A Scalable High-Throughput Deposition and Screening Setup Relevant to Industrial Electrocatalysis

 ,
,
Abstract
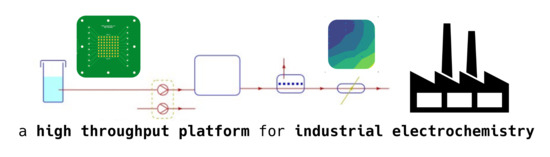
:
1. Introduction
2. Results
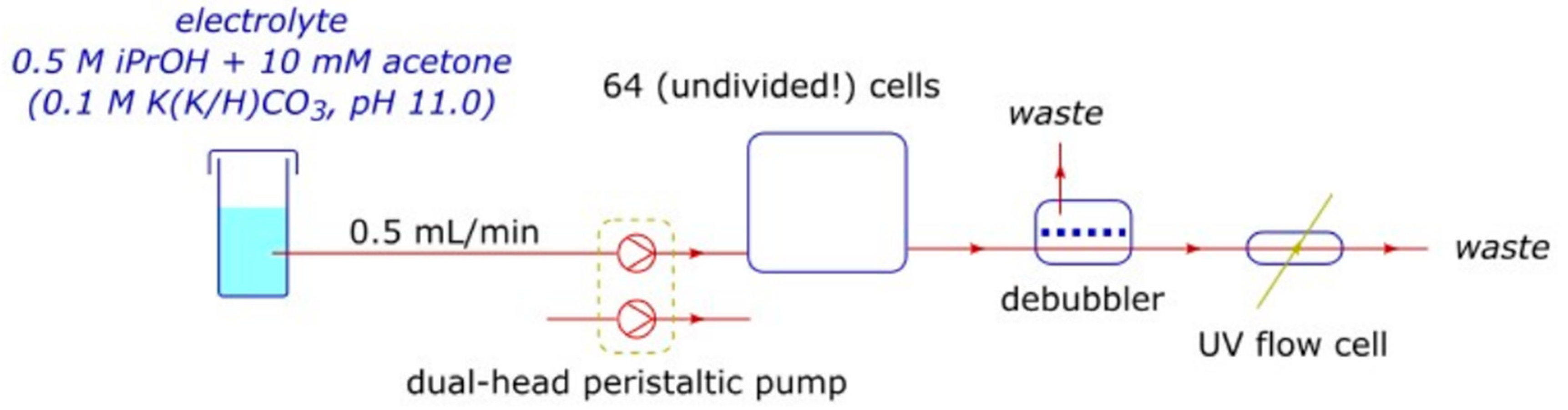
2.1. The High-Throughput Setup
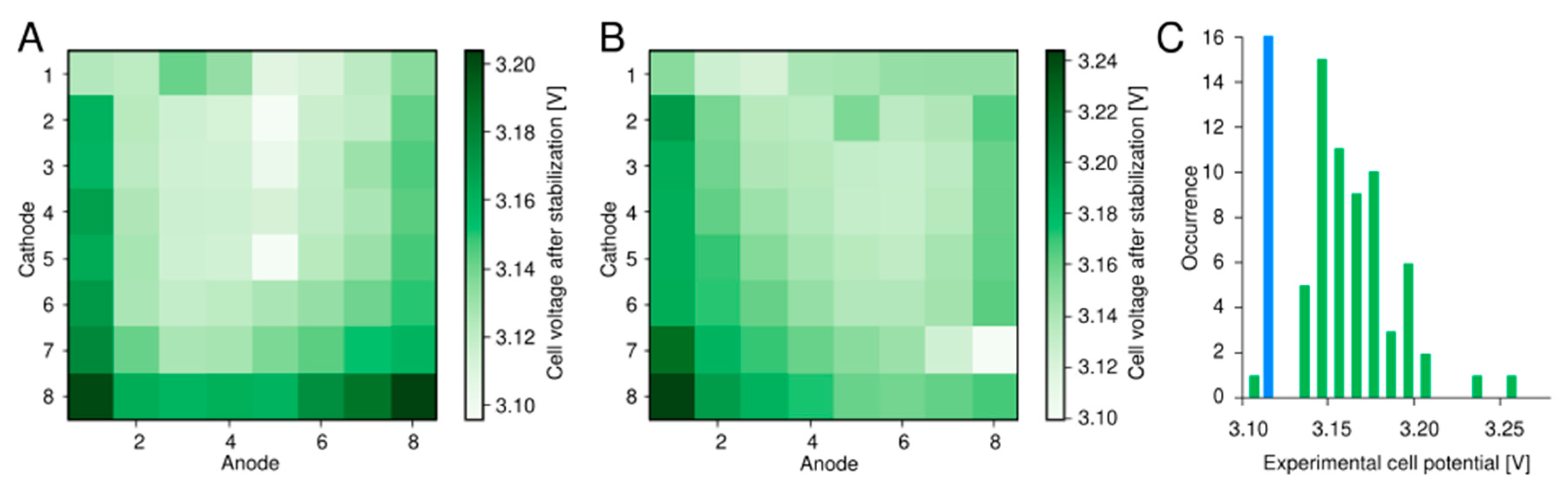
