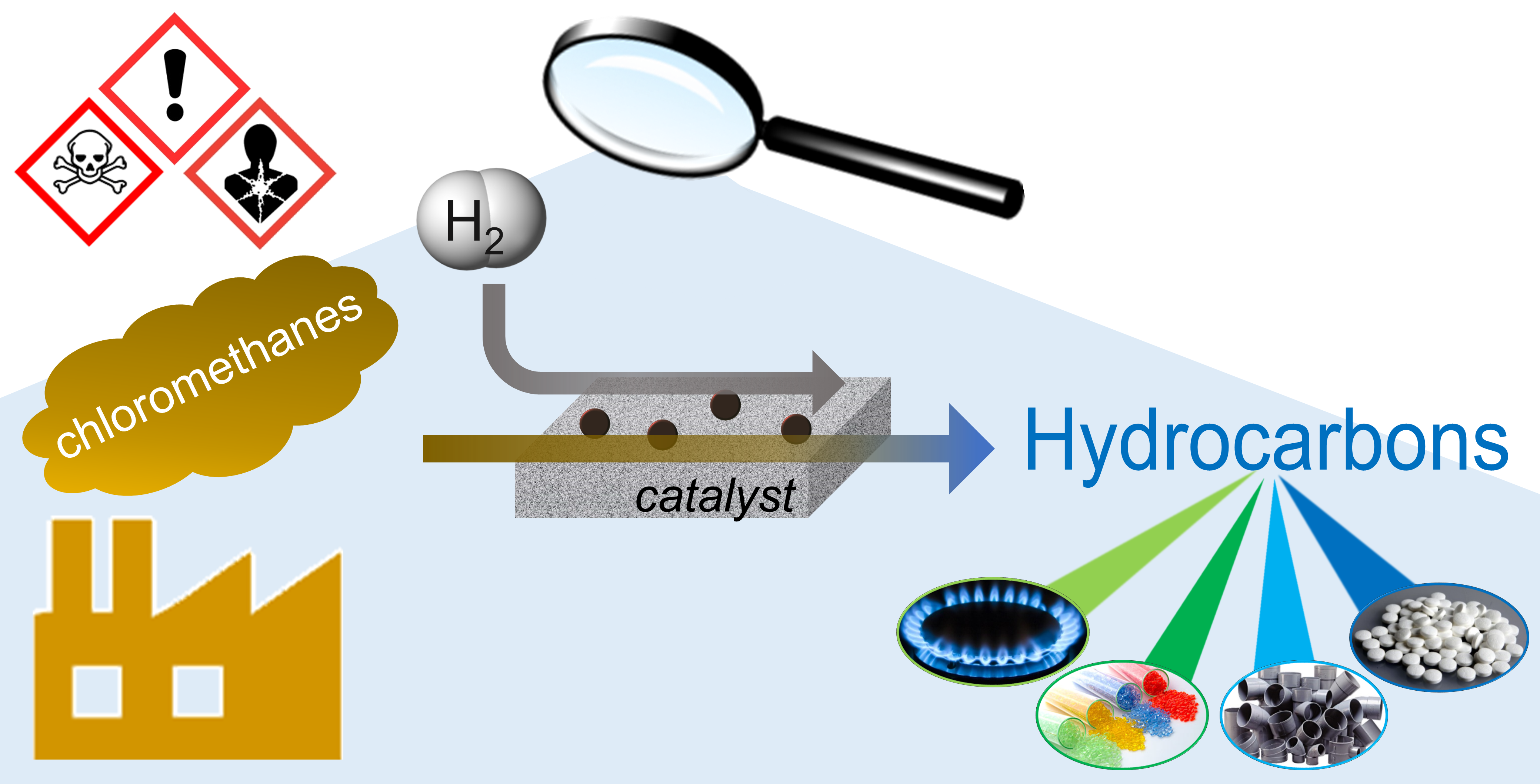
2. Chloromethanes Treated by Hydrodechlorination
3. Hydrodechlorination Catalysts
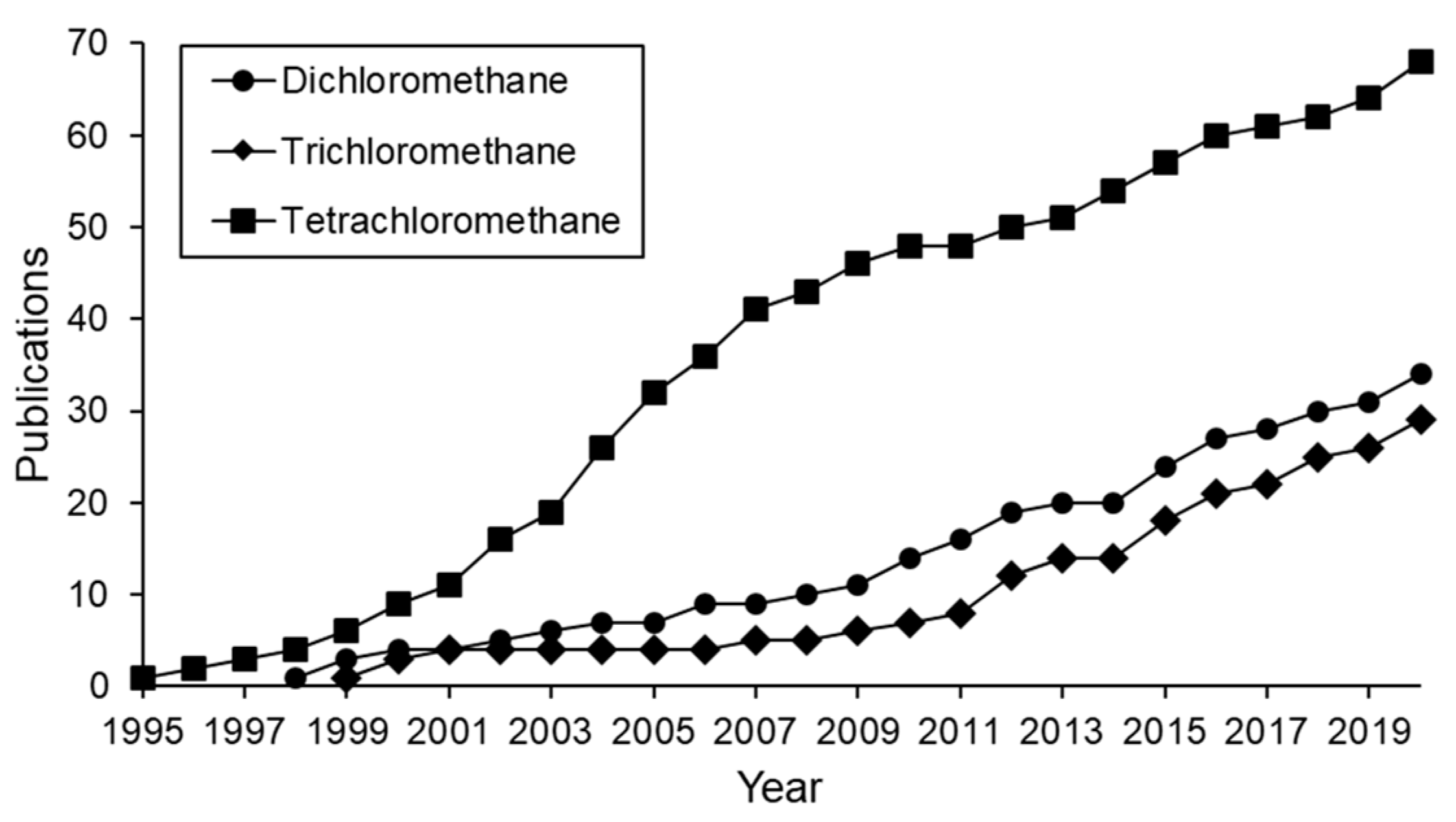
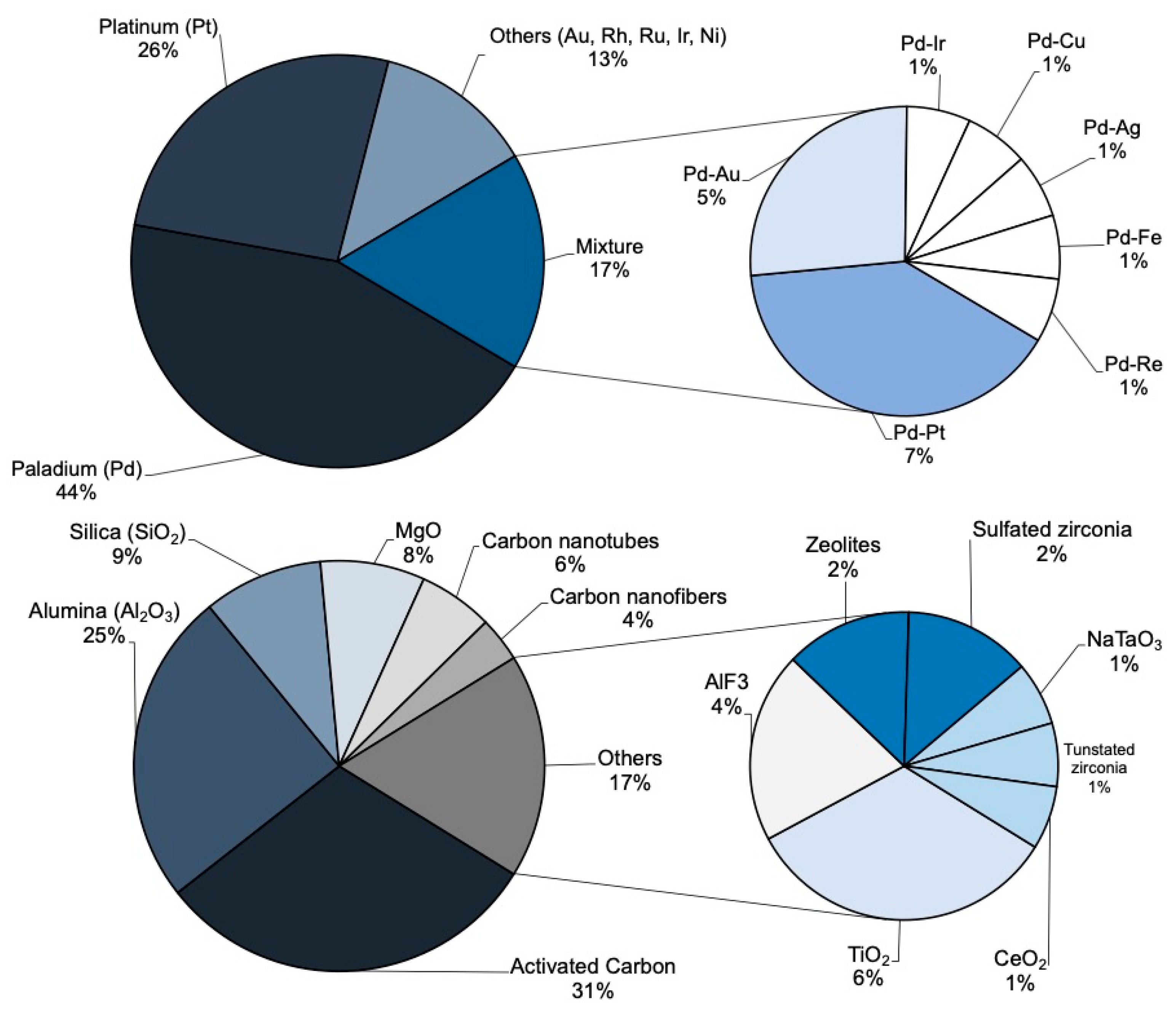
3.1. Synthesis of Catalysts for Hydrodechlorination. Active Phase and Support
3.2. Catalyst Properties and Their Effect on the Catalytic Activity
3.2.1. Metallic Particle Size and Dispersion
3.2.2. Oxidation State of the Active Centers
3.2.3. Surface Acidity
3.2.4. Structure of the Support
3.2.5. Functional Groups and Doping
4. Reaction Systems and Operation Conditions
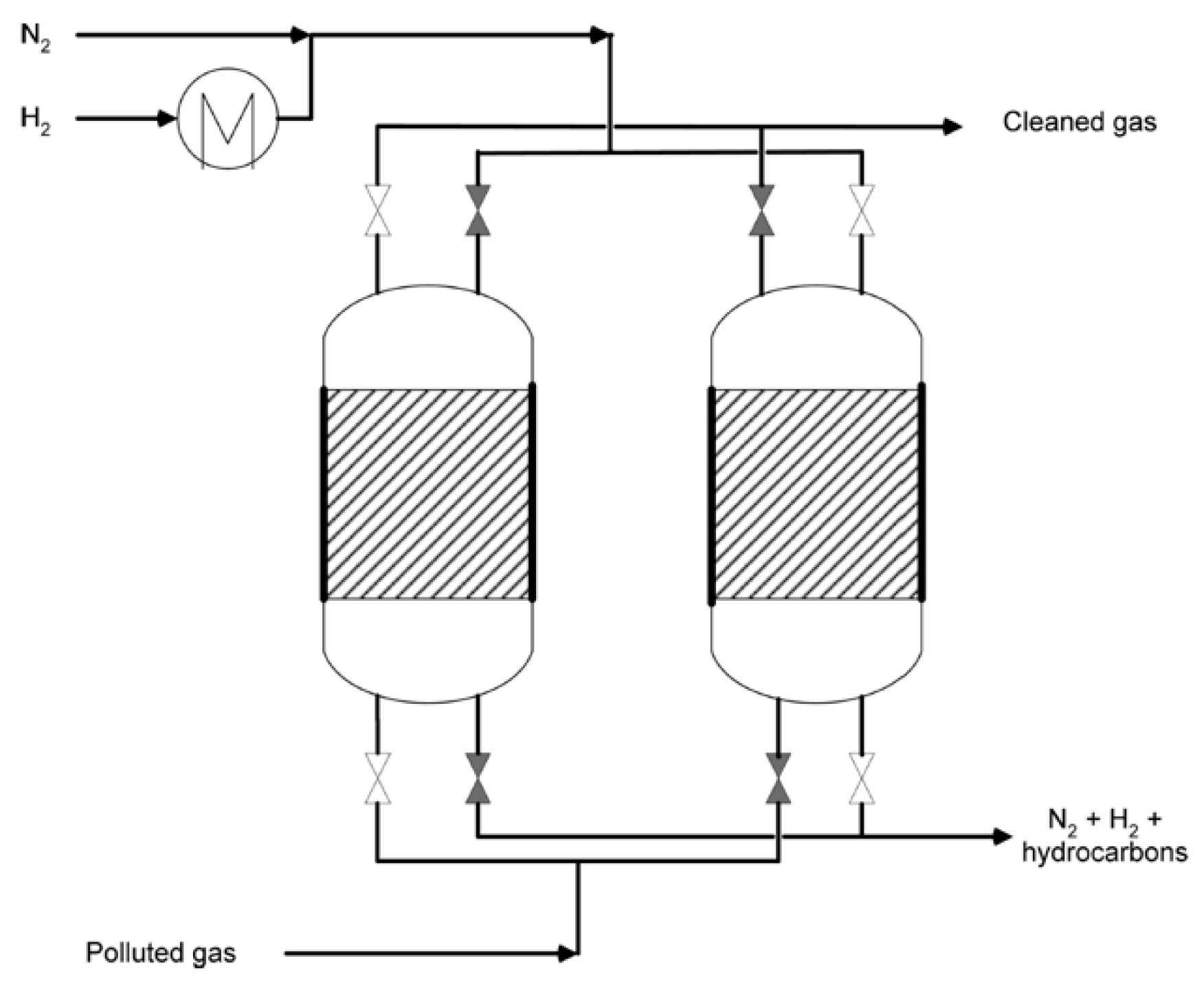
4.1. Reaction Systems
4.2. Effects of Operating Conditions
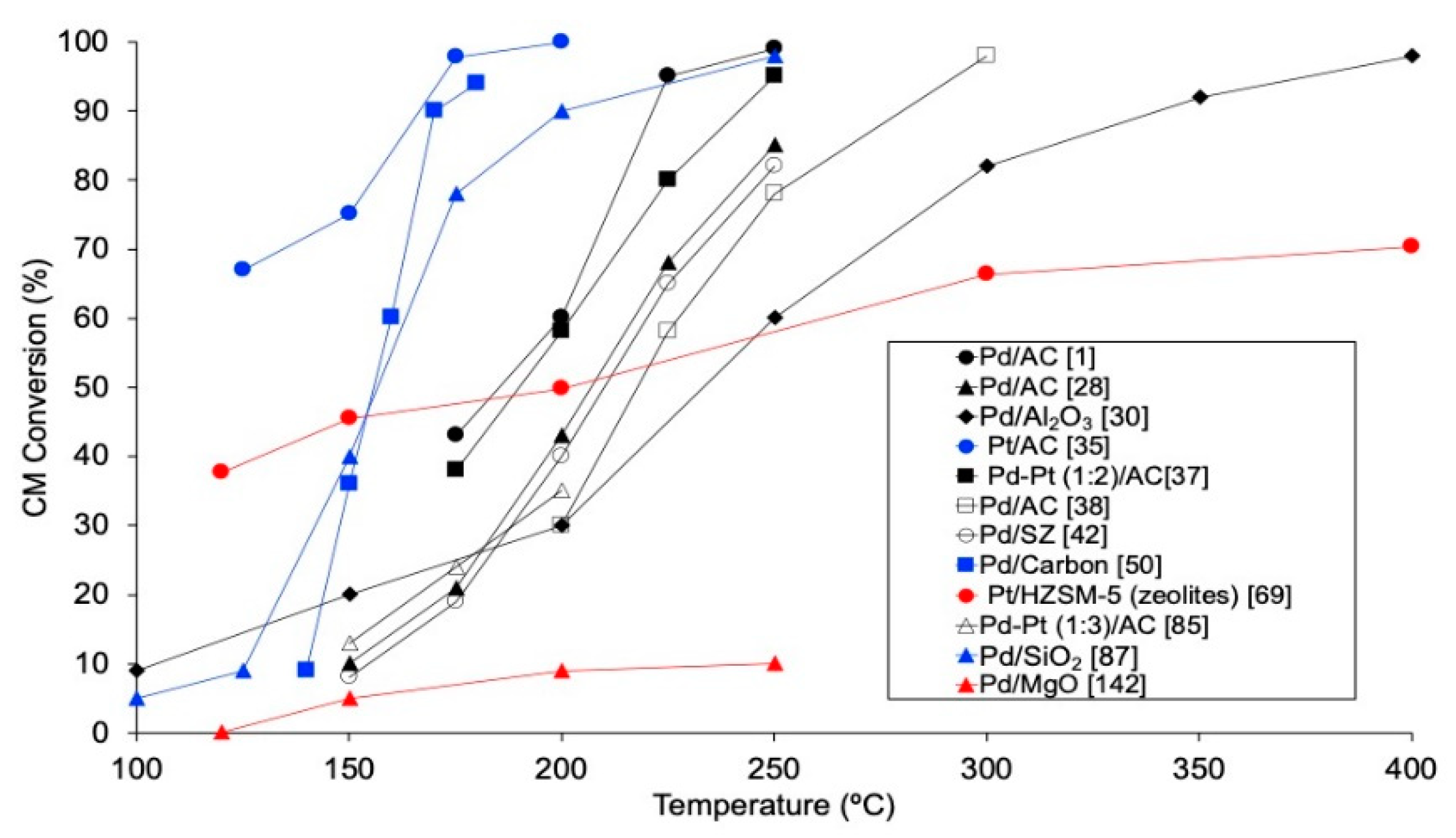
4.2.1. Reaction Temperature
4.2.2. Pressure
4.2.3. Space-Time
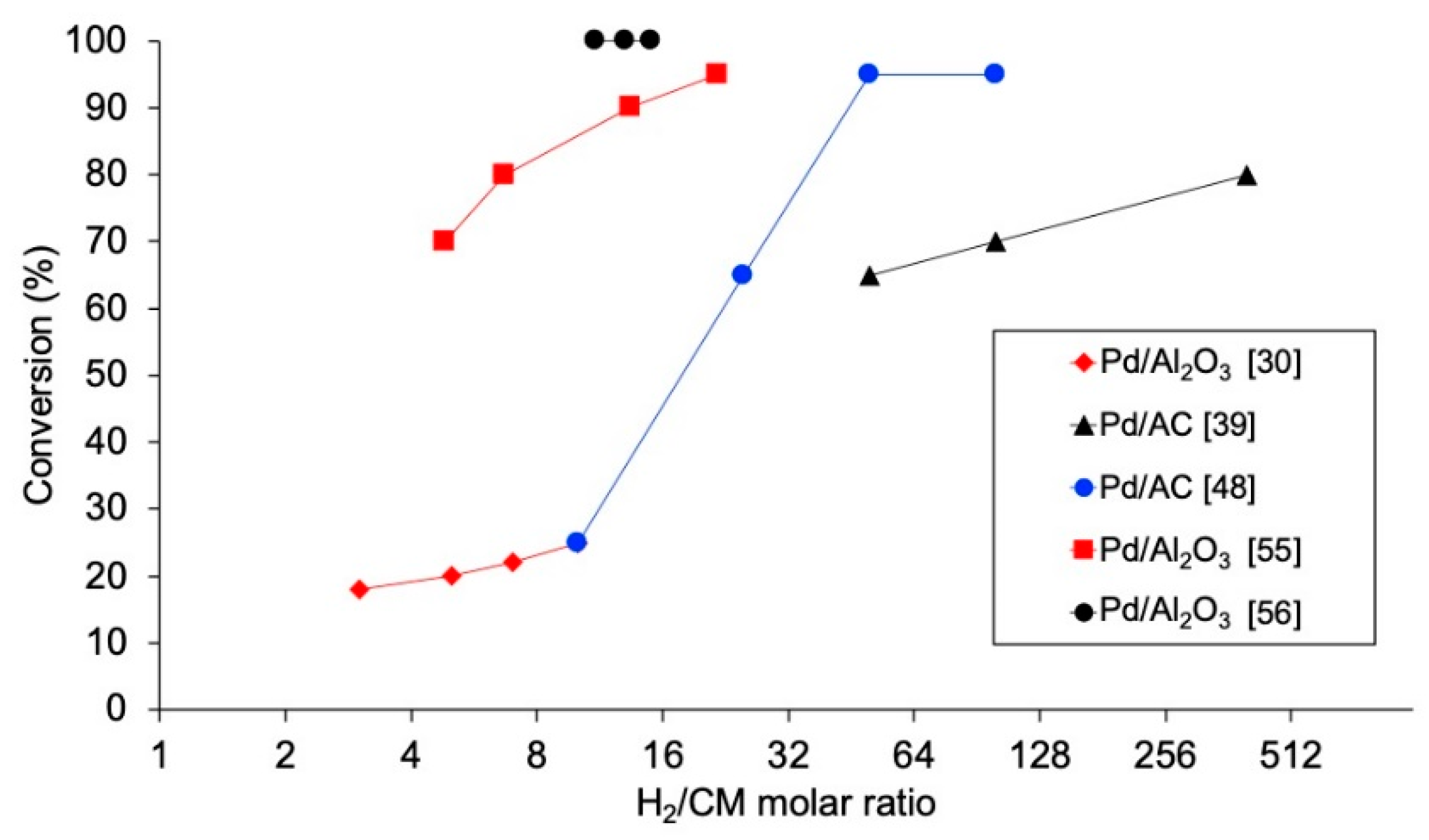
4.2.4. Ratio H2/Chloromethane
5. Mechanism and Kinetics
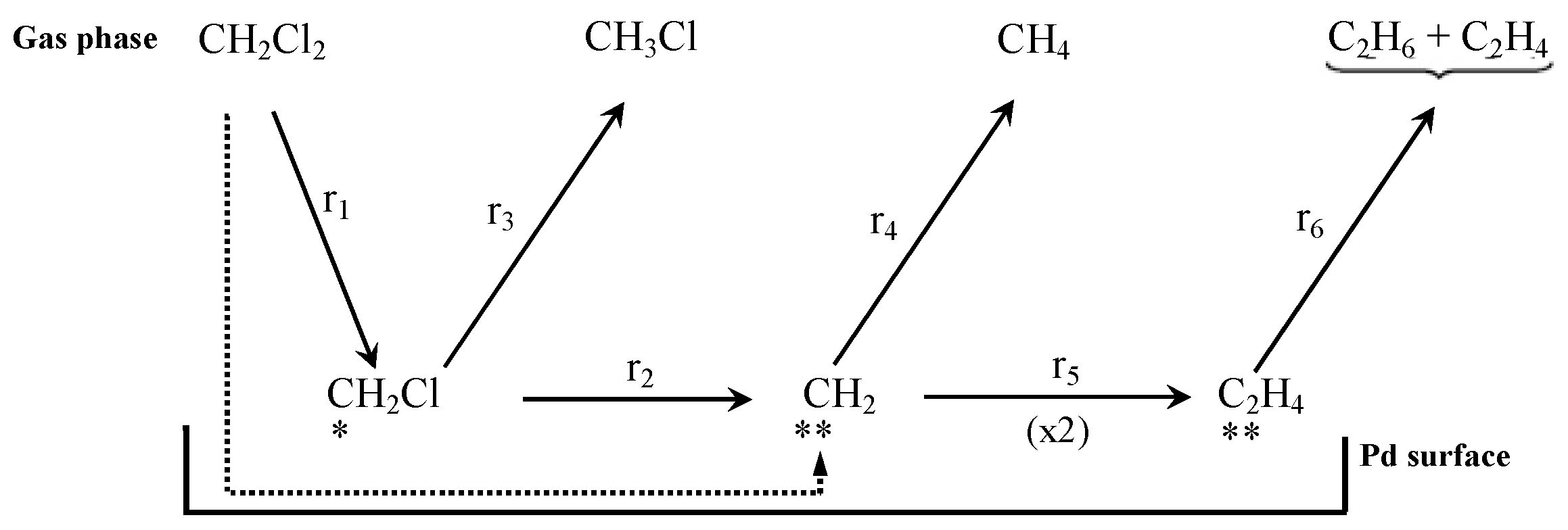
5.1. Mechanism Studies
5.2. Kinetics Studies
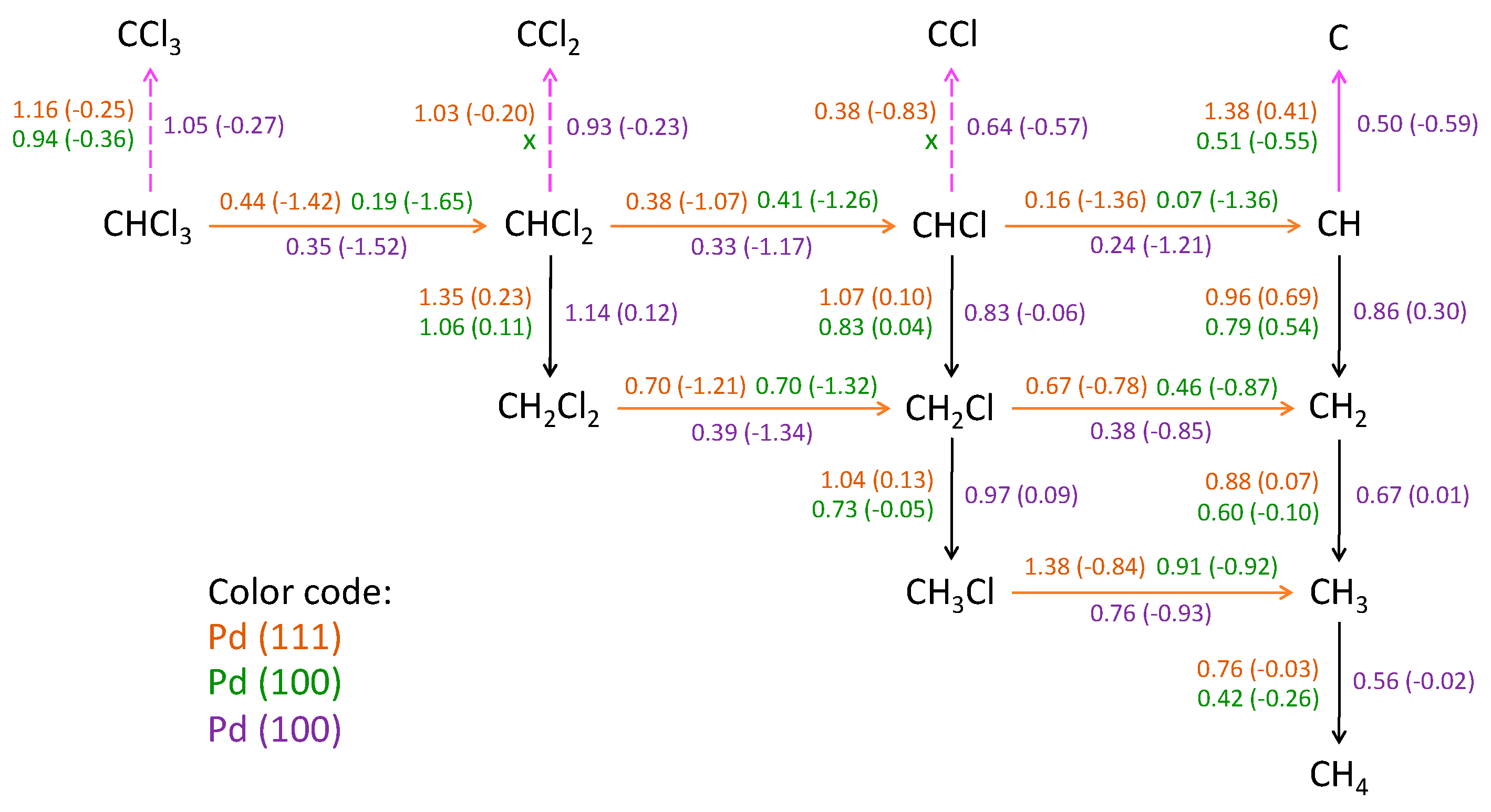
5.3. Activation Energy for the HDC of Chloromethanes
6. Catalysts Stability and Deactivation
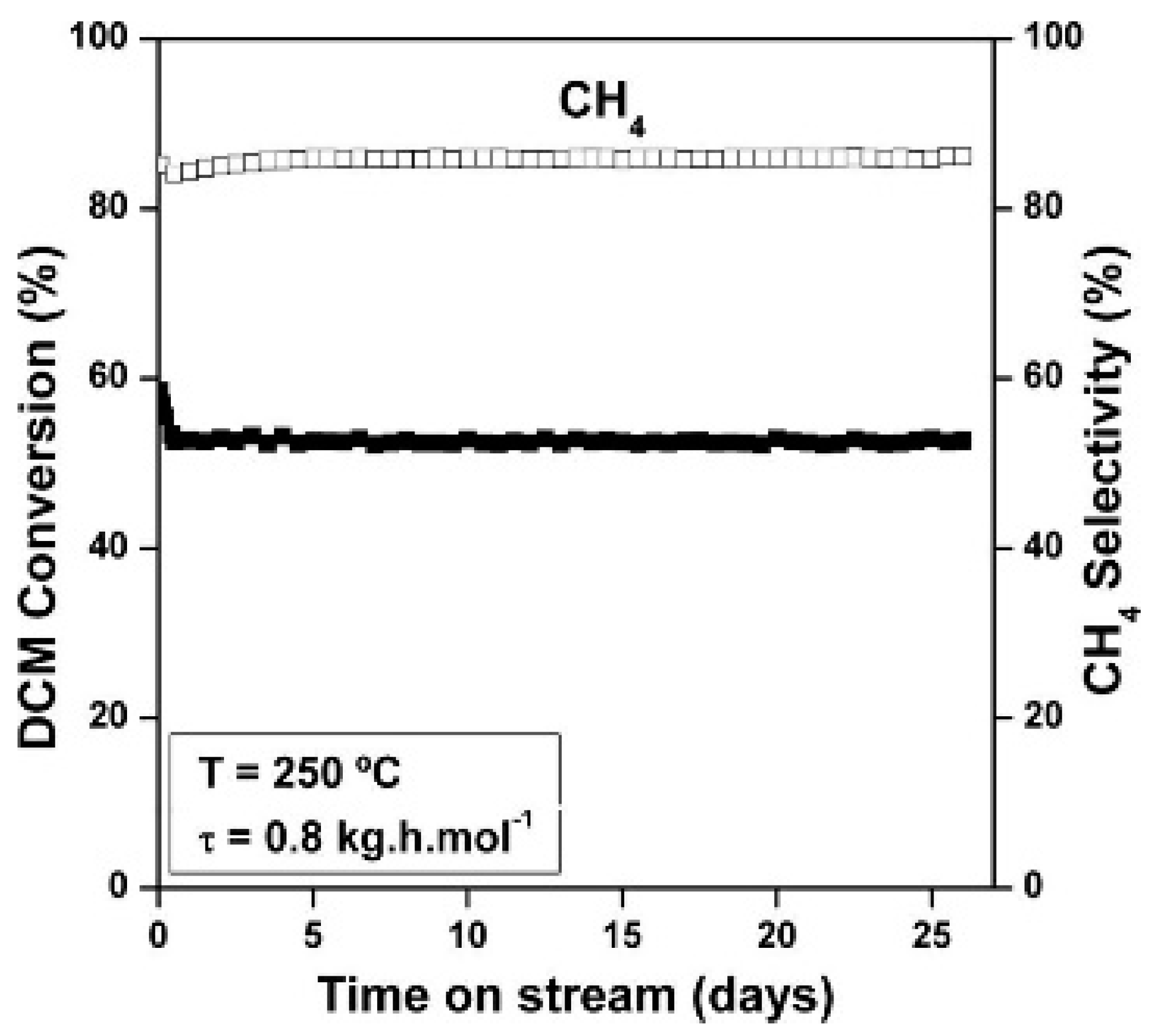
6.1. Stable Catalysts in the HDC of Chloromethanes
6.2. Catalysts Deactivation in the HDC of Chloromethanes
6.2.1. Poisoning of the Catalysts
6.2.2. Sintering of Active Centers
6.2.3. Formation of Carbonaceous Deposits
6.3. Regeneration of Catalysts
7. Summary and Outlook
7.1. Improving Catalytic Stability
7.2. Upgrading of Chloromethanes
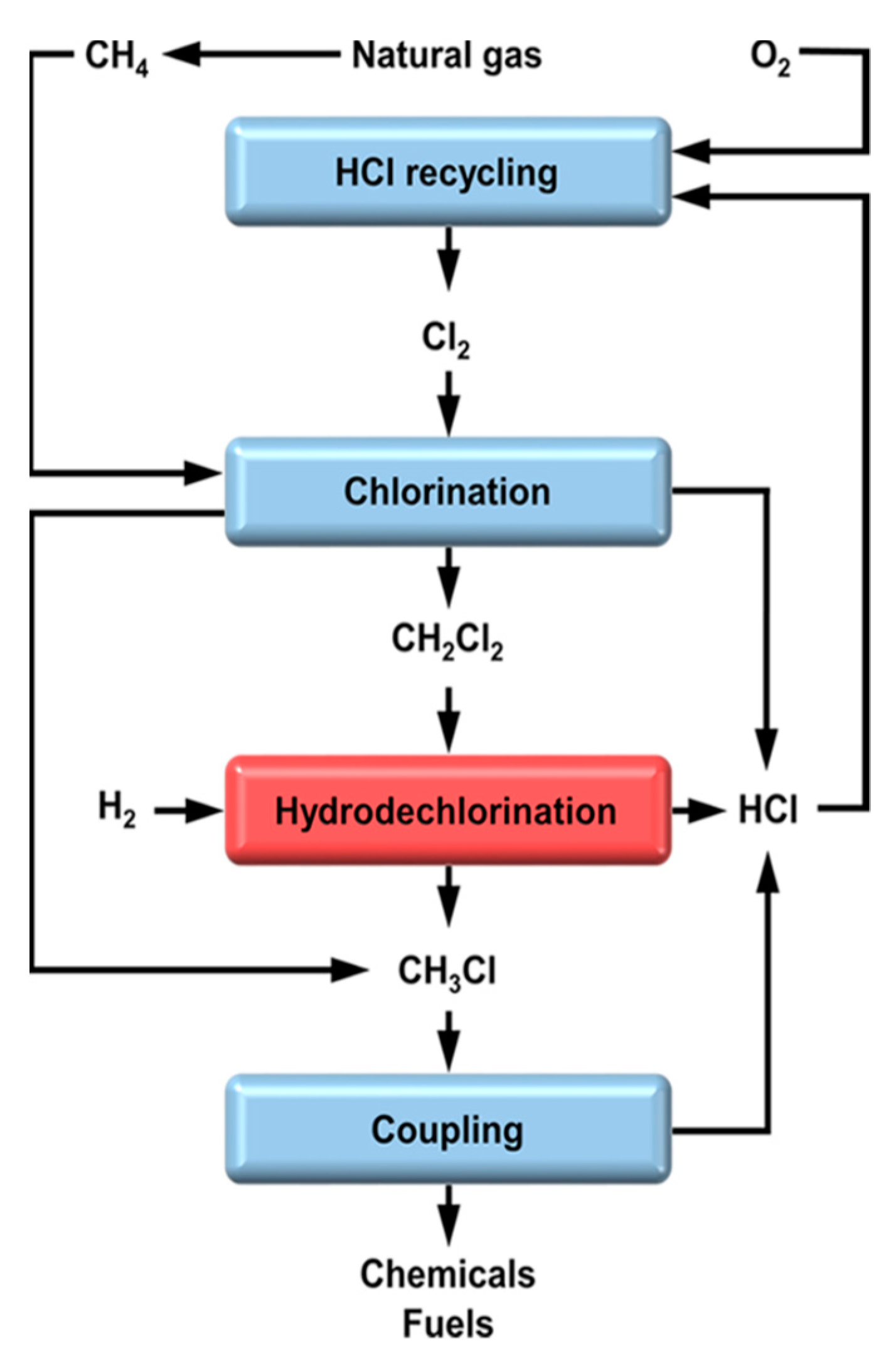
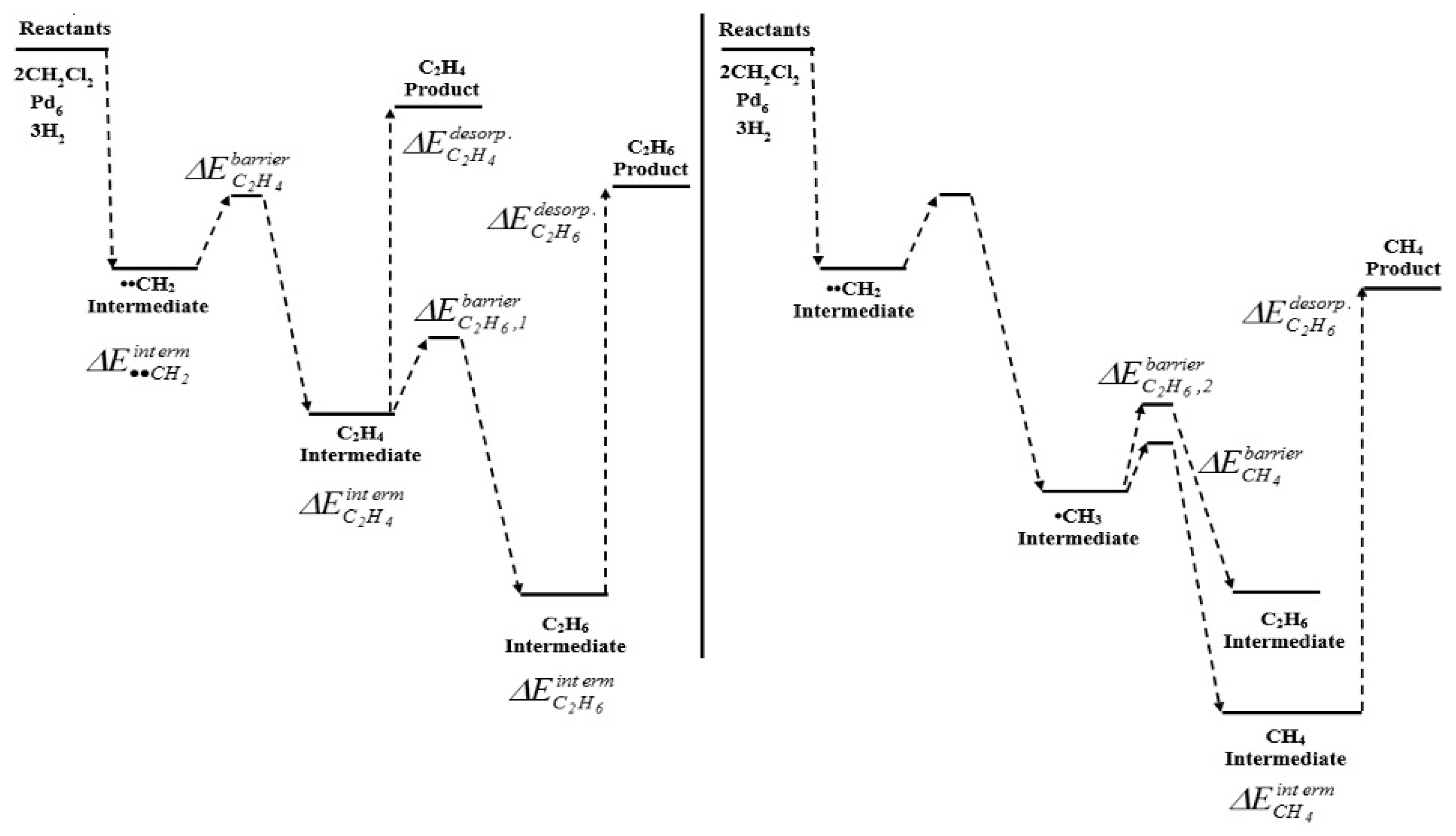
7.2.1. Recycling of Chloromethanes into CH4 Using HDC
7.2.2. An Alternative Route for the Production of Olefins from Waste Gas Streams
Transforming DCM into Olefins:
Transforming TCM into Olefins:
7.3. Application of Gas-Phase HDC in Industry
Author Contributions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
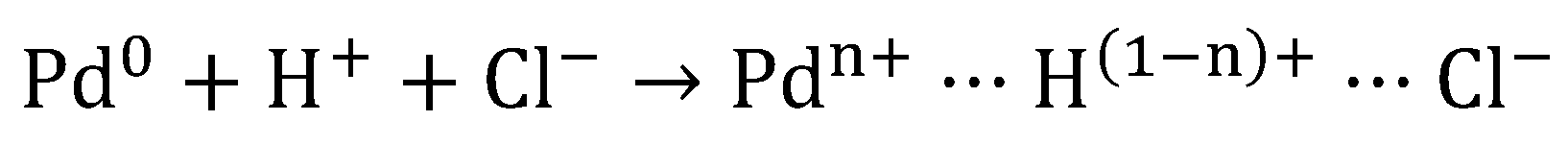{kind=link}
{kind=link}
{kind=link}
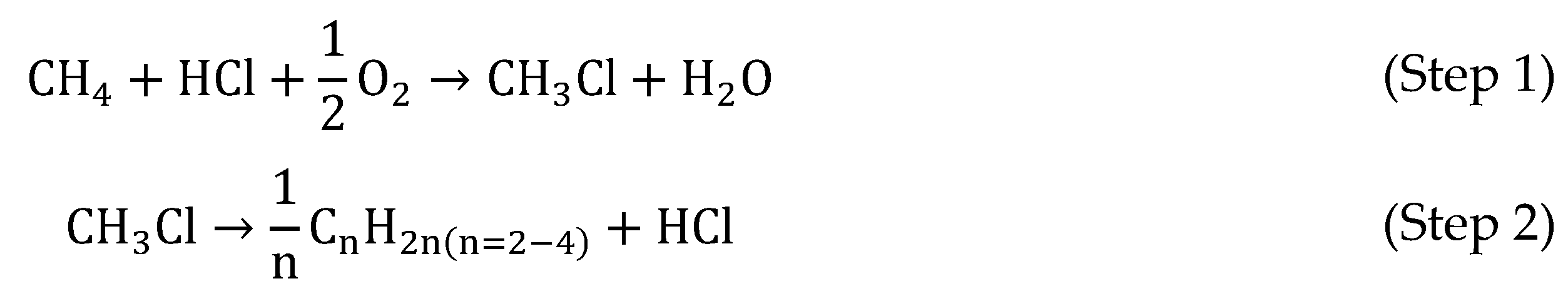{kind=link}
| Catalyst | CM | Operation Conditions | Highest Conversion (%) | Highest Selectivity (%) | Ref. | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T (°C) | P (bar) | τ (*) (kg·h·mol−1) | H2/CM (**) | CH4 | C2 | C3 | C4 | MCM | DCM | TCM | ||||
| Influence of Reaction Temperature | ||||||||||||||
| Pd-Pt(1:1.8)/AC | DCM | 150→250 | 1 | 0.6 | 100 | ca. 20→95 | ca. 83→98 | ca. 17→2 | - | - | [37] | |||
| Pd-Pt(4:1)/AC | DCM | 150→200 | 1 | 0.6 | 100 | ca. 20→53 | 83.2→ 86.7 | 2.4→5 | - | - | 14.4→8.4 | - | - | [85] |
| Pd-Pt(1:1)/AC | DCM | 150→200 | 1 | 0.6 | 100 | ca. 18→56 | 88.2→ 92.2 | 0→0.6 | - | - | 11.8→7.2 | - | - | [85] |
| Pd-Pt(1:3)/AC | DCM | 150→200 | 1 | 0.6 | 100 | ca. 12→50 | 85.8→ 90.2 | - | - | - | 14.2→9.8 | - | - | [85] |
| Pd(1wt.%)/AC | DCM | 150→200 | 1 | 0.6 | 100 | ca. 13→45 | 80.7→ 80.2 | 1.6→8 | - | - | 17.7→11.9 | - | - | [85] |
| Pt(1wt.%)/AC | DCM | 150→200 | 1 | 0.6 | 100 | ca. 9→38 | 79.2→ 83.2 | - | - | - | 20.8→16.8 | - | - | [85] |
| Pd(1wt.%)/AC | MCM | 175→250 | 1 | 1.73 | 100 | ca. 13→35 | n/a | [1] | ||||||
| Pd(1wt.%)/AC | DCM | 175→250 | 1 | 1.73 | 100 | ca. 45→95 | ca. 78→ 68 | ca. 6→22 | ca. 0→2 | - | ca. 16→8 | - | - | [1] |
| Pd(1wt.%)/AC | TCM | 175→250 | 1 | 1.73 | 100 | 100 | ca. 41 | ca. 36 | ca. 14 | ca. 5 | ca. 1 | ca. 3 | - | [1] |
| Pt(1wt.%)/AC | MCM | 175→250 | 1 | 1.73 | 100 | ca. 5→13 | n/a | [1] | ||||||
| Pt(1wt.%)/AC | DCM | 175→250 | 1 | 1.73 | 100 | ca. 35→87 | ca. 80→87 | - | - | - | ca. 20→13 | - | - | [1] |
| Pt(1wt.%)/AC | TCM | 175→250 | 1 | 1.73 | 100 | 100 | ca. 82→92 | - | - | - | ca. 2→1 | ca. 16→7 | - | [1] |
| Rh(1wt.%)/AC | MCM | 175→250 | 1 | 1.73 | 100 | ca. 4→21 | n/a | [1] | ||||||
