Acetonitrile’s Effect on the Efficiency of Ethanol Electrooxidation at a Polycrystalline Pt Electrode in Relation to pH-Dependent Fuel Cell Applications

 ,
,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Electrooxidation of Ethanol by Cyclic Voltammetry in the Absence and Presence of Acetonitrile
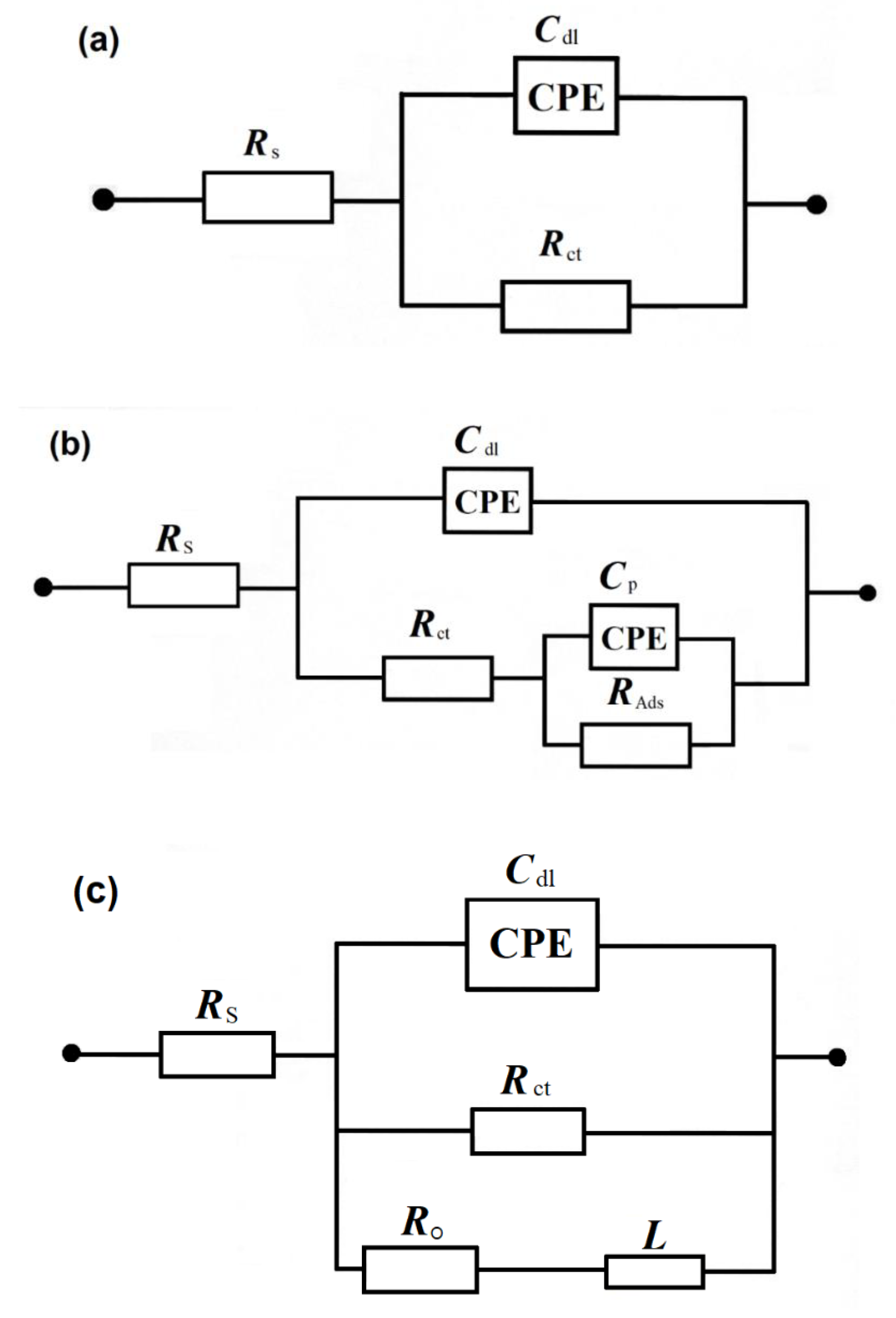
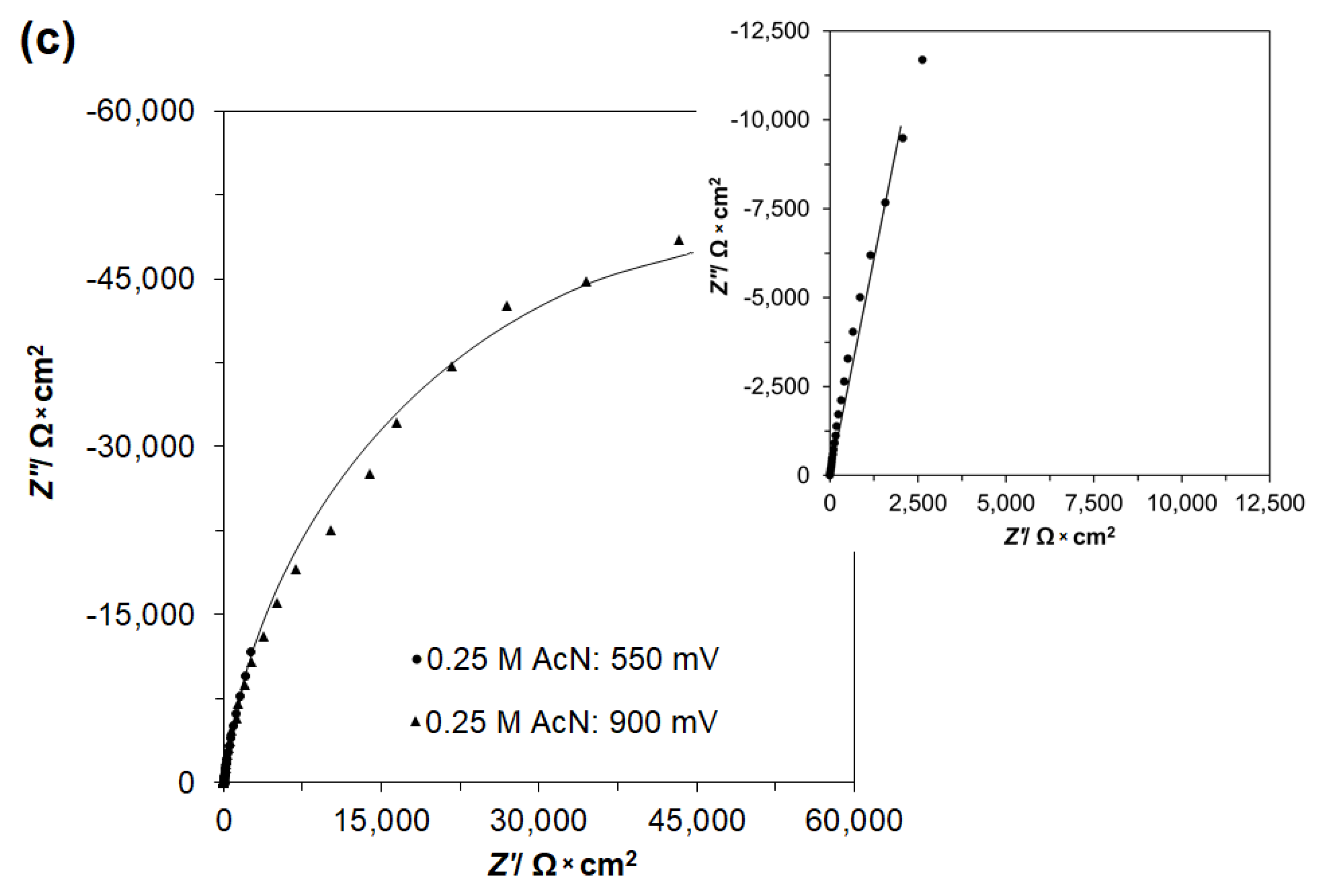
2.2. Influence of AcN on Ethanol Electrooxidation by a.c. Impedance Spectroscopy
2.3. Spectrophotometric UV-VIS Analysis of Ethanol Oxidation Products
3. Materials and Methods
3.1. Solutions and Solutes
3.2. Electrochemical Cell and Electrodes
3.3. Electrochemical and other Equipment
3.4. Assessment of Acetaldehyde and Acetate Contents upon Progress of the Ethanol Electrooxidation Reaction
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rousseau, C.; Countaneceau, C.; Lamy, C.; Leger, J.M. Direct ethanol fuel cell (DEFC): Electrical performances and reaction products distribution under operating conditions with different platinum-based anodes. J. Power Sources 2006, 158, 18–24. [Google Scholar] [CrossRef]
- Song, S.Q.; Zhou, W.J.; Zhou, Z.H.; Jiang, L.H.; Sun, G.Q.; Xin, Q.; Leontidis, V.; Kontou, S.; Tsiakaras, P. Direct ethanol PEM fuel cells: The case of platinum based anodes. Int. J. Hydrog. Energy 2005, 30, 995–1001. [Google Scholar] [CrossRef]
- Abd-El-Latif, A.A.; Mostafa, E.; Huxter, S.; Attard, G.; Baltrischat, H. Electrooxidation of ethanol at polycrystalline and platinum stepped single crystals: A study by differential electrochemical mass spectrometry. Electrochim. Acta 2010, 55, 7951–7960. [Google Scholar] [CrossRef]
- Han, S.B.; Song, Y.J.; Lee, J.M.; Kim, J.Y.; Park, K.W. Platinum nanocube catalysts for methanol and ethanol electrooxidation. Electrochem. Commun. 2008, 10, 1044–1047. [Google Scholar] [CrossRef]
- Shao, M.H.; Adzic, R.R. Electrooxidation of ethanol on a Pt electrode in acid solutions: In situ ATR-SEIRAS study. Electrochim. Acta 2005, 50, 2415–2422. [Google Scholar] [CrossRef]
- Gupta, S.S.; Singh, S.; Datta, J. Temperature effect on the electrode kinetics of ethanol electro-oxidation on Sn modified Pt catalyst through voltammetry and impedance spectroscopy. Mater. Chem. Phys. 2010, 120, 682–690. [Google Scholar] [CrossRef]