2.2. Setup Validation Using PEM Water Electrolysis
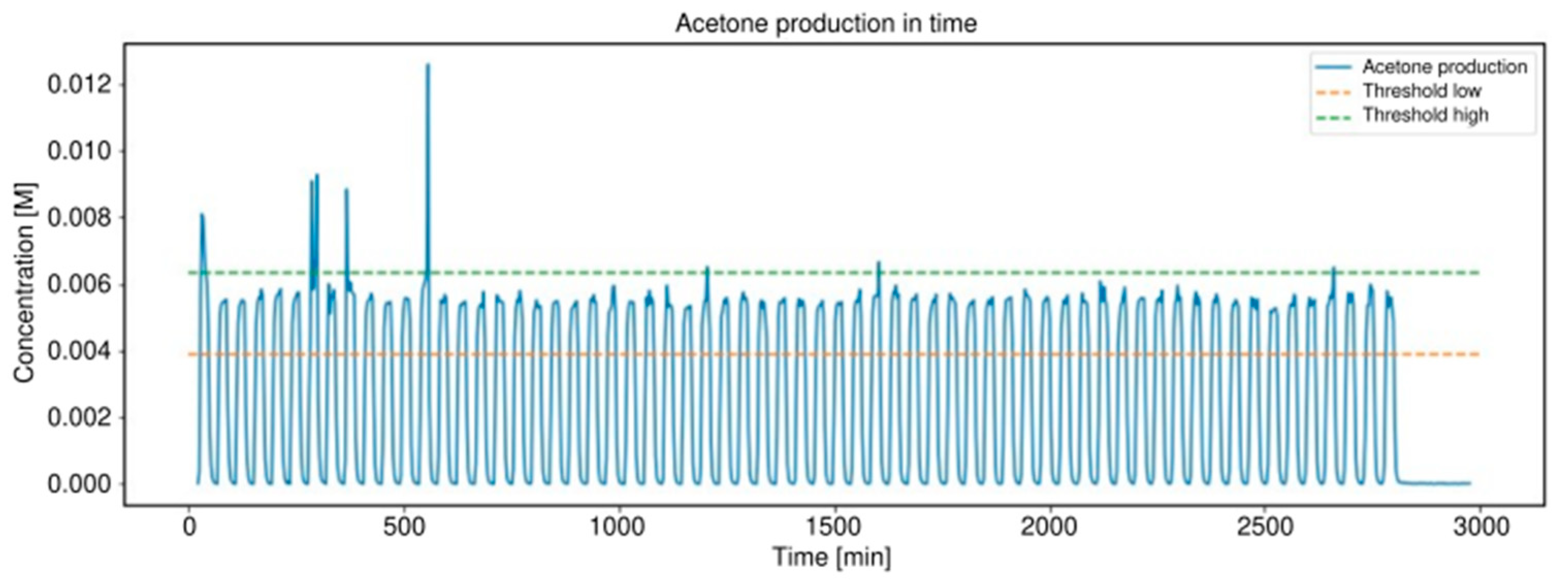
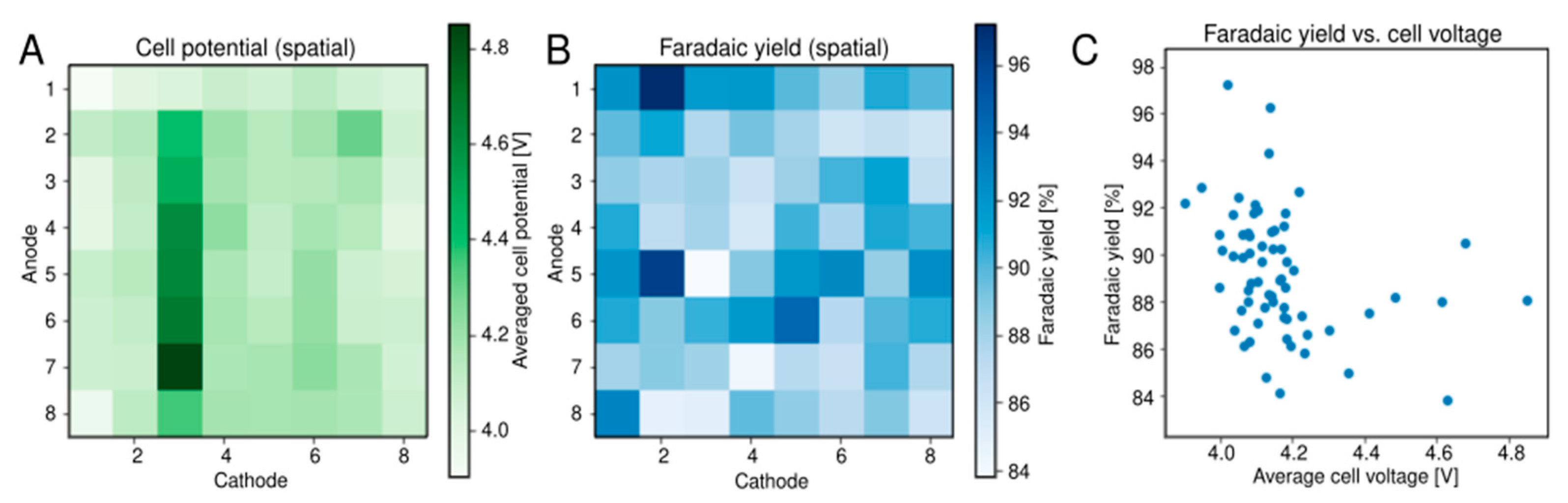
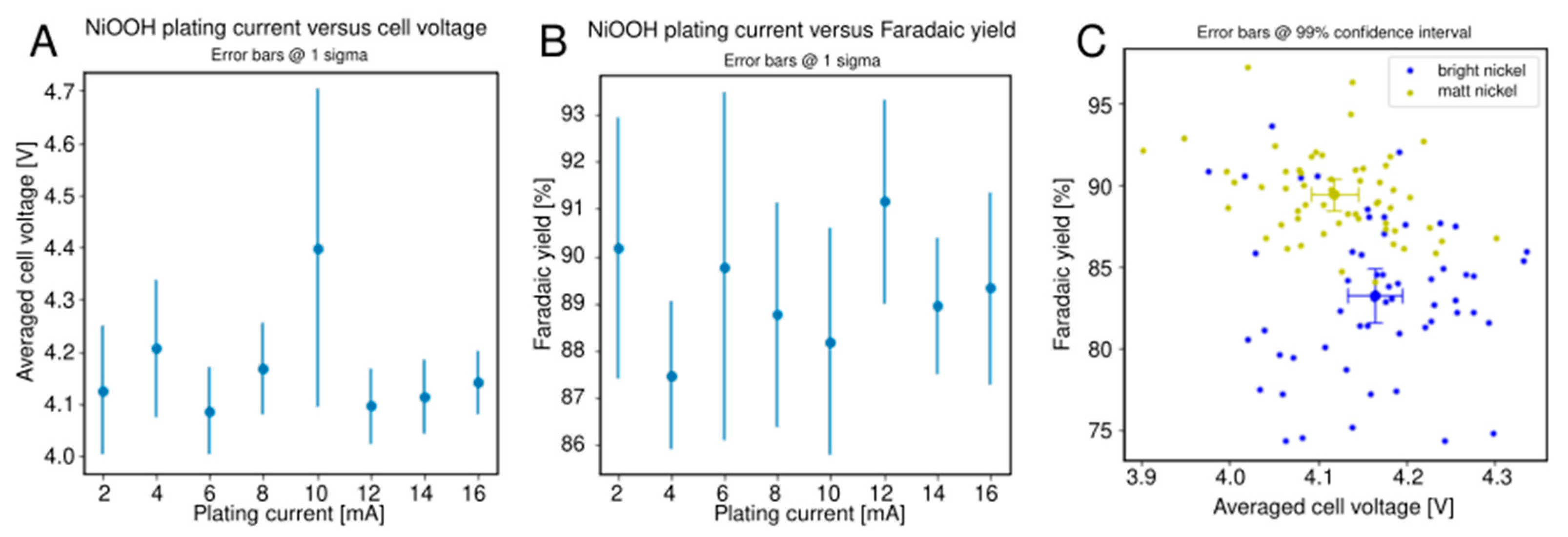
2.3. Alcohol Oxidation
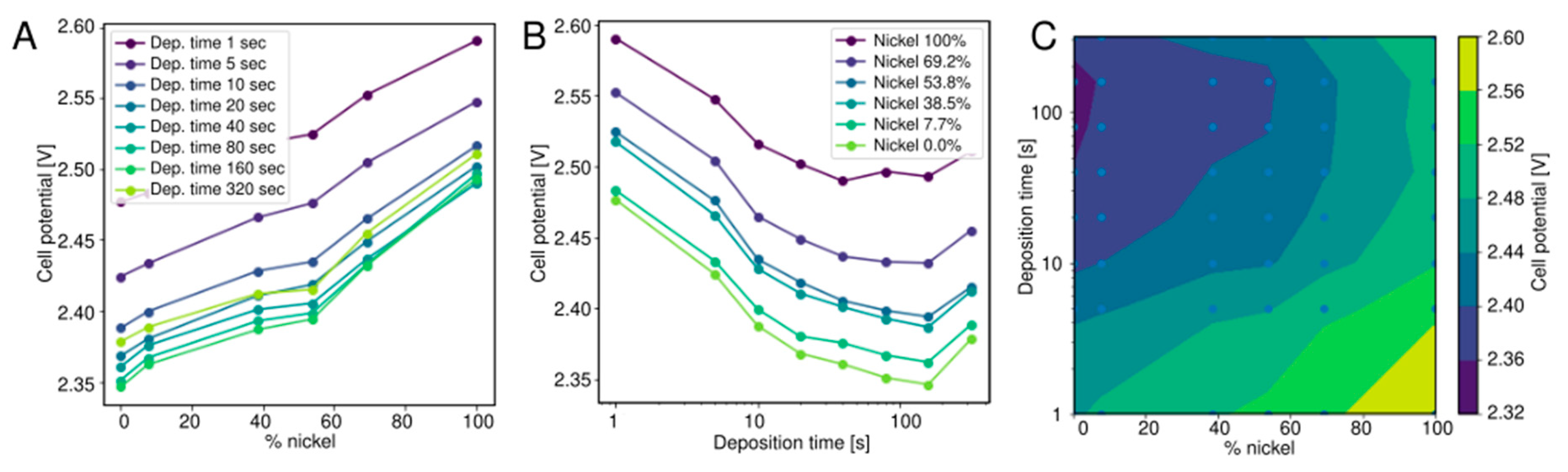
2.4. Alkaline Water Electrolysis
3. Discussion
3.1. The High-Throughput Setup and Its Validation
3.2. Alcohol Oxidation
3.3. Alkaline Water Electrolysis
4. Experimental Section
4.1. Materials and Methods
4.1.1. Terminology
- Measurement: application of current to one cell out of 64 electrochemical cells (anode/cathode combination) while monitoring cell voltage and/or analyzing the reactor outflow.