| Rh(1wt.%)/AC | DCM | 175→250 | 1 | 1.73 | 100 | ca. 38→100 | ca. 76→66 | ca. 10→20 | ca. 2→7 | - | ca. 12→7 | - | - | [1] |
| Rh(1wt.%)/AC | TCM | 175→250 | 1 | 1.73 | 100 | 100 | ca. 25→50 | ca. 15→18 | ca. 15→12 | ca. 0→ 1 | ca. 10→15 | ca. 35→4 | - | [1] |
| Ru(1wt.%)/AC | MCM | 175→250 | 1 | 1.73 | 100 | ca. 1→7 | n/a | [1] | ||||||
| Ru(1wt.%)/AC | DCM | 175→250 | 1 | 1.73 | 100 | ca. 21→87 | ca. 80→56 | ca. 8→17 | ca. 5→14 | - | ca. 7→13 | - | - | [1] |
| Ru(1wt.%)/AC | TCM | 175→250 | 1 | 1.73 | 100 | ca. 80→100 | ca. 8→25 | ca. 1→12 | ca. 10→24 | ca. 0 | ca. 1→10 | ca. 80→29 | - | [1] |
| Pt(0.5wt.%)/WZ | DCM | 150→250 | 1 | 0.8 | 100 | ca. 8→80 | ca. 63→61 | ca. 0→11 | ca. 0→5 | ca. 18→1 | ca. 19→22 | - | - | [42] |
| Pt-Pd(1:1)/WZ | DCM | 150→250 | 1 | 0.8 | 100 | ca. 30→98 | ca. 65→66 | ca. 0→10 | ca. 0→4 | ca. 0→2 | ca. 35→18 | - | - | [42] |
| Pd(0.5wt.%)/WZ | DCM | 150→250 | 1 | 0.8 | 100 | ca. 30→90 | ca. 55→62 | ca. 0→10 | ca. 0→5 | ca. 3→1 | ca. 42→22 | - | - | [42] |
| Pt(0.5wt.%)/SZ | DCM | 150→250 | 1 | 0.8 | 100 | ca. 5→90 | ca. 66→67 | ca. 0→8 | ca. 0→3 | ca. 12→1 | ca. 22→21 | - | - | [42] |
| Pt-Pd(1:1)/SZ | DCM | 150→250 | 1 | 0.8 | 100 | ca. 10→80 | ca. 68→88 | ca. 0→1 | ca. 0 | ca. 5→0 | ca. 27→12 | - | - | [42] |
| Pd(0.5wt.%)/SZ | DCM | 150→250 | 1 | 0.8 | 100 | ca. 8→82 | ca. 65→80 | ca. 0→8 | ca. 0→2 | ca. 12→0 | ca. 23→10 | - | - | [42] |
| Pd(3wt.%)/HY | TTCM | 120→400 | 1 | 10 mg * | 40 ** cm3H2/min | 16.3→91.9 | 9.7→23.8 | 61→50.6 | 23.4→22.1 | 1.3→1.5 | - | 1.0→0.2 | 0.3→0.1 | [69] |
| Pd(3wt.%)/HZSM-5 | TTCM | 120→400 | 1 | 10 mg * | 40 ** cm3H2/min | 32.2→35.6 | 2.0→8.1 | 93→64.3 | 23.4→22.1 | 2.3→22 | - | 0→0.3 | 0.2→2.3 | [69] |
| Pt(3wt.%)/HY | TTCM | 120→400 | 1 | 10 mg * | 40 ** cm3H2/min | 76.1→58.9 | 22.4→67.6 | 48.7→28.8 | 0.9→1.3 | 0 | - | 1.8→0.4 | 25.6→1.7 | [69] |
| Pt(3wt.%)/HZSM-5 | TTCM | 120→400 | 1 | 10 mg * | 40 ** cm3H2/min | 37.6→70.3 | 57.8→93.9 | 3.1→1.9 | 1.6→0.8 | 0→1.7 | - | 0→0.6 | 37.6→0.7 | [69] |
| Pt(1wt.%)/AC | DCM | 150→250 | 1 | 0.8 | 100 | ca. 25→60 | ca. 74→85 | - | - | - | - | - | - | [35] |
| Pt(1wt.%)/AC | TCM | 125→200 | 1 | 0.8 | 100 | 67→ 100 | 88.1→93.8 | - | - | - | 4.2→3.1 | 7.7→3.1 | - | [35] |
| Pd(1wt.%)/AC | MCM | 150→250 | 1 | 0.6 | 100 | ca. 8→25 | n/a | [28] | ||||||
| Pd(1wt.%)/AC | DCM | 150→250 | 1 | 0.6 | 100 | ca. 10→85 | 79.9→64.5 | 3.1→26.6 | 0→1.6 | - | 17→7.3 | - | - | [28] |
| Pd(1wt.%)/AC | TCM | 125→175 | 1 | 0.6 | 100 | ca. 99→100 | 52.7→41.4 | 28.3→37.4 | 8.9→12.9 | 3→4.7 | 2.4→1.1 | 4.7→2.5 | - | [28] |
| Pd(0.5wt.%)/SiO2 | TCM | 100→225 | 1 | 0.1 g * | 9.5 | ca. 2→98 | ca. 75→40 | ca. 18→40 | ca. 5→13 | ca. 2→7 | - | - | - | [87] |
| Pt(0.5wt.%)/AC | TCM | 140→240 | 1 | 8 mg * | 8 *** | 34→93 | 47→68 | - | - | - | - | 53→32 | - | [50] |
| Pt(1wt.%)/η-Al2O3 | TCM | 140→240 | 1 | 8 mg * | 8 *** | 27→89 | 70→77 | - | - | - | - | 30→23 | - | [50] |
| Pt(0.4wt.%)/Vycor | TCM | 240→260 | 1 | 8 mg * | 8 *** | 38→52 | 74→83 | - | - | - | - | 26→17 | - | [50] |
| Pt(1wt.%)/AlF3 | TCM | 180→260 | 1 | 8 mg * | 8 *** | 4→78 | 55→84 | - | - | - | - | 45→16 | - | [50] |
| Pd(0.5wt.%)/AC | TCM | 140→180 | 1 | 2 g * | 7.35 *** | 9→94 | 58→88 | - | - | - | - | 42→12 | - | [50] |
| Pt-Pd(1:1)/SZ | DCM | 150→250 | 1 | 0.8 | 100 | ca. 10→81 | ca. 70→85 | ca. 0→1 | 0 | ca. 5→0 | ca. 25→14 | - | - | [21] |
| Pt-Pd(3:1)/SZ | DCM | 150→250 | 1 | 0.8 | 100 | ca. 8→58 | ca. 62→70 | ca. 1→3 | ca. 0→6 | ca. 7→1 | ca. 30→20 | - | - | [21] |
| Pt-Pd(1:3)/SZ | DCM | 150→250 | 1 | 0.8 | 100 | ca. 8→85 | ca. 69→87 | ca. 1→2 | 0→1 | ca. 1→0 | ca. 29→10 | - | - | [21] |
| Pt-Pd(1:3)/SZ | TCM | 150→250 | 1 | 0.8 | 100 | ca. 70→98 | ca. 82→75 | ca. 10→16 | ca. 3→6 | ca. 0→2 | ca. 0 | ca. 5→1 | - | [21] |