- Switzer, E.E.; Olson, T.S.; Datye, A.K.; Atanassov, P.; Hibbs, M.R.; Cornelius, C.J. Templated Pt–Sn electrocatalysts for ethanol, methanol and CO oxidation in alkaline media. Electrochim. Acta 2009, 54, 989–995. [Google Scholar] [CrossRef]
- Mahapatra, S.S.; Dutta, A.; Datta, J. Temperature effect on the electrode kinetics of ethanol oxidation on Pd modified Pt electrodes and the estimation of intermediates formed in alkali medium. Electrochim. Acta 2010, 55, 9097–9104. [Google Scholar] [CrossRef]
- Hassan, K.M.; Hathoot, A.A.; Maher, R.; Azzem, M.A. Electrocatalytic oxidation of ethanol at Pd, Pt, Pd/Pt and Pt/Pd nano particles supported on poly 1,8-diaminonaphthalene film in alkaline medium. RSC Adv. 2018, 8, 15417–15426. [Google Scholar] [CrossRef] [Green Version]
- Abdel, R.M.H. Nickel Oxide Nanoparticles Supported on Graphitized Carbon for Ethanol Oxidation in NaOH Solution. J. Clust. Sci. 2019, 30, 1003–1016. [Google Scholar] [CrossRef]
- Monyoncho, E.A.; Woo, T.K.; Baranova, E.A. Ethanol electrooxidation reaction in alkaline media for direct ethanol fuel cells. In SPR-Electrochemistry; Banks, C., McIntosh, S., Eds.; Royal Society of Chemistry: London, UK, 2019; Volume 15, pp. 1–57. [Google Scholar]
- Gupta, S.S.; Datta, J. A Comparative Study on Ethanol Oxidation Behavior at Pt and PtRh Electrodeposits. J. Electroanal. Chem. 2006, 594, 65–72. [Google Scholar] [CrossRef]
- Xia, X.H.; Liess, H.D.; Iwasita, T. Early stages in the oxidation of ethanol at low index single crystal platinum electrodes. J. Electroanal. Chem. 1997, 437, 233–240. [Google Scholar] [CrossRef]
- Gomes, J.F.; Busson, B.; Tadjeddine, A.; Tremiliosi-Filho, G. Ethanol electro-oxidation over Pt(h k l): Comparative study on the reaction intermediates probed by FTIR and SFG spectroscopies. Electrochim. Acta 2008, 53, 6899–6905. [Google Scholar] [CrossRef]
- El-Shafei, A.A.; Eiswirth, M. Electrochemical activity of Sn-modified Pt single crystal electrodes for ethanol oxidation. Surf. Sci. 2010, 604, 862–867. [Google Scholar] [CrossRef]
- Pierozynski, B. Kinetic Aspects of Ethanol Electrooxidation on Catalytic Surfaces of Pt in 0.5 M H2SO4. Int. J. Electrochem. Sci. 2012, 7, 3327–3338. [Google Scholar]