- ○
- Experiment: a complete set of 64 measurements, where each measurement is performed at a different electrochemical cell in the 8 × 8 matrix.
- ○
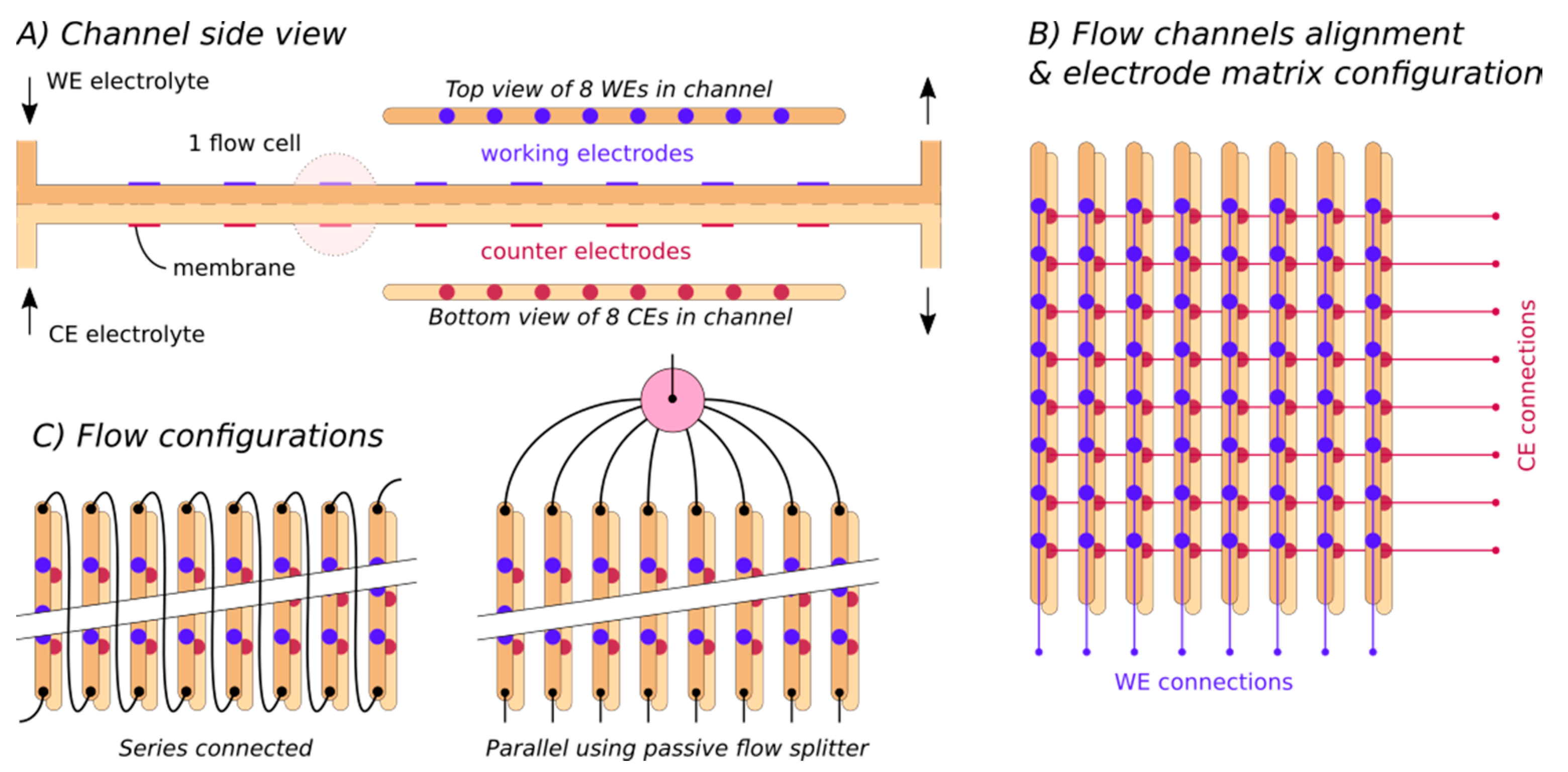
- Row/column: the 8 × 8 electrode matrix is always depicted with the WEs connected vertically and the CEs connected horizontally. Analogous to matrix terminology, the rows are the horizontally connected (counter) electrodes and the columns are the vertically connected (working) electrodes.