| Pt/C | DCM | 200→400 | 1 | 0.8 | 10 | ca. 15→88 | ca. 86→95 | ca. 0 | ca. 0 | ca. 0 | ca. 14→5 | - | - | [33] |
| Pt/C | TCM | 200→400 | 1 | 0.8 | 10 | ca. 96→100 | ca. 96→98 | ca. 0→2 | ca. 0 | ca. 0 | ca. 0 | ca. 4→0 | - | [33] |
| Ru/C | DCM | 150→400 | 1 | 0.8 | 10 | ca. 16→100 | ca. 80→64 | ca. 10→28 | ca. 5→4 | ca. 0 | ca. 5→4 | - | - | [33] |
| Ru/C | TCM | 150→400 | 1 | 0.8 | 10 | ca. 19→95 | ca. 19→45 | ca. 15→35 | ca. 0 | ca. 19→0 | ca. 2→5 | ca. 45→15 | - | [33] |
| Pd/C | DCM | 150→400 | 1 | 0.8 | 10 | ca. 22→95 | ca. 85→54 | ca. 5→44 | ca. 0 | ca. 0 | ca. 10→2 | - | - | [33] |
| Pd/C | TCM | 150→400 | 1 | 0.8 | 10 | ca. 90→100 | ca. 52→20 | ca. 32→75 | ca. 10→1 | ca. 0 | ca. 1→4 | ca. 5→0 | - | [33] |
| Rh/C | DCM | 150→400 | 1 | 0.8 | 10 | ca. 25→100 | ca. 82→67 | ca. 8→27 | ca. 2→6 | ca. 0 | ca. 8→0 | - | - | [33] |
| Rh/C | TCM | 150→400 | 1 | 0.8 | 10 | ca. 55→93 | ca. 45→42 | ca. 16→42 | ca. 16→2 | ca. 0→1 | ca. 8→1 | ca. 15→12 | - | [33] |
| Pt(25wt.%)/CNT | TTCM | 70→90 | 1 | 0.018 g * | 7 | 39→95 | 19.5→22 | 1.4→0.4 | 0.2→0 | - | ca. 0.4→0 | - | 76.5→77 | [138] |
| Pd(2wt.%)/MgO | TTCM | 120→250 | 1 | 0.2 g * | 10 | 0→10 | n/a | [142] | ||||||
| Pd(0.6wt.%)/Al2O3 | DCM | 100→400 | 1 | 0.005 * g min mL−1 | 10 | ca. 10→99 | n/a | [30] | ||||||
| Ni (0.6 wt.%)/Al2O3 | DCM | 100→400 | 1 | 0.005 * g min mL−1 | 10 | ca. 9→99 | n/a | [30] | ||||||
| Influence of Pressure | ||||||||||||||
| Ni-Mo/γ-Al2O3 | DCM | 350 | 20→100 | 1 g * | 3.26 *** | ca. 65→100 | n/a | [40] | ||||||
| Pt(0.5 wt.%)/γ-Al2O3 | TTCM | 130 | 1→9 | 4500 L/kg·h * | 11 | 99.3→99.9 | 32.4→18.4 | - | - | - | - | 0.08→0.6 | 65.2→79.3 | [56] |
| Influence of Space Time | ||||||||||||||
| Pd(1wt.%)/AC | MCM | 250 | 1 | 0.04→1.73 | 100 | ca. 5→35 | - | - | - | - | - | - | - | [1] |
| Pd(1wt.%)/AC | DCM | 250 | 1 | 0.04→1.73 | 100 | ca. 20→95 | ca. 65→68 | ca. 30→22 | ca. 2→1 | - | ca. 3→9 | - | - | [1] |
| Pd(1wt.%)/AC | TCM | 250 | 1 | 0.04→1.73 | 100 | ca. 99→100 | ca. 25→20 | ca. 48→50 | ca. 18→19 | ca. 6→8 | ca. 0 | ca. 3 | - | [1] |
| Pt(1wt.%)/AC | MCM | 250 | 1 | 0.04→1.73 | 100 | ca. 4→13 | n/a | [1] | ||||||
| Pt(1wt.%)/AC | DCM | 250 | 1 | 0.04→1.73 | 100 | ca. 5→85 | ca. 90→89 | - | - | - | ca. 10→11 | - | - | [1] |
| Pt(1wt.%)/AC | TCM | 250 | 1 | 0.04→1.73 | 100 | ca. 40→100 | ca. 92→91 | - | - | - | ca. 1.5 | ca. 6.5→7.5 | - | [1] |
| Rh(1wt.%)/AC | MCM | 250 | 1 | 0.04→1.73 | 100 | ca. 4→23 | n/a | [1] | ||||||
| Rh(1wt.%)/AC | DCM | 250 | 1 | 0.04→1.73 | 100 | ca. 20→100 | ca. 66→65 | ca. 22→23 | ca. 6 | - | ca. 6 | - | - | [1] |
| Rh(1wt.%)/AC | TCM | 250 | 1 | 0.04→1.73 | 100 | ca. 18→100 | ca. 15→50 | ca. 12→18 | ca. 8→12 | ca. 3→0 | ca. 0→15 | ca. 62→5 | - | [1] |
| Ru(1wt.%)/AC | MCM | 250 | 1 | 0.04→1.73 | 100 | ca. 2→7 | n/a | [1] | ||||||
| Ru(1wt.%)/AC | DCM | 250 | 1 | 0.04→1.73 | 100 | ca. 8→85 | ca. 50→51 | ca. 25→26 | ca. 15→10 | - | ca. 10→13 | - | - | [1] |
| Ru(1wt.%)/AC | TCM | 250 | 1 | 0.04→1.73 | 100 | ca. 10→100 | ca. 0→28 | ca. 0→15 | ca. 0→15 | ca. 0 | ca. 0→12 | ca. 100→30 | - | [1] |
| Pd(1wt.%)/AC | MCM | 250 | 1 | 0.08→1.73 | 100 | ca. 5→34 | n/a | [28] | ||||||
| Pd(1wt.%)/AC | DCM | 250 | 1 | 0.08→1.73 | 100 | ca. 28→95 | 62.7→67.9 | 27.9→23.9 | 2.2→1.4 | - | 7.2→6.8 | - | - | [28] |
| Pd(1wt.%)/AC | TCM | 175 | 1 | 0.08→1.73 | 100 | ca. 68→100 | 32.7→42.7 | 38.2→34.5 | 27.1→13.3 | 7.5→5.3 | 1.5→1.2 | 3.0 | - | [28] |
| Pd(0.5wt.%)/AC | DCM | 250 | 1 | 1→6.6 | 400 | ca. 60→92 | ca. 85→80 | ca. 2→5 | - | - | ca. 13→15 | - | - | [39] |
| Pd-Pt(1:1.8)/AC | DCM | 250 | 1 | 0.08→2.5 | 50 | 40→100 | ca. 40→99 | - | - | - | [37] | |||
| Pt(1wt.%)/AC | DCM | 250 | 1 | 0.08→1.73 | 100 | ca. 35→70 | ca. 86→84 | - | - | - | - | - | - | [35] |
