Epoxide Syntheses and Ring-Opening Reactions in Drug Development

 ,
, {kind=link}
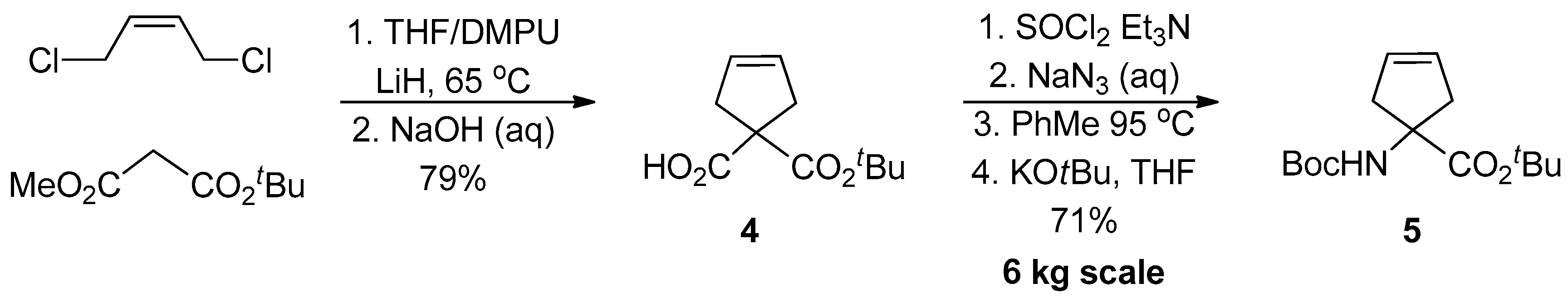