- Sequential operation: the experiment is performed as 64 measurements in series (after each other).
- Semi-parallel operation: the experiment is performed in eight batches of eight fully parallel measurements.
4.1.2. High Throughput (HT) Setup
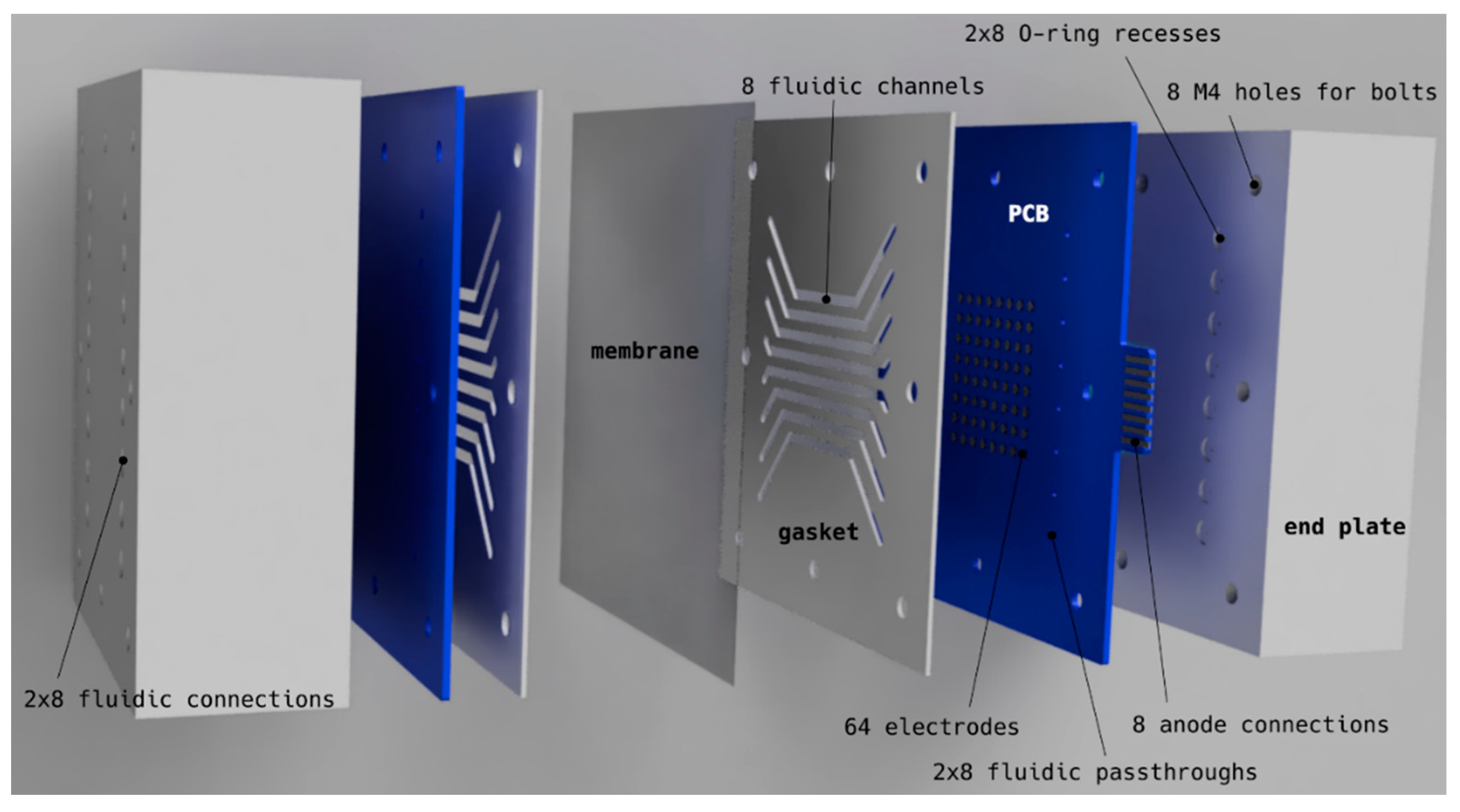
- Electrodes with 2.5 mm diameter (4.9 mm2) were etched onto standard FR4 printed circuit boards (PCBs) with 70 µm copper and an electroless nickel immersion gold (ENIG) finish (Eurocircuit NV, Mechelen, Belgium), containing 16 through holes per PCB for electrolyte inlet (8) and outlet (8). Renderings of the PCBs can be seen in Figure 11.