| Pd(0.5wt.%)/Al2O3 | DCM | 300 | 1 | 0.4→1.8 | 10 | ca. 10→28 | n/a | [19] | ||||||
| Influence of H2/CM Molar Ratio | ||||||||||||||
| Pd-Pt(1:1.8)/AC | DCM | 250 | 1 | 0.6 | 25→200 | ca. 90→98 | ca. 99→95 | ca. 1→5 | - | - | [37] | |||
| Pd(0.5wt.%)/AC | DCM | 250 | 1 | 3.5 | 50→400 | ca. 65→80 | n/a | [39] | ||||||
| Pd(0.5wt.%)/Al2O3-c | DCM | 200 | 1 | 0.005 * g min mL−1 | 3→10 | ca. 50→60 | n/a | [30] | ||||||
| Pd(0.5wt.%)/Al2O3 | DCM | 200 | 1 | 0.005 * g min mL−1 | 3→10 | ca. 40→60 | n/a | [30] | ||||||
| Ni(0.5wt.%)/Al2O3 | DCM | 200 | 1 | 0.005 * g min mL−1 | 3→10 | ca. 28→32 | n/a | [30] | ||||||
| Pd(0.6wt.%)/Al2O3 | DCM | 200 | 1 | 0.005 * g min mL−1 | 3→10 | ca. 22→32 | n/a | [30] | ||||||
| Ni(0.6wt.%)/Al2O3 | DCM | 200 | 1 | 0.005 * g min mL−1 | 3→10 | ca. 28→32 | n/a | [30] | ||||||
| Al2O3 | DCM | 200 | 1 | 0.005 * g min mL−1 | 3→10 | ca. 18→32 | n/a | [30] | ||||||
| Pt(1.5wt.%)/Al2O3 | TTCM | 90 | 1 | 0.26 s * | 6.7→13.4 | ca. 9→11 | ca. 5→8 | ca. 55→2 | ca. 40→90 | [55] | ||||
| Pt(1.5wt.%)/Al2O3 | TTCM | 90 | 1 | 0.26 s * | 21.7→4.8 | ca. 96→46 | ca. 28→22 | ca. 0 | ca. 72→78 | [55] | ||||
| Pt(0.5 wt.%)/γ-Al2O3 | TTCM | 130 | 6 | 4500 L/kg·h * | 9→15 | 100 | 28.4→24.3 | - | - | - | - | 0.75→0.72 | 68.6→73.5 | [56] |
References
- Martin-Martinez, M.; Gómez-Sainero, L.; Alvarez-Montero, M.; Bedia, J.; Rodriguez, J. Comparison of different precious metals in activated carbon-supported catalysts for the gas-phase hydrodechlorination of chloromethanes. Appl. Catal. B Environ. 2013, 132–133, 256–265. [Google Scholar] [CrossRef]
- Huang, B.; Lei, C.; Wei, C.; Zeng, G. Chlorinated volatile organic compounds (Cl-VOCs) in environment—Sources, potential human health impacts, and current remediation technologies. Environ. Int. 2014, 71, 118–138. [Google Scholar] [CrossRef] [PubMed]
- Schlosser, P.M.; Bale, A.S.; Gibbons, C.F.; Wilkins, A.; Cooper, G.S. Human Health Effects of Dichloromethane: Key Findings and Scientific Issues. Environ. Health Perspect. 2015, 123, 114–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolman, K.G.; Dalpiaz, A.S. Occupational and Environmental Hepatotoxicity. In Drug-Induced Liver Disease, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2013; Chapter 36; pp. 659–675. [Google Scholar]
- Derwent, R.G.; Hester, R.E.; Harrison, R.M. Sources, distributions, and fates of VOCs in the atmosphere. In Issues in Environmental Science and Technology; Royal Society of Chemistry (RSC): Cambridge, UK, 2007; pp. 1–16. [Google Scholar]
- The Montreal Protocol on Substances that Deplete the Ozone Layer. Article 2C: Carbon Tetrachlroride. Available online: https://ozone.unep.org/treaties/montreal-protocol/articles/article-2d-carbon-tetrachloride?q=es/treaties/montreal-protocol/articles/articulo-2d-tetracloruro-de-carbono (accessed on 25 April 2020).
- Department of the Environment, Water, Heritage and the Arts. Universal Ratification of the Montreal Protocol. 2009. Available online: https://www.environment.gov.au/protection/ozone/publications/universal-ratification-montreal-protocol (accessed on 10 December 2020).
- Regulation (EC) No 166/2006 of the European Parliament and of the Council of 18 January 2006. Available online: https://eur-lex.europa.eu/legal-content/HR/TXT/?uri=CELEX:32006R0166 (accessed on 5 September 2020).
- CFR Part 63. National Air Emission Standards for Hazardous Air Pollutants: Halogenated Solvent Cleaning; Final Rule of the Environmental Protection Agency of 3 May 2007. 2007. Available online: https://www.epa.gov/stationary-sources-air-pollution/halogenated-solvent-cleaning-national-emission-standards-hazardou-0#rule-summary (accessed on 5 September 2020).