- Pierozynski, B. On the Ethanol Electrooxidation Reaction on Catalytic Surfaces of Pt in 0.1 M NaOH. Int. J. Electrochem. Sci. 2012, 7, 4261–4271. [Google Scholar]
- Fujiwara, N.; Siroma, Z.; Yamazaki, S.; Ioroi, T.; Senoh, H.; Yasuda, K. Direct ethanol fuel cells using an ion exchange membrane. J. Power Sources 2008, 185, 621–626. [Google Scholar] [CrossRef]
- Pierozynski, B. Electrooxidation of Ethanol on PtRh and PtRu Surfaces Studied in 0.5 M H2SO4: Relation to the Behaviour at Polycrystalline Pt. Int. J. Electrochem. Sci. 2012, 7, 4488–4497. [Google Scholar]
- Pierozynski, B. Ethanol Electrooxidation on PtRh and PtRu Catalytic Surfaces in 0.1 M NaOH. Int. J. Electrochem. Sci. 2012, 7, 6406–6416. [Google Scholar]
- Simoes, F.C.; dos Ainos, D.M.; Vigier, F.; Leger, J.M.; Hahn, F.; Coutanceau, C.; Gonzalez, E.R.; Tremiliosi-Filho, G.; de Andrade, A.R.; Olivi, P.; et al. Electroactivity of tin modified platinum electrodes for ethanol electrooxidation. J. Power Sources 2007, 167, 1–10. [Google Scholar] [CrossRef]
- Luo, Z.; Lu, J.; Flox, C.; Nafria, R.; Genc, A.; Arbiol, J.; Llorca, J.; Ibanez, M.; Morante, J.R.; Cabot, A. Pd2Sn [010] nanorods as a highly active and stable ethanol oxidation catalyst. J. Mater. Chem. A 2016, 4, 16706–16713. [Google Scholar] [CrossRef]
- Guo, J.; Chen, R.; Zhu, F.-C.; Sun, S.-G.; Villullas, H.M. New understanding of ethanol oxidation reaction mechanism on Pd/C and Pd2Ru/C catalysts in alkaline direct ethanol fuel cells. Appl. Catal. B Environ. 2018, 224, 602–611. [Google Scholar] [CrossRef] [Green Version]
- Delpeuch, A.B.; Maillard, F.; Chatenet, M.; Soudant, P.; Cremers, C. Ethanol oxidation reaction (EOR) investigation on Pt/C, Rh/C, and Pt-based bi- and tri-metallic electrocatalysts: A DEMS and in situ FTIR study. Appl. Catal. B Environ. 2015, 181, 672–680. [Google Scholar] [CrossRef]
- Yi, J.; Lee, W.H.; Choi, C.H.; Lee, Y.; Park, K.S.; Min, B.K.; Hwang, Y.J.; Oh, H.-S. Effect of Pt introduced on Ru-based electrocatalyst for oxygen evolution activity and stability. Electrochem. Commun. 2019, 104, 1–5. [Google Scholar] [CrossRef]