- Flow channel gaskets were cut out from 1 mm thick polytetrafluoroethylene (PTFE) sheets (Waboplast, Wormer, The Netherlands).
- The membrane used for PEM water splitting was Nafion 115 (127 µm thickness), for alkaline water electrolysis a porous separator made of reinforced PPS (Zirfon Pearl, Agfa, Mortsel, Belgium) and for alcohol oxidation no membrane was used.
- The PCB/gasket/membrane/gasket/PCB stack was pressed together by two 40 mm thick stainless steel 316 end plates, each having 16 fluidic connections (1/4-28 unified fine thread (UNF) flat bottom) on the outside and 16 O-ring grooves (ethylene propylene diene monomer rubber (EPDM) O-rings) on the inside for leak-tight connection to the electrode PCBs.
- Wetted materials for the setup (excluding fluidics) were stainless steel 316 (SS316), EPDM, PCB solder mask (unknown, possibly a UV-cured polymer), PTFE, membrane material and electrode material.
4.1.3. Fluidics
- Tubing was either PTFE or fluorinated ethylene propylene (FEP) (1/8 inch and 1/16 inch), with ethylene tetrafluoroethylene (ETFE) ferrules and polyether ether ketone (PEEK) flow splitters and adapters.
- ○
- Pumps were either PU-2080 high-performance liquid chromatography (HPLC)-type pumps (Jasco, Easton, MD, USA) or 120U/DM2 peristaltic pumps (Watson-Marlow, Wilmington, MA, USA).
- ○
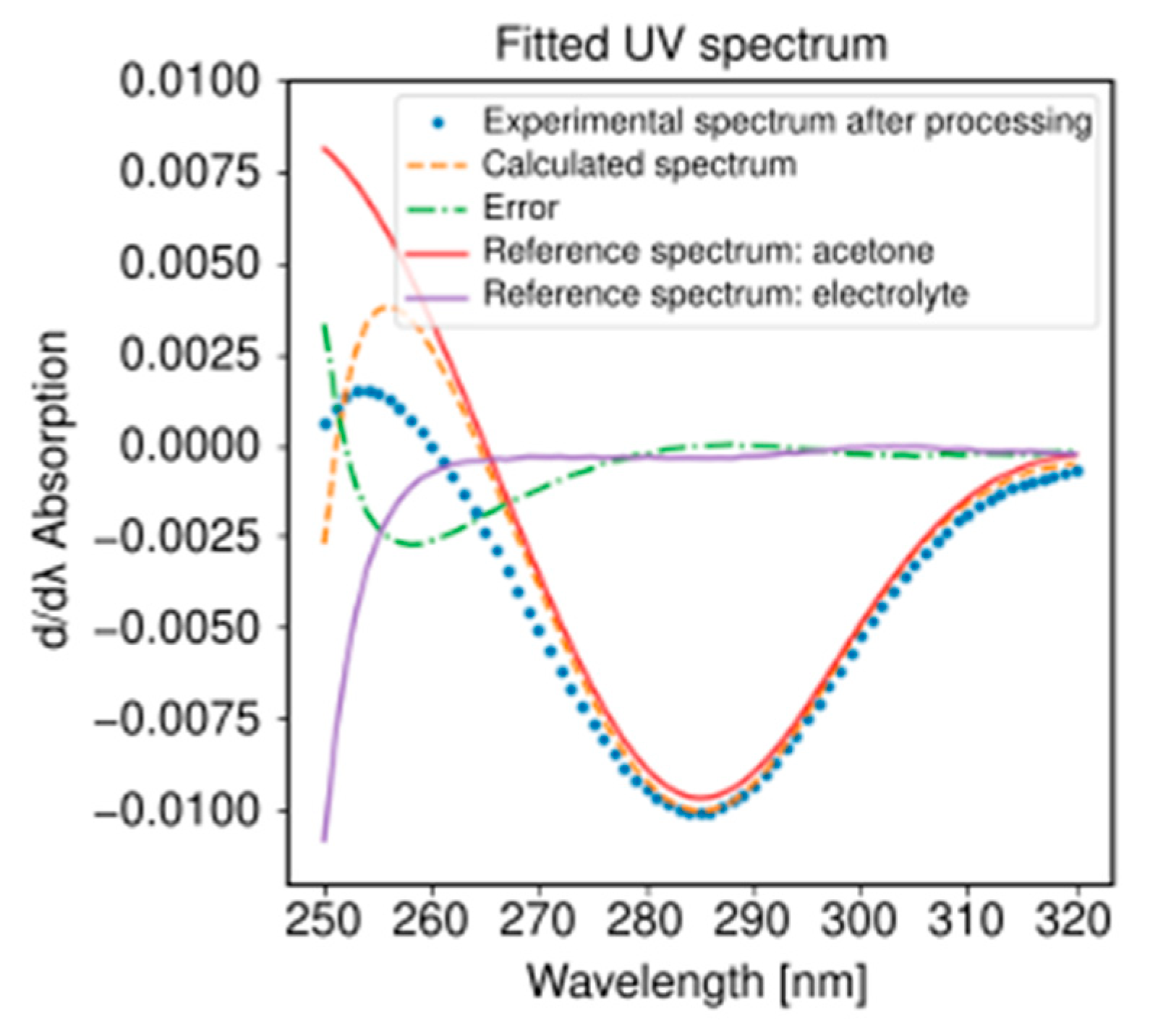
- The UV-visible spectrophotometer was a Lambda 35 (Perkin Elmer, Waltham, MA, USA) equipped with a 176.700-QS flow cell (Hellma, Müllheim, Germany).