- Yuan, G.; Keane, M.A. Liquid phase catalytic hydrodechlorination of chlorophenols at 273 K. Catal. Commun. 2003, 4, 195–201. [Google Scholar] [CrossRef]
- Taylor, P.H.; Dellinger, B. Thermal degradation characteristics of chloromethane mixtures. Environ. Sci. Technol. 1988, 22, 438–447. [Google Scholar] [CrossRef]
- Kovalchuk, V.I.; D’Itri, J.L. Catalytic chemistry of chloro- and chlorofluorocarbon dehalogenation: From macroscopic observations to molecular level understanding. Appl. Catal. A Gen. 2004, 271, 13–25. [Google Scholar] [CrossRef]
- Hannus, I.; Halász, J. Hydrodechlorination over Zeolite Supported Catalysts—Clarification of Reaction Mechanism. J. Jpn. Pet. Inst. 2006, 49, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, C.J. Zeolite Mediated Reactions: Mechanistic Aspects and Environmental Applications. Prog. React. Kinet. Mech. 2008, 33, 1–79. [Google Scholar] [CrossRef]
- Dai, C.; Zhou, Y.; Peng, H.; Huang, S.; Qin, P.; Zhang, J.; Yang, Y.; Luo, L.; Zhang, X. Current progress in remediation of chlorinated volatile organic compounds: A review. J. Ind. Eng. Chem. 2018, 62, 106–119. [Google Scholar] [CrossRef]
- Hu, M.; Liu, Y.; Yao, Z.; Ma, L.; Wang, X. Catalytic reduction for water treatment. Front. Environ. Sci. Eng. 2017, 12. [Google Scholar] [CrossRef]
- Won, Y.S. Thermal stability and reaction mechanism of chloromethanes in excess hydrogen atmosphere. J. Ind. Eng. Chem. 2007, 13, 400–405. [Google Scholar]
- Wu, Y.-P.; Won, Y.-S. Pyrolysis of chloromethanes. Combust. Flame 2000, 122, 312–326. [Google Scholar] [CrossRef]
- López, E. Kinetic study of the gas-phase hydrogenation of aromatic and aliphatic organochlorinated compounds using a Pd/Al2O3 catalyst. J. Hazard. Mater. 2003, 97, 281–294. [Google Scholar] [CrossRef]
- Kartashov, L.M.; Rozanov, V.N.; Treger, Y.A.; Flid, M.R.; Kalyuzhnaya, T.L.; Tkach, D.V. Processing the wastes from the production of methyl chloride in the synthesis of olefins from natural gas. Catal. Ind. 2010, 2, 230–238. [Google Scholar] [CrossRef]
- Bedia, J.; Arevalo-Bastante, A.; Grau, J.; Dosso, L.; Rodriguez, J.; Mayoral, A.; Diaz, I.; Gómez-Sainero, L. Effect of the Pt–Pd molar ratio in bimetallic catalysts supported on sulfated zirconia on the gas-phase hydrodechlorination of chloromethanes. J. Catal. 2017, 352, 562–571. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, W.D.; Kovalchuk, V.I.; McDonald, M.A. Reaction pathways of halocarbon catalytic oligomerization. Catal. Commun. 2012, 18, 98–101. [Google Scholar] [CrossRef]
- Arevalo-Bastante, A.; Álvarez-Montero, M.A.; Bedia, J.; Gómez-Sainero, L.; Rodríguez, J.J. Gas-phase hydrodechlorination of mixtures of chloromethanes with activated carbon-supported platinum catalysts. Appl. Catal. B Environ. 2015, 179, 551–557. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, C.A.G.; Patiño, C.O.M.; De Correa, C.M. Catalytic hydrodechlorination of dichloromethane in the presence of traces of chloroform and tetrachloroethylene. Catal. Today 2008, 133, 520–525. [Google Scholar] [CrossRef]
- González, C.A.; Bartoszek, M.; Martin, A.; De Correa, C.M. Hydrodechlorination of Light Organochlorinated Compounds and Their Mixtures over Pd/TiO2-Washcoated Minimonoliths. Ind. Eng. Chem. Res. 2009, 48, 2826–2835. [Google Scholar] [CrossRef]
- Xu, L.; Bhandari, S.; Chen, J.; Glasgow, J.; Mavrikakis, M. Chloroform Hydrodechlorination on Palladium Surfaces: A Comparative DFT Study on Pd(111), Pd(100), and Pd(211). Top. Catal. 2020, 63, 762–776. [Google Scholar] [CrossRef]
- Ordóñez, S.; Sastre, H.; Díez, F.V. Hydrodechlorination of aliphatic organochlorinated compounds over commercial hydrogenation catalysts. Appl. Catal. B Environ. 2000, 25, 49–58. [Google Scholar] [CrossRef]
- Álvarez-Montero, M.; Gómez-Sainero, L.; Martín-Martínez, M.; Heras, F.; Rodriguez, J. Hydrodechlorination of chloromethanes with Pd on activated carbon catalysts for the treatment of residual gas streams. Appl. Catal. B Environ. 2010, 96, 148–156. [Google Scholar] [CrossRef]
- Liu, S.; Martin-Martinez, M.; Álvarez-Montero, M.A.; Arevalo-Bastante, A.; Rodríguez, J.J.; Gómez-Sainero, L. Recycling of Gas Phase Residual Dichloromethane by Hydrodechlorination: Regeneration of Deactivated Pd/C Catalysts. Catalysts 2019, 9, 733. [Google Scholar] [CrossRef] [Green Version]
- Zuluaga, B.H.A.; González, C.A.; Barrio, I.; Montes, M.; De Correa, C.M. Screening of Pd and Ni supported on sol–gel derived oxides for dichloromethane hydrodechlorination. J. Mol. Catal. A Chem. 2004, 222, 189–198. [Google Scholar] [CrossRef]
- Martin-Martinez, M.; Álvarez-Montero, A.; Gómez-Sainero, L.; Naji, L.; Palomar, J.; Omar, S.; Eser, S.; Rodriguez, J.; Álvarez-Montero, M.A. Deactivation behavior of Pd/C and Pt/C catalysts in the gas-phase hydrodechlorination of chloromethanes: Structure–reactivity relationship. Appl. Catal. B Environ. 2015, 162, 532–543. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Ruiz, C.; Bedia, J.; Grau, J.M.; Romero, A.C.; Rodríguez, D.; Rodríguez, J.J.; Gómez-Sainero, L. Rodríguez Promoting Light Hydrocarbons Yield by Catalytic Hydrodechlorination of Residual Chloromethanes Using Palladium Supported on Zeolite Catalysts. Catalysts 2020, 10, 199. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Sainero, L.M.; Palomar, J.; Omar, S.; Fernández, C.; Bedia, J.; Álvarez-Montero, A.; Rodriguez, J.J. Valorization of chloromethanes by hydrodechlorination with metallic catalysts. Catal. Today 2018, 310, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Martin-Martinez, M.; Rodriguez, J.J.; Baker, R.T.; Gómez-Sainero, L.M. Deactivation and regeneration of activated carbon-supported Rh and Ru catalysts in the hydrodechlorination of chloromethanes into light olefins. Chem. Eng. J. 2020, 397, 125479. [Google Scholar] [CrossRef]
- Álvarez-Montero, M.; Gómez-Sainero, L.; Mayoral, A.; Diaz, I.; Baker, R.; Rodriguez, J. Hydrodechlorination of chloromethanes with a highly stable Pt on activated carbon catalyst. J. Catal. 2011, 279, 389–396. [Google Scholar] [CrossRef]
- Álvarez-Montero, M.A.; Gómez-Sainero, L.; Juan-Juan, J.; Linares-Solano, A.; Rodriguez, J. Gas-phase hydrodechlorination of dichloromethane with activated carbon-supported metallic catalysts. Chem. Eng. J. 2010, 162, 599–608. [Google Scholar] [CrossRef]
- Martin-Martinez, M.; Gómez-Sainero, L.M.; Palomar, J.; Omar, S.; Rodriguez, J.J. Dechlorination of Dichloromethane by Hydrotreatment with Bimetallic Pd-Pt/C Catalyst. Catal. Lett. 2016, 146, 2614–2621. [Google Scholar] [CrossRef]
- De Pedro, Z.M.; Casas, J.A.; Gomez-Sainero, L.M.; Rodriguez, J.J. Hydrodechlorination of dichloromethane with a Pd/AC catalyst: Reaction pathway and kinetics. Appl. Catal. B Environ. 2010, 98, 79–85. [Google Scholar] [CrossRef]
- De Pedro, Z.M.; Gómez-Sainero, L.M.; González-Serrano, E.; Rodríguez, J.J. Gas-Phase Hydrodechlorination of Dichloromethane at Low Concentrations with Palladium/Carbon Catalysts. Ind. Eng. Chem. Res. 2006, 45, 7760–7766. [Google Scholar] [CrossRef]
- Martino, M.; Rosal, R.; Sastre, H.; Díez, F.V. Hydrodechlorination of dichloromethane, trichloroethane, trichloroethylene and tetrachloroethylene over a sulfided Ni/Mo–γ-alumina catalyst. Appl. Catal. B Environ. 1999, 20, 301–307. [Google Scholar] [CrossRef]
- Álvarez-Montero, M.A.; Rodriguez, J.J.; Gómez-Sainero, L.M. Platinum Nanoparticles Supported on Activated Carbon Catalysts for the Gas-Phase Hydrodechlorination of Dichloromethane: Influence of Catalyst Composition and Operating Conditions. Nanomater. Nanotechnol. 2016, 6, 18. [Google Scholar] [CrossRef] [Green Version]
- Bedia, J.; Gómez-Sainero, L.; Grau, J.M.; Busto, M.; Martin-Martinez, M.; Rodriguez, J. Hydrodechlorination of dichloromethane with mono- and bimetallic Pd–Pt on sulfated and tungstated zirconia catalysts. J. Catal. 2012, 294, 207–215. [Google Scholar] [CrossRef]
- Álvarez-Montero, M.A.; Martin-Martinez, M.; Gómez-Sainero, L.M.; Arevalo-Bastante, A.; Bedia, J.; Rodriguez, J.J. Kinetic Study of the Hydrodechlorination of Chloromethanes with Activated-Carbon-Supported Metallic Catalysts. Ind. Eng. Chem. Res. 2015, 54, 2023–2029. [Google Scholar] [CrossRef] [Green Version]
- López, E.; Ordóñez, S.; Diez, F.V. Deactivation of a Pd/Al2O3 catalyst used in hydrodechlorination reactions: Influence of the nature of organochlorinated compound and hydrogen chloride. Appl. Catal. B Environ. 2006, 62, 57–65. [Google Scholar] [CrossRef]
- Saadun, A.J.; Zichittella, G.; Paunovic, V.; Markaide-Aiastui, B.A.; Mitchell, S.; Pérez-Ramírez, J. Epitaxially Directed Iridium Nanostructures on Titanium Dioxide for the Selective Hydrodechlorination of Dichloromethane. ACS Catal. 2019, 10, 528–542. [Google Scholar] [CrossRef]
- Arevalo-Bastante, A.; Martin-Martinez, M.; Álvarez-Montero, M.A.; Rodríguez, J.J.; Gómez-Sainero, L. Properties of Carbon-supported Precious Metals Catalysts under Reductive Treatment and Their Influence in the Hydrodechlorination of Dichloromethane. Catalysts 2018, 8, 664. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.R.; Cho, J.M.; Son, M.; Park, M.-J.; Kim, W.Y.; Kim, S.Y.; Bae, J.W. Selective hydrodechlorination of trichloromethane to dichloromethane over bimetallic Pt-Pd/KIT-6: Catalytic activity and reaction kinetics. Chem. Eng. J. 2018, 331, 556–569. [Google Scholar] [CrossRef]
- Ruiz, C.F.; Bedia, J.; Andreoli, S.; Eser, S.; Rodríguez, J.J.; Gómez-Sainero, L. Selectivity to Olefins in the Hydrodechlorination of Chloroform with Activated Carbon-Supported Palladium Catalysts. Ind. Eng. Chem. Res. 2019, 58, 20592–20600. [Google Scholar] [CrossRef]
- Fernandez-Ruiz, C.; Bedia, J.; Bonal, P.; Rodriguez, J.J.; Gómez-Sainero, L. Chloroform conversion into ethane and propane by catalytic hydrodechlorination with Pd supported on activated carbons from lignin. Catal. Sci. Technol. 2018, 8, 3926–3935. [Google Scholar] [CrossRef]
- Prati, L. Reductive catalytic dehalogenation of light chlorocarbons. Appl. Catal. B Environ. 1999, 23, 135–142. [Google Scholar] [CrossRef]
- Bae, J.W.; Lee, J.S.; Lee, K.H. Hydrodechlorination of CCl4 over Pt/γ-Al2O3 prepared from different Pt precursors. Appl. Catal. A Gen. 2008, 334, 156–167. [Google Scholar] [CrossRef]
- Lokteva, E.S.; Lunin, V.V.; Golubina, E.V.; Simagina, V.I.; Egorova, M.; Stoyanova, I.V. C-C bond formation during hydrodechlorination of CCl4 on Pd-containing catalysts. Adv. Pharmacol. 2000, 130, 1997–2002. [Google Scholar] [CrossRef]
- Bonarowska, M.; Wojciechowska, M.; Zieliński, M.; Kiderys, A.; Zieliński, M.; Winiarek, P.; Karpiński, Z. Hydrodechlorination of Tetrachloromethane over Palladium Catalysts Supported on Mixed MgF2-MgO Carriers. Molecules 2016, 21, 1620. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.C.; Jiang, X.Z. Supported platinum–gallium catalysts for selective hydrodechlorination of CCl4. J. Mol. Catal. A Chem. 2005, 242, 119–128. [Google Scholar] [CrossRef]
- Bonarowska, M.; Karpiński, Z. Hydrodechlorination of Tetrachloromethane Over Supported Platinum Catalysts. Effects of Hydrogen Partial Pressure and Catalyst’s Screening Protocol on the Catalytic Performance. Top. Catal. 2012, 55, 846–852. [Google Scholar] [CrossRef] [Green Version]
- Bae, J.W.; Park, E.D.; Lee, J.S.; Lee, K.H.; Kim, Y.G.; Yeon, S.H.; Sung, B.H. Hydrodechlorination of CCl4 over Pt/γ-Al2O3. Appl. Catal. A Gen. 2001, 217, 79–89. [Google Scholar] [CrossRef]
- Bae, J.W.; Jang, E.J.; Lee, B.I.; Lee, J.S.; Lee, K.H. Effects of Tin on Product Distribution and Catalyst Stability in Hydrodechlorination of CCl4 over Pt-Sn/γ-Al2O3. Ind. Eng. Chem. Res. 2007, 46, 1721–1730. [Google Scholar] [CrossRef]
- Lu, M.; Sun, J.; Zhang, D.; Li, M.; Zhu, J.; Shan, Y. Highly selective hydrodechlorination of CCl4 into CHCl3 on Ag–Pd/carbon catalysts. React. Kinet. Mech. Catal. 2010, 100, 99–103. [Google Scholar] [CrossRef]
- Karpiński, Z.; Bonarowska, M.; Juszczyk, W. Hydrodechlorination of tetrachloromethane over silica-supported palladium-gold alloys. Pol. J. Chem. Technol. 2014, 16, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Bonarowska, M.; Kaszkur, Z.; Łomot, D.; Rawski, M.; Karpiński, Z. Effect of gold on catalytic behavior of palladium catalysts in hydrodechlorination of tetrachloromethane. Appl. Catal. B Environ. 2015, 162, 45–56. [Google Scholar] [CrossRef]
- Bonarowska, M.; Karpiński, Z.; Kosydar, R.; Szumełda, T.; Drelinkiewicz, A. Hydrodechlorination of CCl4 over carbon-supported palladium–gold catalysts prepared by the reverse “water-in-oil” microemulsion method. Comptes Rendus Chim. 2015, 18, 1143–1151. [Google Scholar] [CrossRef]
- Molina, C.B.; Pizarro, A.H.; Casas, J.A.; Rodriguez, J.J. Enhanced Pd pillared clays by Rh inclusion for the catalytic hydrodechlorination of chlorophenols in water. Water Sci. Technol. 2012, 65, 653–660. [Google Scholar] [CrossRef]
- Baeza, J.; Calvo, L.; Rodriguez, J.; Carbo-Argibay, E.; Rivas, J.; Gilarranz, M.A. Activity enhancement and selectivity tuneability in aqueous phase hydrodechlorination by use of controlled growth Pd-Rh nanoparticles. Appl. Catal. B Environ. 2015, 168, 283–292. [Google Scholar] [CrossRef]
- Yuan, G.; Louis, C.; Delannoy, L.; Keane, M.A. Silica- and titania-supported Ni–Au: Application in catalytic hydrodechlorination. J. Catal. 2007, 247, 256–268. [Google Scholar] [CrossRef]
- Legawiec-Jarzyna, M.; Juszczyk, W.; Bonarowska, M.; Kaszkur, Z.; Kępiński, L.; Kowalczyk, Z.; Karpiński, Z. Hydrodechlorination of CCl4 on Pt–Au/Al2O3 Catalysts. Top. Catal. 2009, 52, 1037–1043. [Google Scholar] [CrossRef]
- Choi, H.C.; Choi, S.H.; Lee, J.S.; Lee, K.H.; Kim, Y.G. Effects of Pt Precursors on Hydrodechlorination of Carbon Tetrachloride over Pt/Al2O3. J. Catal. 1997, 166, 284–293. [Google Scholar] [CrossRef]
- Zhang, Z.; Beard, B. Genesis of durable catalyst for selective hydrodechlorination of CCl4 to CHCl3. Appl. Catal. A Gen. 1998, 174, 33–39. [Google Scholar] [CrossRef]
- Legawiec-Jarzyna, M.; Śrębowata, A.; Juszczyk, W. Hydrodechlorination of chloroalkanes on supported platinum catalysts. React. Kinet. Catal. Lett. 2006, 87, 291–296. [Google Scholar] [CrossRef]