- Su, C.; Yang, T.; Zhou, W.; Wang, W.; Xu, X.; Shao, Z. Pt/C-LiCoO2 composites with ultralow Pt loadings as synergistic bifunctional electrocatalysts for oxygen reduction and evolution reaction. J. Matter Chem. A 2016, 4, 4516–4524. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, K.; Marinkovic, N.; Isaacs, H.S.; Adzic, R.R. Synchrotron-Based in situ characterization of Carbon-supported platinum and platinum monolayer electrocatalysts. ACS Catal. 2016, 6, 69–76. [Google Scholar] [CrossRef]
- Da Silva, G.C.; Fernandes, M.R.; Ticianelli, E.A. Activity and Stability of Pt/IrO2 Bifunctional Materials as Catalysts for the Oxygen Evolution/Reduction Reactions. ACS Catal. 2018, 8, 2081–2092. [Google Scholar] [CrossRef]
- Pierozynski, B.; Morin, S.; Conway, B.E. Influence of adsorption of guanidonium cations on H upd at Pt(hkl) surfaces: Lattice-specific anion-mimetic effects. J. Electroanal. Chem. 1999, 467, 30–42. [Google Scholar] [CrossRef]
- Wang, Y.; Zou, S.; Cai, W.B. Recent advances on electro-oxidation of ethanol on Pt- and Pd-based catalysts: From reaction mechanisms to catalytic materials. Catalysts 2015, 5, 1507–1534. [Google Scholar] [CrossRef]
- Hitmi, H.; Belgsir, E.; Léger, J.-M.; Lamy, C.; Lezna, R. A kinetic analysis of the electro-oxidation of ethanol at a platinum electrode in acid medium. Electrochim. Acta 1994, 39, 407–415. [Google Scholar] [CrossRef]
- Reshetenko, T.V.; St-Pierre, J. Study of the acetonitrile poisoning of platinum cathodes on proton exchange membrane fuel cell spatial performance using a segmented cell system. J. Power Sources 2015, 293, 929–940. [Google Scholar] [CrossRef] [Green Version]
- Briega-Martos, V.; Costa-Figueiredo, M.; Orts, J.M.; Rodes, A.; Koper, M.T.M.; Herrero, E.; Feliu, J.M. Acetonitrile adsorption on Pt single-crystal electrodes and its effect on oxygen reduction reaction in acidic and alkaline aqueous solutions. J. Phys. Chem. C 2019, 123, 2300–2313. [Google Scholar] [CrossRef] [Green Version]
- Ahlberg, E.; Friel, M. The cathodic polarization of iron in acidic acetonitrile-water solutions. Electrochim. Acta 1989, 34, 771–780. [Google Scholar] [CrossRef]
- Ledezma-Yanez, I.; Diaz-Morales, O.; Figueiredo, M.C.; Koper, M.T.M. Hydrogen oxidation and hydrogen evolution on a platinum electrode in acetonitrile. ChemElectroChem 2015, 2, 1612–1622. [Google Scholar] [CrossRef]