- The membrane (hydrophobic PTFE, 0.05 μm pore size; SF18004, Tisch Scientific, North Bend, OH, USA) of the bubble trap debubbler (Darwin Microfluidics, Paris, France) was replaced after every experiment.
4.1.4. Electrode Preparation
- Gold electrodes (soft gold, >99.9% Au, 30 μm) were plated by Haveman (Voorburg, The Netherlands) on top of the as-received PCBs.
- Nickel electrodes (100 μm). An as-received PCB was cleaned by rubbing with ethanol and air dried. A bright nickel layer was deposited for 50 min at 100 mA/cm2 and 55 °C using a Watts bath consisting of 200 g/L NiSO4·6H2O, 100 g/L NiCl2·6H2O, 50 g/L boric acid, 3 g/L sodium lauryl sulfate and 1 g/L saccharin (stirred overnight and filtered), using a nickel anode at ca. 7 mm distance without agitation. The PCB was then rinsed with deionised water and ethanol, rubbed with ethanol, air dried and used as such or as base material for deposition of other electrode materials.
- Microrough nickel used for alcohol oxidation. Eight different current densities (linearly spaced between 40–320 mA/cm2) at a total charge passed of 10 C/cm2 was used to plate microrough nickel deposits from a Watts bath at room temperature (40 g/L B(OH)3, 35 g/L NiCl2·6H2O, 300 g/L NiSO4·6H2O) onto the 64 nickel electrodes (randomly spaced).
- ○
- Nickel/iron nanoparticles (alkaline water electrolysis). An 8 × 8 library of Ni/Fe nanoparticle deposits of varying thickness and composition was generated using spark ablation and an aerosol printer. Mixed aerosols were generated by two parallel VSP-G1 spark generators equipped with 2 × Fe (99.9%) and 2 × Ni (99.9%) electrodes, each using 1 slm Ar as carrier gas. The Ni/Fe ratio was varied by changing the relative power (which is proportional to the ablation rate, see Section 3.3) of the individual spark generators from 0–100% in 8 linear steps, while keeping the combined power constant at 13 W. The electrodes were deposited by inertial impaction of the mixed aerosol [13], in a prototype nanomaterial printer equipped with a motorized XYZ stage. For each aerosol composition 8 spots were deposited onto the Ni electrodes of the PCB with varying deposition time, logarithmically spaced from 1–320 s per spot. Using a script to control the nanoparticle print process the electrode array was produced within 1.6 h. Each combination of deposition time and Ni/Fe power ratio was deposited once, giving a total of 64 electrode materials (Table 2).
4.1.5. Experimental Setup, Parameters and Analysis
Chemicals
PEM Water Splitting
- The electrode overpotentials ηanode and ηcathode which are of the form η = α + β log(j) with:
- The pH corrections for both electrode reactions using the Nernst equation: (a) Vanode = 1.23 V − 3.8 × 0.059 = 1.01 V, (b) Vcathode = 0 V − 11 × 0.059 = −0.65 V.
- A negligible membrane potential of 2.5 mV and an iR drop of 70 mV. Membrane and iR potential drop was calculated from V = IR = I × l / (σ × A) where l is either the membrane or fluid layer thickness and σ is the conductivity of the membrane or the electrolyte.