- Fási, A.; Hannus, I.; Halász, J.; Pálinkó, I. Hydrodechlorination Reactions of CCl4 Over HZSM-5- and HY-Supported Pt and Pd Catalysts. Top. Catal. 2012, 55, 853–857. [Google Scholar] [CrossRef]
- Choi, H.C.; Choi, S.H.; Yang, O.B.; Lee, J.S.; Lee, K.H.; Kim, Y.G. Hydrodechlorination of Carbon Tetrachloride over Pt/MgO. J. Catal. 1996, 161, 790–797. [Google Scholar] [CrossRef]
- Garetto, T.F.; Vignatti, C.; Borgna, A.; Monzón, A. Deactivation and regeneration of Pt/Al2O3 catalysts during the hydrodechlorination of carbon tetrachloride. Appl. Catal. B Environ. 2009, 87, 211–219. [Google Scholar] [CrossRef]
- Gómez-Sainero, L.M.; Seoane, X.L.; Arcoya, A. Hydrodechlorination of carbon tetrachloride in the liquid phase on a Pd/carbon catalyst: Kinetic and mechanistic studies. Appl. Catal. B Environ. 2004, 53, 101–110. [Google Scholar] [CrossRef]
- Legawiecjarzyna, M.; Śrębowata, A.; Juszczyk, W.; Karpinski, Z. Hydrodechlorination over Pd–Pt/Al2O3 catalysts. Appl. Catal. A Gen. 2004, 271, 61–68. [Google Scholar] [CrossRef]
- De Souza, A.G.F.; Bentes, A.M.P., Jr.; Rodrigues, A.C.C.; Borges, L.E.P.; Monteiro, J.L.F. Hydrodechlorination of carbon tetrachloride over PtNaX zeolite: Deactivation studies. Catal. Today 2005, 107-108, 493–499. [Google Scholar] [CrossRef]
- Golubina, E.V.; Lokteva, E.S.; Lunin, V.V.; O Turakulova, A.; Simagina, V.I.; Stoyanova, I.V. Modification of the supported palladium catalysts surface during hydrodechlorination of carbon tetrachloride. Appl. Catal. A Gen. 2003, 241, 123–132. [Google Scholar] [CrossRef]
- Santo, V.D.; Dossi, C.; Recchia, S.; Colavita, P.; Vlaic, G.; Psaro, R. Carbon tetrachloride hydrodechlorination with organometallics-based platinum and palladium catalysts on MgO. J. Mol. Catal. A Chem. 2002, 182–183, 157–166. [Google Scholar] [CrossRef]
- Machynskyy, O.; Śrębowata, A.; Kemnitz, E.; Karpiński, Z. Hydrodechlorination of Carbon Tetrachloride and 1,2-Dichloroethane on Palladium Nanoparticles Supported on Metal Fluorides. Int. J. Green Energy 2014, 12, 780–786. [Google Scholar] [CrossRef]
- Lu, M.; Li, X.; Chen, B.; Li, M.; Xin, H.; Song, L. Catalytic dechlorination of carbon tetrachloride in liquid phase with methanol as H-donor over Ag/C catalyst. J. Nanosci. Nanotechnol. 2014, 14, 7315–7318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delannoy, L.; Giraudon, J.-M.; Grangerc, P.; Leclercq, L.; Leclercq, G. Hydrodechlorination of CCl4 over group VI transition metal carbides. Appl. Catal. B Environ. 2002, 37, 161–173. [Google Scholar] [CrossRef]
- Bonarowska, M.; Zieliński, M.; Matus, K.; Sá, J.; Śrębowata, A. Influence of microwave activation on the catalytic behavior of Pd-Au/C catalysts employed in the hydrodechlorination of tetrachloromethane. React. Kinet. Mech. Catal. 2018, 124, 375–388. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Choi, H.C.; Yanga, O.B.; Lee, K.H.; Lee, J.S.; Kim, Y.G. Hydrodechlorination of tetrachloromethane over supported Pt catalysts. J. Chem. Soc. Chem. Commun. 1995, 21, 2169. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Sainero, L.M.; Seoane, X.L.; Tijero, E.; Arcoya, A. Hydrodechlorination of carbon tetrachloride to chloroform in the liquid phase with a Pd/carbon catalyst. Study of the mass transfer steps. Chem. Eng. Sci. 2002, 57, 3565–3574. [Google Scholar] [CrossRef]
- Gomez-Sainero, L.M.; Cortés, A.; Seoane, X.L.; Arcoya, A. Hydrodechlorination of Carbon Tetrachloride to Chloroform in the Liquid Phase with Metal-Supported Catalysts. Effect of the Catalyst Components. Ind. Eng. Chem. Res. 2000, 39, 2849–2854. [Google Scholar] [CrossRef]
- Ahmadzai, H.; Bock, R.P.; Burholder, J.B.; Butler, J.H.; Chatteriee, A.; Chipperfield, M.P.; Daniel, J.S.; Derek, N.; Fleming, E.L.; Fraser, P.J.; et al. SPARC Report on the Mystery of Carbon Tetrachloride; No. 7; WCRP: Geneva, Switzerland, 2016; pp. 1–67. [Google Scholar]
- Martin-Martinez, M.; Gómez-Sainero, M.L.; Bedia, J.; Arevalo-Bastante, A.; Rodríguez, J.J. Enhanced activity of carbon-supported Pd–Pt catalysts in the hydrodechlorination of dichloromethane. Appl. Catal. B Environ. 2016, 184, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Keane, M.A. Supported Transition Metal Catalysts for Hydrodechlorination Reactions. ChemCatChem 2011, 3, 800–821. [Google Scholar] [CrossRef]
- Mori, T.; Kikuchi, T.; Kubo, J.; Morikawa, Y. Hydrodechlorination of Trichloromethane to Higher Hydrocarbons over Pd/SiO2 Catalyst. Chem. Lett. 2001, 30, 936–937. [Google Scholar] [CrossRef]
- Ali, A.M.; Podila, S.; Daous, M.A.; Al-Zahrani, A.A.; Mahpudz, A. Highly efficient hydrotalcite supported palladium catalyst for hydrodechlorination of 1,2,4-tri chlorobenzene: Influence of Pd loading. J. Chem. Sci. 2020, 132, 1–10. [Google Scholar] [CrossRef]
- Gómez-Sainero, L.M.; Seoane, X.L.; Fierro, J.L.; Arcoya, A. Liquid-Phase Hydrodechlorination of CCl4 to CHCl3 on Pd/Carbon Catalysts: Nature and Role of Pd Active Species. J. Catal. 2002, 209, 279–288. [Google Scholar] [CrossRef]
- Jujjuri, S.; Ding, E.; Hommel, E.; Shore, S.; Keane, M.A. Synthesis and characterization of novel silica-supported Pd/Yb bimetallic catalysts: Application in gas-phase hydrodechlorination and hydrogenation. J. Catal. 2006, 239, 486–500. [Google Scholar] [CrossRef]
- Cao, Y.C.; Li, Y. In situ synthesis of supported palladium complexes: Highly stable and selective supported palladium catalysts for hydrodechlorination of CCl2F2. Appl. Catal. A Gen. 2005, 294, 298–305. [Google Scholar] [CrossRef]
- Bonarowska, M.; Matus, K.; Śrębowata, A.; Sá, J. Application of silica-supported Ir and Ir-M (M = Pt, Pd, Au) catalysts for low-temperature hydrodechlorination of tetrachloromethane. Sci. Total. Environ. 2018, 644, 287–297. [Google Scholar] [CrossRef]
- Golubina, E.V.; Lokteva, E.S.; Lazareva, T.S.; Kostyuk, B.G.; Lunin, V.V.; Simagina, V.I.; Stoyanova, I.V. Hydrodechlorination of Tetrachloromethane in the Vapor Phase in the Presence of Pd–Fe/Sibunit Catalysts. Kinet. Catal. 2004, 45, 183–188. [Google Scholar] [CrossRef]
- Rodríguez-Reinoso, F.; Rodríguez-Ramos, I.; Moreno-Castilla, C.; Guerrero-Ruiz, A.; López-González, J.D. Platinum catalyst supported on activated carbons: I. Preparation and characterization. J. Catal. 1983, 99, 171–183. [Google Scholar] [CrossRef]
- Ruiz-Garcia, C.; Heras, F.; Calvo, L.; Alonso-Morales, N.; Rodriguez, J.; Gilarranz, M. Improving the activity in hydrodechlorination of Pd/C catalysts by nitrogen doping of activated carbon supports. J. Environ. Chem. Eng. 2020, 8, 103689. [Google Scholar] [CrossRef]
- Bonarowska, M.; Burda, B.; Juszczyk, W.; Pielaszek, J.; Kowalczyk, Z.; Karpinski, Z. Hydrodechlorination of CCl2F2 (CFC-12) over Pd-Au/C catalysts. Appl. Catal. B Environ. 2001, 35, 13–20. [Google Scholar] [CrossRef]
- Amorim, C.; Yuan, G.; Patterson, P.M.; Keane, M.A. Catalytic hydrodechlorination over Pd supported on amorphous and structured carbon. J. Catal. 2005, 234, 268–281. [Google Scholar] [CrossRef]
- Orellana, F.; Pecchi, G.; Reyes, P. Selective hydrodechlorination of 1,2-dichloroethane over Pd-Sn/SiO2 catalysts. J. Chil. Chem. Soc. 2005, 50, 431–434. [Google Scholar] [CrossRef]
- Ning, X.; Sun, Y.; Fu, H.; Qu, X.; Xu, Z.; Zheng, S. N-doped porous carbon supported Ni catalysts derived from modified Ni-MOF-74 for highly effective and selective catalytic hydrodechlorination of 1,2-dichloroethane to ethylene. Chemosphere 2020, 241, 124978. [Google Scholar] [CrossRef] [PubMed]
- Merte, L.R.; Peng, G.; Bechstein, R.; Rieboldt, F.; Farberow, C.A.; Grabow, L.C.; Kudernatsch, W.; Wendt, S.; Laegsgaard, E.; Mavrikakis, M.; et al. Water-Mediated Proton Hopping on an Iron Oxide Surface. Science 2012, 336, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Karim, W.; Spreafico, C.; Kleibert, A.; Gobrecht, J.; Vandevondele, C.S.J.; Ekinci, W.K.J.G.Y.; Van Bokhoven, W.K.J.A. Catalyst support effects on hydrogen spillover. Nat. Cell Biol. 2017, 541, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Im, J.; Shin, H.; Jang, H.; Kim, H.; Choi, M. Maximizing the catalytic function of hydrogen spillover in platinum-encapsulated aluminosilicates with controlled nanostructures. Nat. Commun. 2014, 5, 3370. [Google Scholar] [CrossRef] [PubMed]
- Haber, J.; Block, J.H.; Delmon, B. Manual of methods and procedures for catalyst characterization (Technical Report). Pure Appl. Chem. 1995, 67, 1257–1306. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, P.; Gaigneaux, E.; De Vos, D.E.; Martens, J.A.; Poncelet, G.; Jacobs, P.A. Scientific Bases for the Preparation of Heterogeneous Catalysts, 1st ed.; Elsevier Science: Amsterdam, The Netherlands, 2002. [Google Scholar]
- Huang, Y.; Schwarz, J. The effect of catalyst preparation on catalytic activity. Appl. Catal. 1987, 30, 239–253. [Google Scholar] [CrossRef]
- Gurbani, A.; Ayastuy, J.; González-Marcos, M.; Herrero, J.; Guil, J.; Gutiérrez-Ortiz, M.A. Comparative study of CuO–CeO2 catalysts prepared by wet impregnation and deposition–precipitation. Int. J. Hydrog. Energy 2009, 34, 547–553. [Google Scholar] [CrossRef]
- Wu, Z.; Tang, N.; Xiao, L.; Liu, Y.; Wang, H. MnOx/TiO2 composite nanoxides synthesized by deposition-precipitation method as a superior catalyst for NO oxidation. J. Colloid Interface Sci. 2010, 352, 143–148. [Google Scholar] [CrossRef]
- Bonarowska, M.; Karpiński, Z. Characterization of supported Pd-Pt catalysts by chemical probes. Catal. Today 2008, 137, 498–503. [Google Scholar] [CrossRef]
- Imre, B.; Hannus, I.; Kiricsi, I. Comparative IR spectroscopic study of Pt- and Pd-containing zeolites in the hydrodechlorination reaction of carbon tetrachloride. J. Mol. Struct. 2005, 744-747, 501–506. [Google Scholar] [CrossRef]
- Bradu, C.; Căpăţ, C.; Papa, F.; Frunza, L.; Olaru, E.-A.; Crini, G.; Morin-Crini, N.; Euvrard, É.; Balint, I.; Zgura, I.; et al. Pd-Cu catalysts supported on anion exchange resin for the simultaneous catalytic reduction of nitrate ions and reductive dehalogenation of organochlorinated pollutants from water. Appl. Catal. A Gen. 2019, 570, 120–129. [Google Scholar] [CrossRef]
- Liu, Y.; Diao, X.; Tao, F.; Yang, C.; Wang, H.; Takaoka, M.; Sun, Y. Insight into the low-temperature decomposition of Aroclor 1254 over activated carbon-supported bimetallic catalysts obtained with XANES and DFT calculations. J. Hazard. Mater. 2019, 366, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.; Kim, H.; Oh, J.; Ahn, B.J. Hydrodechlorination of chlorophenols over Pd catalysts supported on zeolite Y, MCM-41 and graphene. Res. Chem. Intermed. 2018, 44, 3835–3847. [Google Scholar] [CrossRef]
- Kowalewski, E.; Kamińska, I.I.; Słowik, G.; Lisovytskiy, D.; Śrębowata, A. Effect of metal precursor and pretreatment conditions on the catalytic activity of Ni/C in the aqueous phase hydrodechlorination of 1,1,2-trichloroethene. React. Kinet. Mech. Catal. 2017, 121, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Terekhov, A.V.; Zanaveskin, L.N.; Zanaveskin, K.L.; Konorev, O.A. Catalytic hydrodechlorination of chlorinated hydrocarbons in a medium of sodium hydroxide solutions. I: Conversion of carbon tetrachloride. Catal. Ind. 2013, 5, 32–41. [Google Scholar] [CrossRef]
- Ren, Y.; Fan, G.; Jiang, W.; Xu, B.; Liu, F. Effective hydrodechlorination of 4-chlorophenol catalysed by magnetic palladium/reduced graphene oxide under mild conditions. RSC Adv. 2014, 4, 25440–25446. [Google Scholar] [CrossRef]
- Choi, E.-K.; Park, K.-H.; Lee, H.-B.; Cho, M.; Ahn, S. Formic acid as an alternative reducing agent for the catalytic nitrate reduction in aqueous media. J. Environ. Sci. 2013, 25, 1696–1702. [Google Scholar] [CrossRef]
- Kopinke, F.-D.; MacKenzie, K.; Koehler, R.; Georgi, A. Alternative sources of hydrogen for hydrodechlorination of chlorinated organic compounds in water on Pd catalysts. Appl. Catal. A Gen. 2004, 271, 119–128. [Google Scholar] [CrossRef]
- Liu, H.; Long, L.; Xu, Z.; Zheng, S. Pd-NCQD composite confined in SBA-15 as highly active catalyst for aqueous phase catalytic hydrodechlorination of 2,4-dichlorophenoxyacetic acid. Chem. Eng. J. 2020, 400, 125987. [Google Scholar] [CrossRef]
- Feng, Z.; Gao, C.; Ma, X.; Zhan, J. Well-dispersed Pd nanoparticles on porous ZnO nanoplates via surface ion exchange for chlorobenzene-selective sensor. RSC Adv. 2019, 9, 42351–42359. [Google Scholar] [CrossRef] [Green Version]
- Baeza, J.; Calvo, L.; Gilarranz, M.A.; Rodriguez, J. Effect of size and oxidation state of size-controlled rhodium nanoparticles on the aqueous-phase hydrodechlorination of 4-chlorophenol. Chem. Eng. J. 2014, 240, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Bonarowska, M.; Kaszkur, Z.; Kępiński, L.; Karpiński, Z. Hydrodechlorination of tetrachloromethane on alumina- and silica-supported platinum catalysts. Appl. Catal. B Environ. 2010, 99, 248–256. [Google Scholar] [CrossRef]
- Ramos, A.L.D.; Alves, P.D.S.; Aranda, D.A.G.; Schmal, M. Characterization of carbon supported palladium catalysts: Inference of electronic and particle size effects using reaction probes. Appl. Catal. A Gen. 2004, 277, 71–81. [Google Scholar] [CrossRef]
- Baeza, J.A.; Calvo, L.; Murzin, D.Y.; Rodriguez, J.J.; Gilarranz, M.A. Kinetic Analysis of 4-Chlorophenol Hydrodechlorination Catalyzed by Rh Nanoparticles Based on the Two-Step Reaction and Langmuir–Hinshelwood Mechanisms. Catal. Lett. 2014, 144, 2080–2085. [Google Scholar] [CrossRef]
- Baeza, J.; Calvo, L.; Gilarranz, M.; Mohedano, A.; Casas, J.; Rodriguez, J. Catalytic behavior of size-controlled palladium nanoparticles in the hydrodechlorination of 4-chlorophenol in aqueous phase. J. Catal. 2012, 293, 85–93. [Google Scholar] [CrossRef]
- Bae, J.W.; Kim, I.G.; Lee, J.S.; Lee, K.H.; Jang, E.J. Hydrodechlorination of CCl4 over Pt/Al2O3: Effects of platinum particle size on product distribution. Appl. Catal. A Gen. 2003, 240, 129–142. [Google Scholar] [CrossRef]
- Gómez-Quero, S.; Cárdenas-Lizana, F.; Keane, M.A. Effect of Metal Dispersion on the Liquid-Phase Hydrodechlorination of 2,4-Dichlorophenol over Pd/Al2O3. Ind. Eng. Chem. Res. 2008, 47, 6841–6853. [Google Scholar] [CrossRef]