- Smiljanić, M.L.; Srejić, I.L.; Marinović, V.M.; Rakočević, Z.L.; Štrbac, S.B. Inhibiting effect of acetonitrile on oxygen reduction on polycrystalline Pt electrode in sodium chloride solution. Hem. Ind. 2012, 66, 327–333. [Google Scholar] [CrossRef] [Green Version]
- Srejić, I.; Smiljanić, M.; Rakočević, Z.; Štrbac, S. Oxygen reduction on polycrystalline Pt and Au electrodes in perchloric acid solution in the presence of acetonitrile. Int. J. Electrochem. Sci. 2011, 6, 3344–3354. [Google Scholar]
- Mikolajczyk, T.; Luba, M.; Pierozynski, B.; Smoczynski, L. A detrimental effect of acetonitrile on the kinetics of underpotentially deposited hydrogen and hydrogen evolution reaction, examined on Pt electrode in H2SO4 and NaOH solutions. Catalysts 2020, 10, 625. [Google Scholar] [CrossRef]
- Ma, B.; Dong, L. Electrochemical impedance analysis of methanol oxidation on carbon nanotube-supported Pt and Pt-Ru nanoparticles. J. Solid State Electrochem. 2013, 17, 2783–2788. [Google Scholar] [CrossRef]
- Danaee, I.; Jafarian, M.; Forouzandeh, F.; Gobal, F.; Mahjani, M.G. Electrochemical impedance studies of methanol oxidation on GC/Ni and GC/NiCu electrode. Int. J. Hydrog. Energy 2009, 34, 859–869. [Google Scholar] [CrossRef]
- Bommersbach, P.; Mohamedi, M.; Guay, D. Electro-oxidation of Ethanol at Sputter-Deposited Platinum-Tin Catalysts. J. Electrochem. Soc. 2007, 154, B876–B882. [Google Scholar] [CrossRef]
- Benetton, X.D.; Navarro-Avila, S.G.; Carrera-Figueiras, C. Electrochemical Evaluation of Ti/TiO2-polyaniline Anodes for Microbial Fuel Cells using Hypersaline Microbial Consortia for Synthetic-wastewater Treatment. J. New Mater. Electrochem. Syst. 2010, 13, 1–6. [Google Scholar]
- Han, L.; Ju, H.; Xu, Y. Ethanol electro-oxidation: Cyclic voltammetry, electrochemical impedance spectroscopy and galvanostatic oscillation. Int. J. Hydrog. Energy 2012, 37, 15156–15163. [Google Scholar] [CrossRef]
- Lasia, A.; Rami, A. Kinetics of hydrogen evolution on Ni-Al alloy electrodes. J. Appl. Electrochem. 1992, 22, 376–382. [Google Scholar]
- Chen, L.; Lasia, A. Study of the Kinetics of Hydrogen Evolution Reaction on Nickel-Zinc Alloy Electrodes. J. Electrochem. Soc. 1991, 138, 3321–3328. [Google Scholar] [CrossRef]