Alcohol Oxidation
Alkaline Water Electrolysis
4.2. Hardware Description and Experimental Sequence
4.2.1. Sequential Operation
4.2.2. Semi-Parallel Operation
4.2.3. Connections, Matrix Selection, Towards Real High-Throughput
- For 64 cells, eight rows and eight columns are required, with a total of 16 connections
- For N cells, √N rows and √N columns are required, with a total of 2 × √N connections
- For 4096 cells, 64 rows and 64 columns are required, with a total of 128 connections
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maier, W.F.; Stowe, K.; Sieg, S. Combinatorial and high-throughput materials science. Angew. Chem. Int. Ed. 2007, 46, 6016–6067. [Google Scholar] [CrossRef] [PubMed]
- Potyrailo, R.; Rajan, K.; Stoewe, K.; Takeuchi, I.; Chisholm, B.; Lam, H. Combinatorial and high-Throughput screening of materials libraries: Review of state of the art. ACS Comb. Sci. 2011, 13, 579–633. [Google Scholar] [CrossRef] [PubMed]
- Muster, T.H.; Trinchi, A.; Markley, T.A.; Lau, D.; Martin, P.; Bradbury, A.; Bendavid, A.; Dligatch, S. A Review of high throughput and combinatorial electrochemistry. Electrochim. Acta 2011, 56, 9679–9699. [Google Scholar] [CrossRef]
- Adams, N.; Schubert, U.S. Software solutions for combinatorial and high-throughput materials and polymer research. Macromol. Rapid Commun. 2004, 25, 48–58. [Google Scholar] [CrossRef]
- D’Argenio, V. The high-throughput analyses era: Are we ready for the data struggle? High Throughput 2018, 7, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manandhar, K.; Pletcher, D. The preparation of high surface area nickel oxide electrodes for synthesis. J. Appl. Electrochem. 1979, 9, 707–713. [Google Scholar] [CrossRef]
- Lyalin, B.V.; Petrosyan, V.A. Oxidation of organic compounds on NiOOH electrode. Russ. J. Electrochem. 2010, 46, 1199–1214. [Google Scholar] [CrossRef]
- Wang, D.; Wang, P.; Wang, S.; Chen, Y.-H.; Zhang, H.; Lei, A. Direct electrochemical oxidation of alcohols with hydrogen evolution in continuous-flow reactor. Nat. Commun. 2019, 10, 2796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaramillo, T.F.; Ivanovskaya, A.; McFarland, E.W. High-throughput screening system for catalytic hydrogen-producing materials. J. Comb. Chem. 2002, 4, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Haber, J.A.; Cai, Y.; Jung, S.; Xiang, C.; Mitrovic, S.; Jin, J.; Bell, A.T.; Gregoire, J.M. Discovering Ce-rich oxygen evolution catalysts, from high throughput screening to water electrolysis. Energy Environ. Sci. 2014, 7, 682–688. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, T.V.; Feng, J.; Schmidt-Ott, A. New developments in spark production of nanoparticles. Adv. Powder Technol. 2014, 25, 56–70. [Google Scholar] [CrossRef]
- Lu, X.; Zhao, C. Electrodeposition of hierarchically structured three-dimensional nickel–iron electrodes for efficient oxygen evolution at high current densities. Nat. Commun. 2015, 6, 6616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández de la Mora, J.; Rao, N.; McMurry, P.H. Inertial impaction of fine particles at moderate Reynolds numbers and in the transonic regime with a thin-plate orifice nozzle. J. Aerosol Sci. 1990, 21, 889–909. [Google Scholar] [CrossRef]
- Barnartt, S. The oxygen-evolution reaction at gold anodes: II. Overpotential measurements and reaction mechanism in sulfuric acid solutions. J. Electrochem. Soc. 1959, 106, 991–994. [Google Scholar] [CrossRef]
- Brown, A.P.; Krumpelt, M.; Loutfy, R.O.; Yao, N.P. Kinetics of the hydrogen evolution reaction on mild steel and nickel cathodes in concentrated sodium hydroxide solutions. J. Electrochem. Soc. 1982, 129, 2481–2487. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bright Nickel (Control) | Microrough Nickel | |||||
|---|---|---|---|---|---|---|
| Metric | Mean | Stdev | 99% ci | Mean | Stdev | 99% ci |
| Cell potential | 4.16 V | 0.08 V | 0.03 V | 4.12 V | 0.07 V | 0.03 V |
| Faradaic yield | 83.3% | 4.6% | 1.7% | 89.4% | 2.6% | 0.9% |
| Deposition Time [s] | Rel. Power G1 Ni [%] | Rel. Power G1 Fe [%] |
|---|---|---|
| 1 | 100 | 0 |
| 5 | 85 | 15 |
| 10 | 69 | 31 |
| 20 | 54 | 46 |
| 40 | 38 | 62 |
| 80 | 23 | 77 |
| 160 | 8 | 92 |
| 320 | 0 | 100 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becker, R.; Weber, K.; Pfeiffer, T.V.; Kranendonk, J.v.; Schouten, K.J. A Scalable High-Throughput Deposition and Screening Setup Relevant to Industrial Electrocatalysis. Catalysts 2020, 10, 1165. https://doi.org/10.3390/catal10101165
Becker R, Weber K, Pfeiffer TV, Kranendonk Jv, Schouten KJ. A Scalable High-Throughput Deposition and Screening Setup Relevant to Industrial Electrocatalysis. Catalysts. 2020; 10(10):1165. https://doi.org/10.3390/catal10101165
Chicago/Turabian StyleBecker, René, Katharina Weber, Tobias V. Pfeiffer, Jan van Kranendonk, and Klaas Jan Schouten. 2020. "A Scalable High-Throughput Deposition and Screening Setup Relevant to Industrial Electrocatalysis" Catalysts 10, no. 10: 1165. https://doi.org/10.3390/catal10101165