- Omar, S.; Palomar, J.; Gómez-Sainero, L.M.; Álvarez-Montero, M.A.; Martin-Martinez, M.; Rodriguez, J.J. Density Functional Theory Analysis of Dichloromethane and Hydrogen Interaction with Pd Clusters: First Step to Simulate Catalytic Hydrodechlorination. J. Phys. Chem. C 2011, 115, 14180–14192. [Google Scholar] [CrossRef]
- Lv, X.; Prastistho, W.; Yang, Q.; Tokoro, C. Application of nano-scale zero-valent iron adsorbed on magnetite nanoparticles for removal of carbon tetrachloride: Products and degradation pathway. Appl. Organomet. Chem. 2020, 34, e5592. [Google Scholar] [CrossRef]
- Lowry, G.V.; Reinhard, M. Hydrodehalogenation of 1- to 3-Carbon Halogenated Organic Compounds in Water Using a Palladium Catalyst and Hydrogen Gas. Environ. Sci. Technol. 1999, 33, 1905–1910. [Google Scholar] [CrossRef]
- Huang, C.-C.; Lo, S.-L.; Lien, H.-L. Zero-valent copper nanoparticles for effective dechlorination of dichloromethane using sodium borohydride as a reductant. Chem. Eng. J. 2012, 203, 95–100. [Google Scholar] [CrossRef]
- Huang, C.-C.; Lo, S.-L.; Lien, H.-L. Vitamin B12-mediated hydrodechlorination of dichloromethane by bimetallic Cu/Al particles. Chem. Eng. J. 2015, 273, 413–420. [Google Scholar] [CrossRef]
- Karpiński, Z. Catalysis by Supported, Unsupported, and Electron-Deficient Palladium. Adv. Catal. 1990, 37, 45–100. [Google Scholar] [CrossRef]
- He, Y.; Fan, J.; Feng, J.; Luo, C.; Yang, P.; Li, D. Pd nanoparticles on hydrotalcite as an efficient catalyst for partial hydrogenation of acetylene: Effect of support acidic and basic properties. J. Catal. 2015, 331, 118–127. [Google Scholar] [CrossRef]
- Cecilia, J.A.; Infantes-Molina, A.; Rodríguez-Castellón, E.; Ilyina, A. Gas phase catalytic hydrodechlorination of chlorobenzene over cobalt phosphide catalysts with different P contents. J. Hazard. Mater. 2013, 260, 167–175. [Google Scholar] [CrossRef]
- Padmasri, A.; Venugopal, A.; Kumar, V.S.; Shashikala, V.; Nagaraja, B.; Seetharamulu, P.; Sreedhar, B.; Raju, B.D.; Rao, P.K.; Rao, K.R. Role of hydrotalcite precursors as supports for Pd catalysts in hydrodechlorination of CCl2F2. J. Mol. Catal. A Chem. 2004, 223, 329–337. [Google Scholar] [CrossRef]
- Celik, G.; Ailawar, S.A.; Gunduz, S.; Edmiston, P.L.; Ozkan, U.S. Formation of carbonaceous deposits on Pd-based hydrodechlorination catalysts: Vibrational spectroscopy investigations over Pd/Al2O3 and Pd/SOMS. Catal. Today 2019, 323, 129–140. [Google Scholar] [CrossRef]
- Gerber, I.C.; Oubenali, M.; Bacsa, R.R.; Durand, J.; Gonçalves, A.G.; Pereira, M.; Jolibois, F.; Perrin, L.; Poteau, R.; Serp, P. Theoretical and Experimental Studies on the Carbon-Nanotube Surface Oxidation by Nitric Acid: Interplay between Functionalization and Vacancy Enlargement. Chem A Eur. J. 2011, 17, 11467–11477. [Google Scholar] [CrossRef]
- Bonarowska, M.; Lin, K.N.; Jarzyna, M.L.; Stobinski, L.; Juszczyk, W.; Kaszkur, Z.; Karpiński, Z.; Lin, H.M. Multi-Wall Carbon Nanotubes as a Support for Platinum Catalysts for the Hydrodechlorination of Carbon Tetrachloride and Dichlorodifluoromethane. Solid State Phenom. 2007, 128, 261–271. [Google Scholar] [CrossRef]
- Rosas, J.M.; Bedia, J.; Rodríguez-Mirasol, J.; Cordero, T. Preparation of Hemp-Derived Activated Carbon Monoliths. Adsorption of Water Vapor. Ind. Eng. Chem. Res. 2008, 47, 1288–1296. [Google Scholar] [CrossRef]
- Bedia, J.; Rosas, J.M.; Márquez, J.; Rodríguez-Mirasol, J.; Cordero, T. Preparation and characterization of carbon based acid catalysts for the dehydration of 2-propanol. Carbon 2009, 47, 286–294. [Google Scholar] [CrossRef]
- Valero-Romero, M.; García-Mateos, F.; Rodríguez-Mirasol, J.; Cordero, T. Role of surface phosphorus complexes on the oxidation of porous carbons. Fuel Process. Technol. 2017, 157, 116–126. [Google Scholar] [CrossRef]
- Góralski, J.; Szczepaniak, B.; Grams, J.; Maniukiewicz, W.; Paryjczak, T. Characteristic of physicochemical properties of Pd/MgO catalysts used in the hydrodechlorination process with CCI4. Pol. J. Chem. Technol. 2007, 9, 77–80. [Google Scholar] [CrossRef]
- Lan, L.; Liu, Y.; Liu, S.; Ma, X.; Li, X.; Dong, Z.; Xia, C. Effect of the supports on catalytic activity of Pd catalysts for liquid-phase hydrodechlorination/hydrogenation reaction. Environ. Technol. 2019, 40, 1615–1623. [Google Scholar] [CrossRef]
- Netskina, O.; Komova, O.; Tayban, E.; Oderova, G.; Mukha, S.; Kuvshinov, G.; Simagina, V.I. The influence of acid treatment of carbon nanofibers on the activity of palladium catalysts in the liquid-phase hydrodechlorination of dichlorobenzene. Appl. Catal. A Gen. 2013, 467, 386–393. [Google Scholar] [CrossRef]
- Gerber, I.C.; Serp, P. A Theory/Experience Description of Support Effects in Carbon-Supported Catalysts. Chem. Rev. 2020, 120, 1250–1349. [Google Scholar] [CrossRef]
- Karousis, N.; Tagmatarchis, N.; Tasis, D. Current Progress on the Chemical Modification of Carbon Nanotubes. Chem. Rev. 2010, 110, 5366–5397. [Google Scholar] [CrossRef]
- Gu, X.; Qi, W.; Xu, X.; Sun, Z.; Zhang, L.; Liu, W.; Pan, X.; Su, D. Covalently functionalized carbon nanotube supported Pd nanoparticles for catalytic reduction of 4-nitrophenol. Nanoscale 2014, 6, 6609–6616. [Google Scholar] [CrossRef]
- Kundu, S.; Wang, Y.; Xia, W.; Muhler, M. Thermal Stability and Reducibility of Oxygen-Containing Functional Groups on Multiwalled Carbon Nanotube Surfaces: A Quantitative High-Resolution XPS and TPD/TPR Study. J. Phys. Chem. C 2008, 112, 16869–16878. [Google Scholar] [CrossRef]
- Shen, W.; Fan, W. Nitrogen-containing porous carbons: Synthesis and application. J. Mater. Chem. A 2013, 1, 999–1013. [Google Scholar] [CrossRef]
- Cao, Y.; Mao, S.; Li, M.; Chen, Y.; Wang, Y. Metal/Porous Carbon Composites for Heterogeneous Catalysis: Old Catalysts with Improved Performance Promoted by N-Doping. ACS Catal. 2017, 7, 8090–8112. [Google Scholar] [CrossRef]
- An, N.; Zhang, M.; Zhang, Z.; Dai, Y.; Shen, Y.; Tang, C.; Yuan, X.; Zhou, W. High-performance palladium catalysts for the hydrogenation toward dibenzylbiotinmethylester: Effect of carbon support functionalization. J. Colloid Interface Sci. 2018, 510, 181–189. [Google Scholar] [CrossRef]
- Lu, C.; Wang, M.; Feng, Z.; Qi, Y.; Feng, F.; Ma, L.; Zhang, Q.; Li, X. A phosphorus–carbon framework over activated carbon supported palladium nanoparticles for the chemoselective hydrogenation of para-chloronitrobenzene. Catal. Sci. Technol. 2017, 7, 1581–1589. [Google Scholar] [CrossRef]
- An, N.; Dai, Y.; Tang, C.; Yuan, X.; Dong, J.; Shen, Y.; Zhou, W. Design and preparation of a simple and effective palladium catalyst and the hydrogenation performance toward dibenzylbiotinmethylester. J. Colloid Interface Sci. 2016, 470, 56–61. [Google Scholar] [CrossRef]
- Diaz, E.; Mohedano, A.F.; Casas, J.A.; Rodriguez, J.J. Analysis of the deactivation of Pd, Pt and Rh on activated carbon catalysts in the hydrodechlorination of the MCPA herbicide. Appl. Catal. B Environ. 2016, 181, 429–435. [Google Scholar] [CrossRef]
- Bedia, J.; Rosas, J.; Rodríguez-Mirasol, J.; Cordero, T. Pd supported on mesoporous activated carbons with high oxidation resistance as catalysts for toluene oxidation. Appl. Catal. B Environ. 2010, 94, 8–18. [Google Scholar] [CrossRef]
- Guillén, E.; Rico, R.; Lopezromero, J.M.; Bedia, J.; Rosas, J.M.; Rodríguez-Mirasol, J.; Cordero, T. Pd-activated carbon catalysts for hydrogenation and Suzuki reactions. Appl. Catal. A Gen. 2009, 368, 113–120. [Google Scholar] [CrossRef]
- Chary, K.V.; Rao, P.V.R.; Vishwanathan, V. Synthesis and high performance of ceria supported nickel catalysts for hydrodechlorination reaction. Catal. Commun. 2006, 7, 974–978. [Google Scholar] [CrossRef]
- Li, F.; Liu, Y.; Ma, T.; Xu, D.; Li, X.; Gong, G. Catalysis of the hydrodechlorination of 4-chlorophenol and the reduction of 4-nitrophenol by Pd/Fe3O4@C. New J. Chem. 2017, 41, 4014–4021. [Google Scholar] [CrossRef]
- Concibido, N.C.; Okuda, T.; Nishijima, W.; Okada, M. Deactivation and reactivation of Pd/C catalyst used in repeated batch hydrodechlorination of PCE. Appl. Catal. B Environ. 2007, 71, 64–69. [Google Scholar] [CrossRef]
- Bonarowska, M.; Malinowski, A.; Karpinski, Z. Hydrogenolysis of C–C and C–Cl bonds by Pd–Re/Al2O3 catalysts. Appl. Catal. A Gen. 1999, 188, 145–154. [Google Scholar] [CrossRef]
- Chen, N.; Rioux, R.M.; Barbosa, L.A.M.M.; Ribeiro, F.H. Kinetic and Theoretical Study of the Hydrodechlorination of CH4−xClx (x = 1−4) Compounds on Palladium†. Langmuir 2010, 26, 16615–16624. [Google Scholar] [CrossRef]
- Reeves, C.; Meyer, R.; Mullins, C.B. Dissociative adsorption and hydrodechlorination of CCl4 on Ir(1 1 0). J. Mol. Catal. A Chem. 2003, 202, 135–146. [Google Scholar] [CrossRef]
- Thomas, J.M.; Thomas, W.J. The Dynamics of Selective and Polyfunctional Catalysis. In Introduction to the Principles of Heterogeneous Catalysts; Thomas, J.M., Thomas, W.J., Eds.; Academic Press: New York, NY, USA, 1969; Chapter 7. [Google Scholar]
- Centi, G. Elementary Reaction Steps in Heterogeneous Catalysis; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1993; p. 93. [Google Scholar]
- Converti, A.; Zilli, M.; De Faveri, D.M.; Ferraiolo, G. Hydrogenolysis of organochlorinated pollutants: Kinetics and thermodynamics. J. Hazard. Mater. 1991, 27, 127–135. [Google Scholar] [CrossRef]
- Forni, P.; Prati, L.; Rossi, M. Catalytic dehydrohalogenation of polychlorinated biphenyls Part II: Studies on a continuous process. Appl. Catal. B Environ. 1997, 14, 49–53. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Mechanisms of catalyst deactivation. Appl. Catal. A Gen. 2001, 212, 17–60. [Google Scholar] [CrossRef]
- Ordonez, S. Characterisation of the deactivation of platinum and palladium supported on activated carbon used as hydrodechlorination catalysts. Appl. Catal. B Environ. 2001, 31, 113–122. [Google Scholar] [CrossRef]
- Urbano, F.J.; Marinas, J. Hydrogenolysis of organohalogen compounds over palladium supported catalysts. J. Mol. Catal. A Chem. 2001, 173, 329–345. [Google Scholar] [CrossRef]
- Heinrichs, B. Palladium–silver sol–gel catalysts for selective hydrodechlorination of 1,2-dichloroethane into ethylene IV. Deactivation mechanism and regeneration. J. Catal. 2003, 220, 215–225. [Google Scholar] [CrossRef]
- Chakraborty, D.; Kulkarni, P.P.; Kovalchuk, V.I.; D’Itri, J.L. Dehalogenative oligomerization of dichlorodifluoromethane catalyzed by activated carbon-supported Pt–Cu catalysts: Effect of Cu to Pt atomic ratio. Catal. Today 2004, 88, 169–181. [Google Scholar] [CrossRef]
- Ordóñez, S.; Díaz, E.; Bueres, R.F.; Asedegbega-Nieto, E.; Sastre, H. Carbon nanofibre-supported palladium catalysts as model hydrodechlorination catalysts. J. Catal. 2010, 272, 158–168. [Google Scholar] [CrossRef]
- Lapierre, R.B.; Wu, D.; Kranich, W.L.; Weiss, A.H. Hydrodechlorination of 1,1-bis(p-chlorophenyl)-2,2-dichloroethylene (p,p′-DDE) in the vapor phase. J. Catal. 1978, 52, 59–71. [Google Scholar] [CrossRef]
- Coq, B. Conversion of chlorobenzene over palladium and rhodium catalysts of widely varying dispersion. J. Catal. 1986, 101, 434–445. [Google Scholar] [CrossRef]
- Fung, S. Hydrogenolysis of methyl chloride on metals. J. Catal. 1987, 103, 220–223. [Google Scholar] [CrossRef]
- Gampine, A.; Eyman, D.P. Catalytic Hydrodechlorination of Chlorocarbons. 2. Ternary Oxide Supports for Catalytic Conversions of 1,2-Dichlorobenzene. J. Catal. 1998, 179, 315–325. [Google Scholar] [CrossRef]
- Ordóñez, S.; Sastre, H.; Díez, F. Thermogravimetric determination of coke deposits on alumina-supported noble metal catalysts used as hydrodechlorination catalysts. Thermochim. Acta 2001, 379, 25–34. [Google Scholar] [CrossRef]
- Aramendía, M.; Boráu, V.; García, I.; Jiménez, C.; Lafont, F.; Marinas, A.; Marinas, J.; Urbano, F.J. Liquid-phase hydrodechlorination of chlorobenzene over palladium-supported catalysts. J. Mol. Catal. A Chem. 2002, 184, 237–245. [Google Scholar] [CrossRef]
- Tavoularis, G.; Keane, M.A. Gas phase catalytic dehydrochlorination and hydrodechlorination of aliphatic and aromatic systems. J. Mol. Catal. A Chem. 1999, 142, 187–199. [Google Scholar] [CrossRef]
- Barrabés, N.; Föttinger, K.; Llorca, J.; Dafinov, A.; Medina, F.; Sá, J.; Hardacre, C.; Rupprechter, G.; Föttinger, K. Pretreatment Effect on Pt/CeO2 Catalyst in the Selective Hydrodechlorination of Trichloroethylene. J. Phys. Chem. C 2010, 114, 17675–17682. [Google Scholar] [CrossRef]
- Kalmykov, P.A.; Magdalinova, N.A.; Klyuev, M.V. Liquid-Phase Hydrogenation of Halobenzenes in the Presence of Palladium-Containing Nanodiamonds. Pet. Chem. 2018, 58, 1206–1212. [Google Scholar] [CrossRef]
- Rajagopal, S.; Spatola, A.F. Mechanism of Palladium-Catalyzed Transfer Hydrogenolysis of Aryl Chlorides by Formate Salts. J. Org. Chem. 1995, 60, 1347–1355. [Google Scholar] [CrossRef]
- Aramendía, M.; Borau, V.; García, I.; Jimenez, C.; Marinas, J.; Urbano, F.J. Influence of the reaction conditions and catalytic properties on the liquid-phase hydrodebromination of bromobenzene over palladium supported catalysts: Activity and deactivation. Appl. Catal. B Environ. 1999, 20, 101–110. [Google Scholar] [CrossRef]
- Park, C.; Menini, C.; Valverde, J.; Keane, M.A. Carbon–Chlorine and Carbon–Bromine Bond Cleavage in the Catalytic Hydrodehalogenation of Halogenated Aromatics. J. Catal. 2002, 211, 451–463. [Google Scholar] [CrossRef]
- Dodson, D.A.; Rase, H.F. Methylene Chloride from Chloroform by Hydrochlorination. Ind. Eng. Chem. Prod. Res. Dev. 1978, 17, 236–240. [Google Scholar] [CrossRef]
- Ordóñez, S.; Diez, F.V.; Sastre, H. Hydrodechlorination of tetrachloroethylene over vanadium-modified Pt/Al2O3 catalysts. Catal. Lett. 2001, 72, 177–182. [Google Scholar] [CrossRef]
- Ordóñez, S.; Sastre, H.; Díez, F.V. Hydrodechlorination of tetrachloroethene over Pd/Al2O3: Influence of process conditions on catalyst performance and stability. Appl. Catal. B Environ. 2003, 40, 119–130. [Google Scholar] [CrossRef]
- Moon, D.J.; Chung, M.J.; Park, K.Y.; Hong, S.I. Deactivation of Pd catalysts in the hydrodechlorination of chloropentafluoroethane. Appl. Catal. A Gen. 1998, 168, 159–170. [Google Scholar] [CrossRef]