- Pajkossy, T. Impedance of rough capacitive electrodes. J. Electroanal. Chem. 1994, 364, 111–125. [Google Scholar] [CrossRef]
- Conway, B.E. Impedance Spectroscopy: Theory, Experiment, and Applications; Barsoukov, E., Macdonald, J.R., Eds.; Wiley: Hoboken, NJ, USA, 2005; p. 494. [Google Scholar]
- Pell, W.G.; Zolfaghari, A.; Conway, B.E. Capacitance of the double-layer at polycrystalline Pt electrodes bearing a surface-oxide film. J. Electroanal. Chem. 2002, 532, 13–23. [Google Scholar] [CrossRef]
- Briega-Martos, V.; Solla-Gullon, J.; Koper, M.T.M.; Herrero, E. Electrocatalytic enhancement of formic acid oxidation reaction by acetonitrile on well-defined platinum surfaces. Electrochim. Acta 2019, 295, 835–845. [Google Scholar] [CrossRef]
- Macdonald, J.R. Impedance Spectroscopy: Emphasizing Solid Materials and Systems; Wiley: New York, NY, USA, 1987. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| E/mV | Rct/Ω × cm2 | Cdl/µF × cm−2 | RAds/Ω × cm2 | Cp/ µF × cm−2 |
|---|---|---|---|---|
| 0.5 M H2SO4 + 0.25 M C2H5OH | ||||
| 550 | 1524 ± 34 | 29.3 ± 0.9 | - | - |
| 650 | 2831 ± 243 | 53.2 ± 0.4 | −3371 ± 263 | 69.1 ± 3.1 |
| 700 | 948 ± 60 | 37.9 ± 0.5 | −1055 ± 37 | 97.0 ± 5.2 |
| 800 | −439 ± 11 | 31.1 ± 0.5 | −301 ± 1 | 60.2 ± 1.1 |
| 900 | −1043 ± 83 | 36.9 ± 0.6 | −854 ± 6 | 48.8 ± 0.6 |
| 0.5 M H2SO4 + 0.25 M C2H5OH + 1.85 × 10−5 M AcN | ||||
| 550 | 1587 ± 39 | 19.6 ± 0.4 | - | - |
| 650 | 3852 ± 505 | 42.6 ± 0.3 | −7697 ± 387 | 61.1 ± 1.3 |
| 700 | 2294 ± 219 | 33.3 ± 0.4 | −2188 ± 56 | 67.7 ± 1.6 |
| 800 | −451 ± 17 | 22.9 ± 0.4 | −442 ± 3 | 22.6 ± 0.6 |
| 900 | −1408 ± 112 | 28.7 ± 0.4 | −1021 ± 7 | 40.3 ± 0.8 |
| 0.5 M H2SO4 + 0.25 M C2H5OH + 1.85 × 10−4 M AcN | ||||
| 550 | 1330 ± 14 | 42.1 ± 0.5 | - | - |
| 650 | 3000 ± 131 | 54.6 ± 1.0 | - | - |
| 700 | 1664 ± 26 | 32.7 ± 0.5 | −1000 ± 111 | 664.1 ± 46.7 |
| 800 | −2506 ± 459 | 20.8 ± 0.4 | −379 ± 4 | 108.2 ± 1.7 |
| 900 | −784 ± 124 | 35.6 ± 0.5 | −214 ± 4 | 254.7 ± 11.0 |
| 0.5 M H2SO4 + 0.25 M C2H5OH + 1.85 × 10−3 M AcN | ||||
| 550 | 2460 ± 61 | 71.6 ± 1.4 | - | - |
| 650 | 3028 ± 62 | 50.5 ± 0.4 | - | - |
| 700 | 4192 ± 167 | 37.7 ± 0.4 | −12,716 ± 2406 | 101.4 ± 9.0 |