{kind=link}
{kind=link}
{kind=link}
{kind=link}
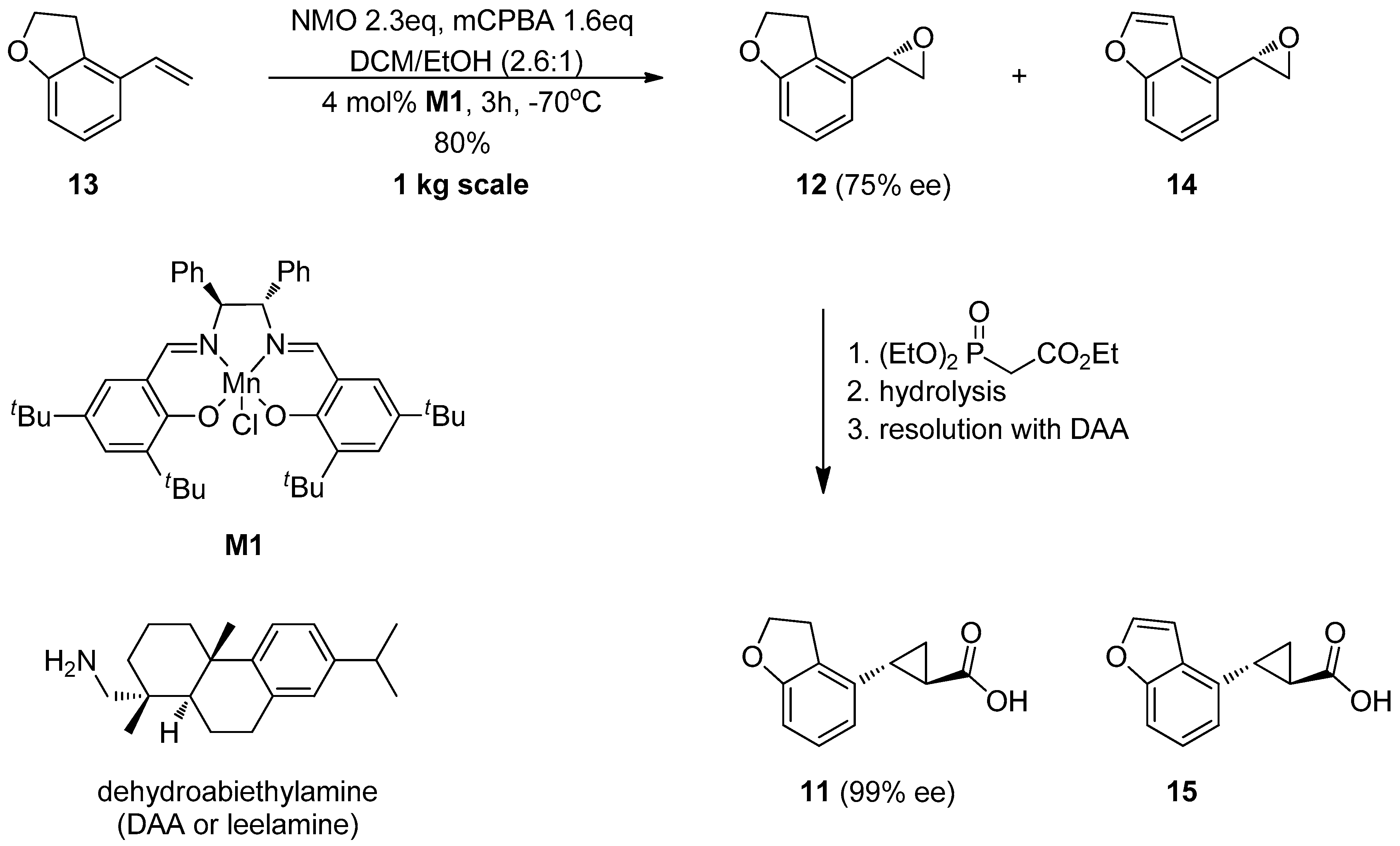{kind=link}