- Mori, T.; Yasuoka, T.; Morikawa, Y. Hydrodechlorination of 1,1,2-trichloro-1,2,2-trifluoroethane (CFC-113) over supported ruthenium and other noble metal catalysts. Catal. Today 2004, 88, 111–120. [Google Scholar] [CrossRef]
- Coq, B.; Hub, S.; Figueras, F.; Tournigant, D. Conversion under hydrogen of dichlorodifluoromethane over bimetallic palladium catalysts. Appl. Catal. A Gen. 1993, 101, 41–50. [Google Scholar] [CrossRef]
- Creyghton, E.; Burgers, M.; Jansen, J.; Van Bekkum, H. Vapour-phase hydrodehalogenation of chlorobenzene over platinum/H-BEA zeolite. Appl. Catal. A Gen. 1995, 128, 275–288. [Google Scholar] [CrossRef]
- Van De Sandt, E.J.; Wiersma, A.; Makkee, M.; Van Bekkum, H.; Moulijn, J.A. Selection of activated carbon for the selective hydrogenolysis of CCl2F2 (CFC-12) into CH2F2 (HFC-32) over palladium-supported catalysts. Appl. Catal. A Gen. 1998, 173, 161–173. [Google Scholar] [CrossRef]
- Mori, T.; Kubo, J.; Morikawa, Y. Hydrodechlorination of 1,1,1-trichloroethane over silica-supported palladium catalyst. Appl. Catal. A Gen. 2004, 271, 69–76. [Google Scholar] [CrossRef]
- Ordóñez, S.; Díaz, E.; Díez, F.V.; Sastre, H. Regeneration of Pd/Al2O3 catalysts used for tetrachloroethylene hydrodechlorination. React. Kinet. Catal. Lett. 2007, 90, 101–106. [Google Scholar] [CrossRef]
- Nieto-Márquez, A.; Valverde, J.; Keane, M.A. Catalytic growth of structured carbon from chloro-hydrocarbons. Appl. Catal. A Gen. 2007, 332, 237–246. [Google Scholar] [CrossRef]
- Patrick, J.; Barranco, R. Carbon deposits: Formation, Nature and Characterization. In Proceedings of the COMA/CRF Meeting, Scunthorpe, UK, 27 April 2006. [Google Scholar]
- Wiersma, A.; Van De Sandt, E.; Makkee, M.; Luteijn, C.; Van Bekkum, H.; Moulijn, J. Process for the selective hydrogenolysis of CCl2F2 (CFC-12) into CH2F2 (HFC-32). Catal. Today 1996, 27, 257–264. [Google Scholar] [CrossRef]
- Makkee, M.; Van De Sandt, E.; Wiersma, A.; Moulijn, J. Development of a satisfactory palladium on activated carbon catalyst for the selective hydrogenolysis of CCl2F2 (CFC-12) into CH2F2 (HFC-32). J. Mol. Catal. A Chem. 1998, 134, 191–200. [Google Scholar] [CrossRef]
- Kang, Y.; Han, Y.; Tian, M.; Huang, C.; Wang, C.; Lin, J.; Hou, B.; Su, Y.; Li, L.; Wang, J.; et al. Promoted methane conversion to syngas over Fe-based garnets via chemical looping. Appl. Catal. B Environ. 2020, 278, 119305. [Google Scholar] [CrossRef]
- Yasuda, S.; Osuga, R.; Kunitake, Y.; Kato, K.; Fukuoka, A.; Kobayashi, H.; Gao, M.; Hasegawa, J.-Y.; Manabe, R.; Shima, H.; et al. Zeolite-supported ultra-small nickel as catalyst for selective oxidation of methane to syngas. Commun. Chem. 2020, 3, 1–8. [Google Scholar] [CrossRef]
- Schwarz, H. Chemistry with Methane: Concepts Rather than Recipes. Angew. Chem. Int. Ed. 2011, 50, 10096–10115. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, J.; Wang, T. Enhancing the Acetylene Yield from Methane by Decoupling Oxidation and Pyrolysis Reactions: A Comparison with the Partial Oxidation Process. Ind. Eng. Chem. Res. 2016, 55, 8383–8394. [Google Scholar] [CrossRef]
- Dinh, D.K.; Lee, D.H.; Song, Y.-H.; Jo, S.; Kim, K.-T.; Iqbal, M.; Kang, H. Efficient methane-to-acetylene conversion using low-current arcs. RSC Adv. 2019, 9, 32403–32413. [Google Scholar] [CrossRef] [Green Version]
- Velazquez, J.C.; Leekumjorn, S.; Nguyen, Q.X.; Fang, Y.-L.; Heck, K.N.; Hopkins, G.D.; Reinhard, M.; Wong, M.S. Chloroform hydrodechlorination behavior of alumina-supported Pd and PdAu catalysts. AIChE J. 2013, 59, 4474–4482. [Google Scholar] [CrossRef]
- Xu, L.; Yao, X.; Khan, A.; Mavrikakis, M. Chloroform Hydrodechlorination over Palladium-Gold Catalysts: A First-Principles DFT Study. ChemCatChem 2016, 8, 1739–1746. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, S.; Zhi, Y.; Wei, Y.; Liu, Z. Methylcyclopentenyl cation mediated reaction route in methanol-to-olefins reaction over H-RUB-50 with small cavity. J. Energy Chem. 2020, 45, 25–30. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi, A.; Haghighi, M.; Aghamohammadi, S. Effect of calcination temperature and composition on the spray-dried microencapsulated nanostructured SAPO-34 with kaolin for methanol conversion to ethylene and propylene in fluidized bed reactor. Microporous Mesoporous Mater. 2020, 297, 110046. [Google Scholar] [CrossRef]
- Holmen, A. Direct conversion of methane to fuels and chemicals. Catal. Today 2009, 142, 2–8. [Google Scholar] [CrossRef]
- Salih, H.A.; Muraza, O.; Abussaud, B.; Al-Shammari, T.K.; Yokoi, T. Catalytic Enhancement of SAPO-34 for Methanol Conversion to Light Olefins Using in Situ Metal Incorporation. Ind. Eng. Chem. Res. 2018, 57, 6639–6646. [Google Scholar] [CrossRef]
- Wei, Y.; Zhang, D.; Liu, Z.; Su, B.-L. Methyl Halide to Olefins and Gasoline over Zeolites and SAPO Catalysts: A New Route of MTO and MTG. Chin. J. Catal. 2012, 33, 11–21. [Google Scholar] [CrossRef]
- Zheng, J.; Jin, D.; Liu, Z.; Zhu, K.; Zhou, X.; Yuan, W.-K. Synthesis of Nanosized SAPO-34 via an Azeotrope Evaporation and Dry Gel Conversion Route and Its Catalytic Performance in Chloromethane Conversion. Ind. Eng. Chem. Res. 2018, 57, 548–558. [Google Scholar] [CrossRef]
- Gosh, A.; Gosh, K.; Khanmamedova, A.; Mier, M.; Banke, J. Silicoaluminophosphate Catalyst for Chloromethane Conversion. Patent WO 2016/099775, 23 June 2016. [Google Scholar]
- Fickel, D.W.; Sabnis, K.D.; Li, L.; Kulkarni, N.; Winter, L.R.; Yan, B.; Chen, J.G. Chloromethane to olefins over H-SAPO-34: Probing the hydrocarbon pool mechanism. Appl. Catal. A Gen. 2016, 527, 146–151. [Google Scholar] [CrossRef]
- Zheng, J.; Zhang, W.; Liu, Z.; Huo, Q.; Zhu, K.; Zhou, X.; Yuan, W. Unraveling the non-classic crystallization of SAPO-34 in a dry gel system towards controlling meso-structure with the assistance of growth inhibitor: Growth mechanism, hierarchical structure control and catalytic properties. Microporous Mesoporous Mater. 2016, 225, 74–87. [Google Scholar] [CrossRef]
- Kong, L.-T.; Shen, B.; Jiang, Z.; Zhao, J.-G.; Liu, J.-C. Synthesis of SAPO-34 with the presence of additives and their catalytic performance in the transformation of chloromethane to olefins. React. Kinet. Mech. Catal. 2015, 114, 697–710. [Google Scholar] [CrossRef]
- Jiang, Z.; Shen, B.-X.; Zhao, J.-G.; Wang, L.; Kong, L.-T.; Xiao, W.-G. Enhancement of Catalytic Performances for the Conversion of Chloromethane to Light Olefins over SAPO-34 by Modification with Metal Chloride. Ind. Eng. Chem. Res. 2015, 54, 12293–12302. [Google Scholar] [CrossRef]
- Kong, L.-T.; Jiang, Z.; Zhao, J.; Liu, J.; Shen, B. The Synthesis of Hierarchical SAPO-34 and its Enhanced Catalytic Performance in Chloromethane Conversion to Light Olefins. Catal. Lett. 2014, 144, 1609–1616. [Google Scholar] [CrossRef]
- Olsbye, U.; Saure, O.V.; Muddada, N.B.; Bordiga, S.; Lamberti, C.; Nilsen, M.H.; Lillerud, K.P.; Svelle, S. Methane conversion to light olefins—How does the methyl halide route differ from the methanol to olefins (MTO) route? Catal. Today 2011, 171, 211–220. [Google Scholar] [CrossRef]
- Shin, Y.H.; Kweon, S.; Park, M.B.; Chae, H.-J. Comparative study of CHA- and AEI-type zeolytic catalysts for the conversion of chloromethane into light olefins. Korean J. Chem. Eng. 2018, 35, 1433–1440. [Google Scholar] [CrossRef]
- Gamero, M.; Aguayo, A.T.; Ateka, A.; Pérez-Uriarte, P.; Gayubo, A.G.; Bilbao, J. Role of Shape Selectivity and Catalyst Acidity in the Transformation of Chloromethane into Light Olefins. Ind. Eng. Chem. Res. 2015, 54, 7822–7832. [Google Scholar] [CrossRef]
- Kong, L.-T.; Shen, B.-X.; Zhao, J.-G.; Liu, J.-C. Comparative Study on the Chloromethane to Olefins Reaction over SAPO-34 and HZSM-22. Ind. Eng. Chem. Res. 2014, 53, 16324–16331. [Google Scholar] [CrossRef]
- Xu, T.; Liu, H.; Zhao, Q.; Cen, S.; Du, L.; Tang, Q. Conversion of chloromethane to propylene over fluoride-treated H-ZSM-35 zeolite catalysts. Catal. Commun. 2019, 119, 96–100. [Google Scholar] [CrossRef]
- Huang, J.; Wang, W.; Fei, Z.; Liu, Q.; Chen, X.; Zhang, Z.; Tang, J.; Cui, M.; Qiao, X. Enhanced Light Olefin Production in Chloromethane Coupling over Mg/Ca Modified Durable HZSM-5 Catalyst. Ind. Eng. Chem. Res. 2019, 58, 5131–5139. [Google Scholar] [CrossRef]
- Wen, D.; Liu, Q.; Fei, Z.; Yang, Y.; Zhang, Z.; Chen, X.; Tang, J.; Cui, M.; Qiao, X. Organosilane-Assisted Synthesis of Hierarchical Porous ZSM-5 Zeolite as a Durable Catalyst for Light-Olefins Production from Chloromethane. Ind. Eng. Chem. Res. 2018, 57, 446–455. [Google Scholar] [CrossRef]
- Gamero, M.; Valle, B.; Castano, P.; Aguayo, A.T.; Bilbao, J. Reaction network of the chloromethane conversion into light olefins using a HZSM-5 zeolite catalyst. J. Ind. Eng. Chem. 2018, 61, 427–436. [Google Scholar] [CrossRef]
- Liu, Q.; Wen, D.; Yang, Y.; Fei, Z.; Zhang, Z.; Chen, X.; Tang, J.; Cui, M.; Qiao, X. Enhanced catalytic performance for light-olefins production from chloromethane over hierarchical porous ZSM-5 zeolite synthesized by a growth-inhibition strategy. Appl. Surf. Sci. 2018, 435, 945–952. [Google Scholar] [CrossRef]
- Gamero, M.; Valle, B.; Gayubo, A.G.; Castano, P.; Aguayo, A.T.; Bilbao, J. Kinetic Model for the Conversion of Chloromethane into Hydrocarbons over a HZSM-5 Zeolite Catalyst. Ind. Eng. Chem. Res. 2018, 57, 908–919. [Google Scholar] [CrossRef]
- Ibáñez, M.; Gamero, M.; Ruiz-Martínez, J.J.; Weckhuysen, B.M.; Aguayo, A.T.; Bilbao, J.; Castano, P. Simultaneous coking and dealumination of zeolite H-ZSM-5 during the transformation of chloromethane into olefins. Catal. Sci. Technol. 2016, 6, 296–306. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Song, H.; Deng, W.; Zhang, Q.; Wang, Y. Catalytic conversion of methyl chloride to lower olefins over modified H-ZSM-34. Chin. J. Catal. 2013, 34, 2047–2056. [Google Scholar] [CrossRef]
- Xu, T.; Zhang, Q.; Song, H.; Wang, Y. Fluoride-treated H-ZSM-5 as a highly selective and stable catalyst for the production of propylene from methyl halides. J. Catal. 2012, 295, 232–241. [Google Scholar] [CrossRef]
- He, J.; Xu, T.; Wang, Z.; Zhang, Q.; Deng, W.; Wang, Y. Transformation of Methane to Propylene: A Two-Step Reaction Route Catalyzed by Modified CeO2 Nanocrystals and Zeolites. Angew. Chem. Int. Ed. 2012, 51, 2438–2442. [Google Scholar] [CrossRef]
- Lokteva, E.S.; Simagina, V.I.; Golubina, E.V.; Stoyanova, I.V.; Lunin, V.V. Formation of C1–C5 Hydrocarbons from CCl4 in the Presence of Carbon-Supported Palladium Catalysts. Kinet. Catal. 2000, 41, 776–781. [Google Scholar] [CrossRef]
- Mori, T.; Hirose, K.; Kikuchi, T.; Kubo, J.; Morikawa, Y. Formation of Higher Hydrocarbons from Chloromethanes via Hydrodechlorination over Pd/SiO2 Catalyst. J. Jpn. Pet. Inst. 2002, 45, 256–259. [Google Scholar] [CrossRef] [Green Version]
- Elola, A.; Díaz, E.; Ordóñez, S.; Díaz, E. A New Procedure for the Treatment of Organochlorinated Off-Gases Combining Adsorption and Catalytic Hydrodechlorination. Environ. Sci. Technol. 2009, 43, 1999–2004. [Google Scholar] [CrossRef] [PubMed]













| Kinetic Model | Kinetic Expression | Equation | Ref. |
|---|---|---|---|
| Pseudo-first order, catalytic fixed-bed reactor | (2) | [38] | |
| LH model | (3) | [161] | |
| LH model with DCM adsorption control, catalytic fixed-bed reactor | (4) | [38] | |
| (5) | |||
| (6) | |||
| (7) | |||
| LH model with adsorption control, catalytic fixed-bed reactor | (8) | [43] | |
| LH model with chemical reaction control, catalytic fixed-bed reactor | (9) | ||
| LH model with desorption control, catalytic fixed-bed reactor | (10) |
| Catalyst | Method | Ea (kJ·mol−1) | Ref. | |
|---|---|---|---|---|
| MCM | 5% Pd/C | DFT | 62.0 | [161] |
| 5% Pd/C | Arrhenius Arrhenius | 64.0 | [161] | |
| - | 184.5 * | [165] | ||
| DCM | 5% Pd/C | DFT | 56.0 | [161] |
| 5% Pd/C | Arrhenius | 58.0 | [161] | |
| 1% Pd/C | 50.9 | [1] | ||
| 1% Pd/C | 50.0 | [43] | ||
| 0.5% Pd/C | 52.3 | [38] | ||
| 1% Pt/C | 52.5 | [1] | ||
| 1% Pt/C | 49.0 | [43] | ||
| 1% Rh/C | 50.3 | [1] | ||
| 1% Rh/C | 39.0 | [43] | ||
| 1% Ru/C | 44.4 | [1] | ||
| 1% Ru/C | 64.0 | [43] | ||
| 0.3% Pd/γ-Al2O3 | 130.2 | [24] | ||
| 0.4% Pd/γ-Al2O3 | 114.7 | [24] | ||
| 0.6% Pd/γ-Al2O3 | 92.5 | [24] | ||
| 0.3% Pd/TiO2 | 130.5 | [24] | ||
| 0.4% Pd/TiO2 | 129.3 | [24] | ||
| 0.7% Pd/TiO2 | 97.5 | [24] | ||
| - | 237.6 * | [165] | ||
| TCM | 5% Pd/C | DFT | 46.0 | [161] |
| 5% Pd/C | Arrhenius | 54.0 | [161] | |
| 1% Pd/C | 32.4 | [1] | ||
| 1% Pd/C | 52.0 | [43] | ||
| 1% Pt/C | 32.4 | [1] | ||
| 1% Pt/C | 29.0 | [43] | ||
| 1% Rh/C | 17.1 | [1] | ||
| 1% Ru/C | 41.4 | [1] | ||
| - | 243.5 * | [165] | ||
| Pt/K6 | Akaike’s Information Criteria | 18–60 | [47] | |
| Pd/K6 | ||||
| PtPd/K6(30) | ||||
| PtPd/K6(40) | ||||
| PtPd/K6(50) | ||||
| PtPd/SiC@K6(40) | ||||
| PtPd/SiC | ||||
| TTCM | 5% Pd/C | DFT | 40.0 | [161] |
| 5% Pd/C | Arrhenius | 48.0 | [161] | |
| - | 256.5 * | [165] | ||
| 3% Pd/MgF2-SolGel | 38.3 | [53] | ||
| 3% Pd/MgF2-Carb | 42.1 | [53] | ||
| 3% Pd/MgO-SolGel | 56.7 | [53] | ||
| 3% Pd/MgO-Carb | 48.5 | [53] | ||
| 1% Pd/MgF2-Carb | 46.3 | [53] | ||
| 1% Pd/MgO-Carb | 65.0 | [53] | ||
| 1.5% Pt/Al2O3 | 57.7 | [55] | ||
| 1.5% Pt/SiO2 | 53.8 | [55] | ||
| 2.8% Pd/Sibunit carbon | 51.9 | [60] | ||
| 6.2% Pd60-Au40/Sibunit carbon | 59.9 | [60] | ||
| 1% Pt/Al2O3 | 56.0 | [65] | ||
| 1% Pt95-Au5/Al2O3 | 22.9 | [65] | ||
| 1.4% Pt70-Au30/Al2O3 | 26.4 | [65] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Otero, J.A.; Martin-Martinez, M.; Rodriguez-Franco, D.; Rodriguez, J.J.; Gómez-Sainero, L.M. Understanding Hydrodechlorination of Chloromethanes. Past and Future of the Technology. Catalysts 2020, 10, 1462. https://doi.org/10.3390/catal10121462
Liu S, Otero JA, Martin-Martinez M, Rodriguez-Franco D, Rodriguez JJ, Gómez-Sainero LM. Understanding Hydrodechlorination of Chloromethanes. Past and Future of the Technology. Catalysts. 2020; 10(12):1462. https://doi.org/10.3390/catal10121462
Chicago/Turabian StyleLiu, Sichen, Javier A. Otero, Maria Martin-Martinez, Daniel Rodriguez-Franco, Juan J. Rodriguez, and Luisa M. Gómez-Sainero. 2020. "Understanding Hydrodechlorination of Chloromethanes. Past and Future of the Technology" Catalysts 10, no. 12: 1462. https://doi.org/10.3390/catal10121462