| 800 | 9168 ± 2328 | 17.2 ± 0.2 | −20,223 ± 1957 | 27.8 ± 1.2 |
| 900 | −5929 ± 1895 | 28.4 ± 0.7 | −5936 ± 105 | 30.2 ± 0.5 |
| 0.5 M H2SO4 + 0.25 M C2H5OH + 0.25 M AcN | ||||
| 550 | 131,404 ± 11,769 | 57.9 ± 0.5 | - | - |
| 900 | 52,759 ± 3174 | 43.6 ± 0.8 | - | - |
| E/mV | Rct/Ω × cm2 | Cdl/µF × cm−2 | RO/ Ω × cm2 | L/H |
|---|---|---|---|---|
| 0.1 M NaOH + 0.25 M C2H5OH | ||||
| 600 | 2211 ± 91 | 26.4 ± 1.1 | 604 ± 47 | 247 ± 10 |
| 675 | 2842 ± 257 | 27.5 ± 1.9 | 494 ± 38 | 155 ± 8 |
| 800 | 121 ± 1 | 69.3 ± 0.3 | - | - |
| 0.1 M NaOH + 0.25 M C2H5OH + 1.85 × 10−5 M AcN | ||||
| 600 | 1648 ± 53 | 28.0 ± 1.2 | 713 ± 55 | 328 ± 16 |
| 675 | 1428 ± 102 | 30.4 ± 2.9 | 482 ± 100 | 264 ± 26 |
| 800 | 141 ± 1 | 81.4 ± 1.8 | - | - |
| 0.1 M NaOH + 0.25 M C2H5OH + 1.85 × 10−4 M AcN | ||||
| 600 | 1558 ± 61 | 27.9 ± 1.4 | 1123 ± 95 | 315 ± 25 |
| 675 | 659 ± 18 | 26.6 ± 1.7 | - | - |
| 800 | 101 ± 1 | 85.0 ± 1.7 | - | - |
| 0.1 M NaOH + 0.25 M C2H5OH + 1.85 × 10−3 M AcN | ||||
| 600 | 598 ± 7 | 28.5 ± 0.9 | 805 ± 70 | 380 ± 22 |
| 675 | 215 ± 2 | 34.2 ± 1.2 | - | - |
| 800 | 61 ± 1 | 109.4 ± 5.4 | - | - |
| 0.1 M NaOH + 0.25 M C2H5OH + 0.25 M AcN | ||||
| 600 | 1770 ± 13 | 19.0 ± 0.3 | - | - |
| 800 | 16,046 ± 461 | 24.3 ± 0.3 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pierożynski, B.; Mikołajczyk, T.; Łuba, M.; Wojtacha, P.; Smoczyński, L. Acetonitrile’s Effect on the Efficiency of Ethanol Electrooxidation at a Polycrystalline Pt Electrode in Relation to pH-Dependent Fuel Cell Applications. Catalysts 2020, 10, 1286. https://doi.org/10.3390/catal10111286
Pierożynski B, Mikołajczyk T, Łuba M, Wojtacha P, Smoczyński L. Acetonitrile’s Effect on the Efficiency of Ethanol Electrooxidation at a Polycrystalline Pt Electrode in Relation to pH-Dependent Fuel Cell Applications. Catalysts. 2020; 10(11):1286. https://doi.org/10.3390/catal10111286
Chicago/Turabian StylePierożynski, Bogusław, Tomasz Mikołajczyk, Mateusz Łuba, Paweł Wojtacha, and Lech Smoczyński. 2020. "Acetonitrile’s Effect on the Efficiency of Ethanol Electrooxidation at a Polycrystalline Pt Electrode in Relation to pH-Dependent Fuel Cell Applications" Catalysts 10, no. 11: 1286. https://doi.org/10.3390/catal10111286