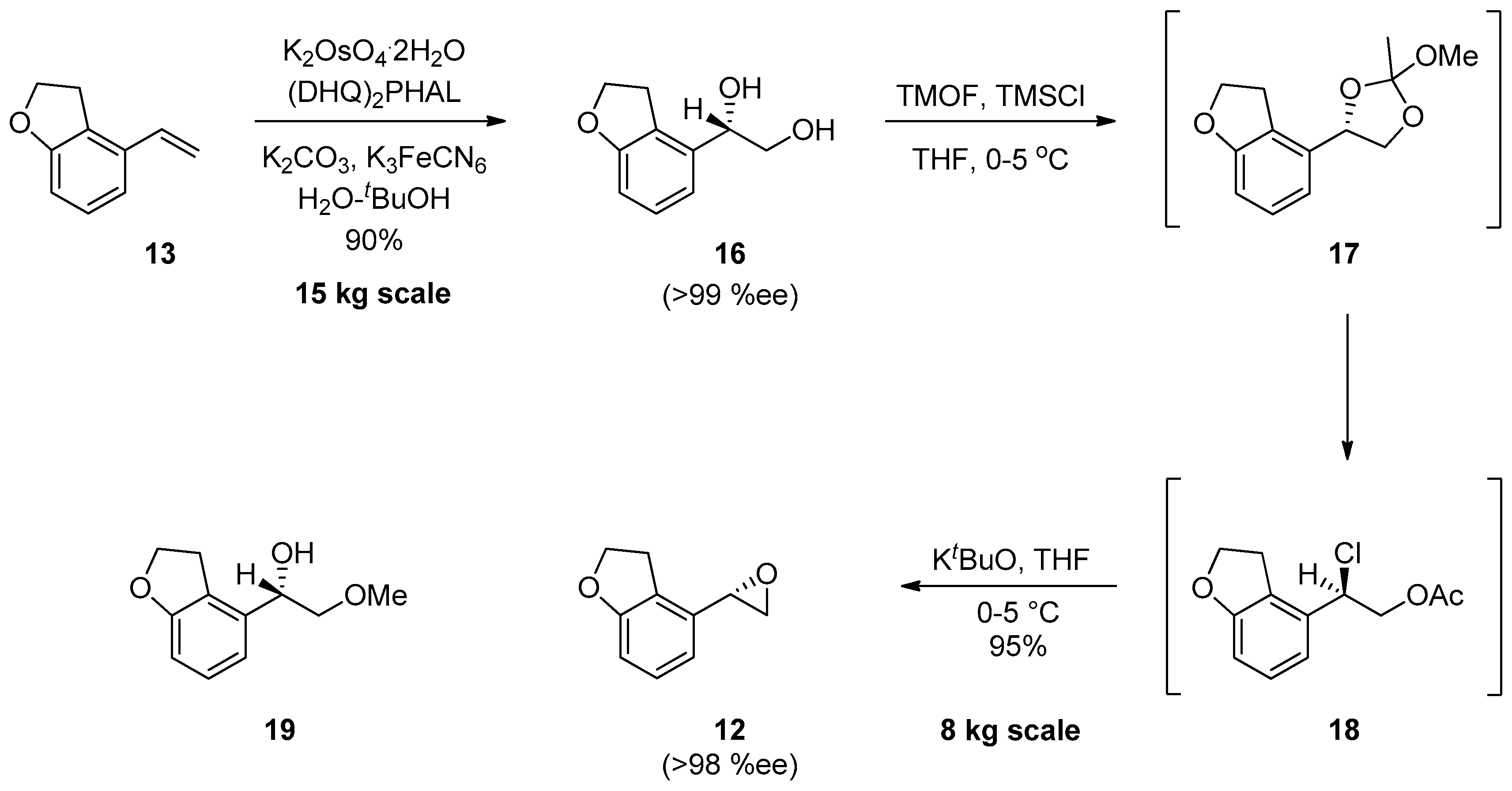{kind=link}
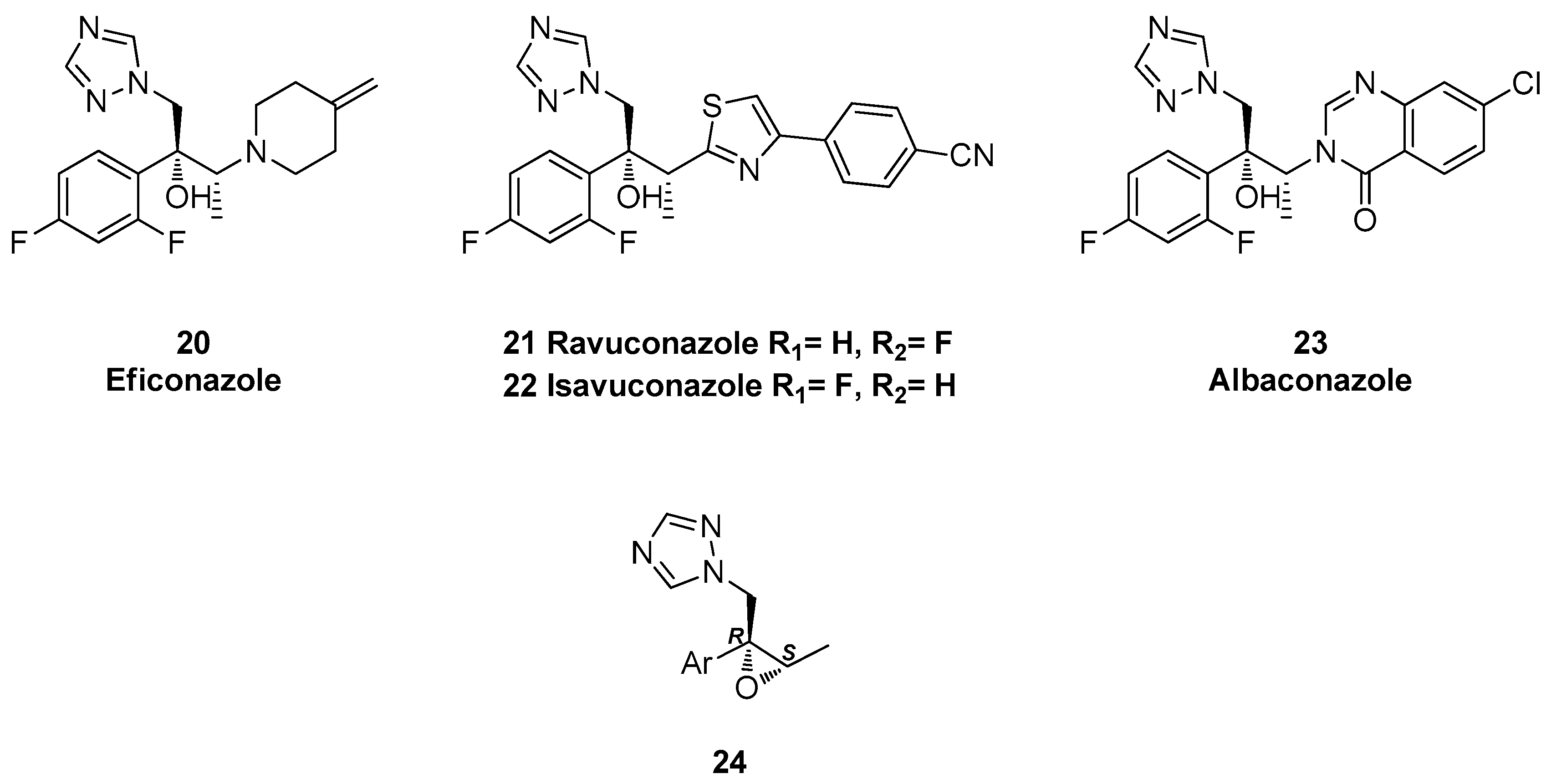{kind=link}
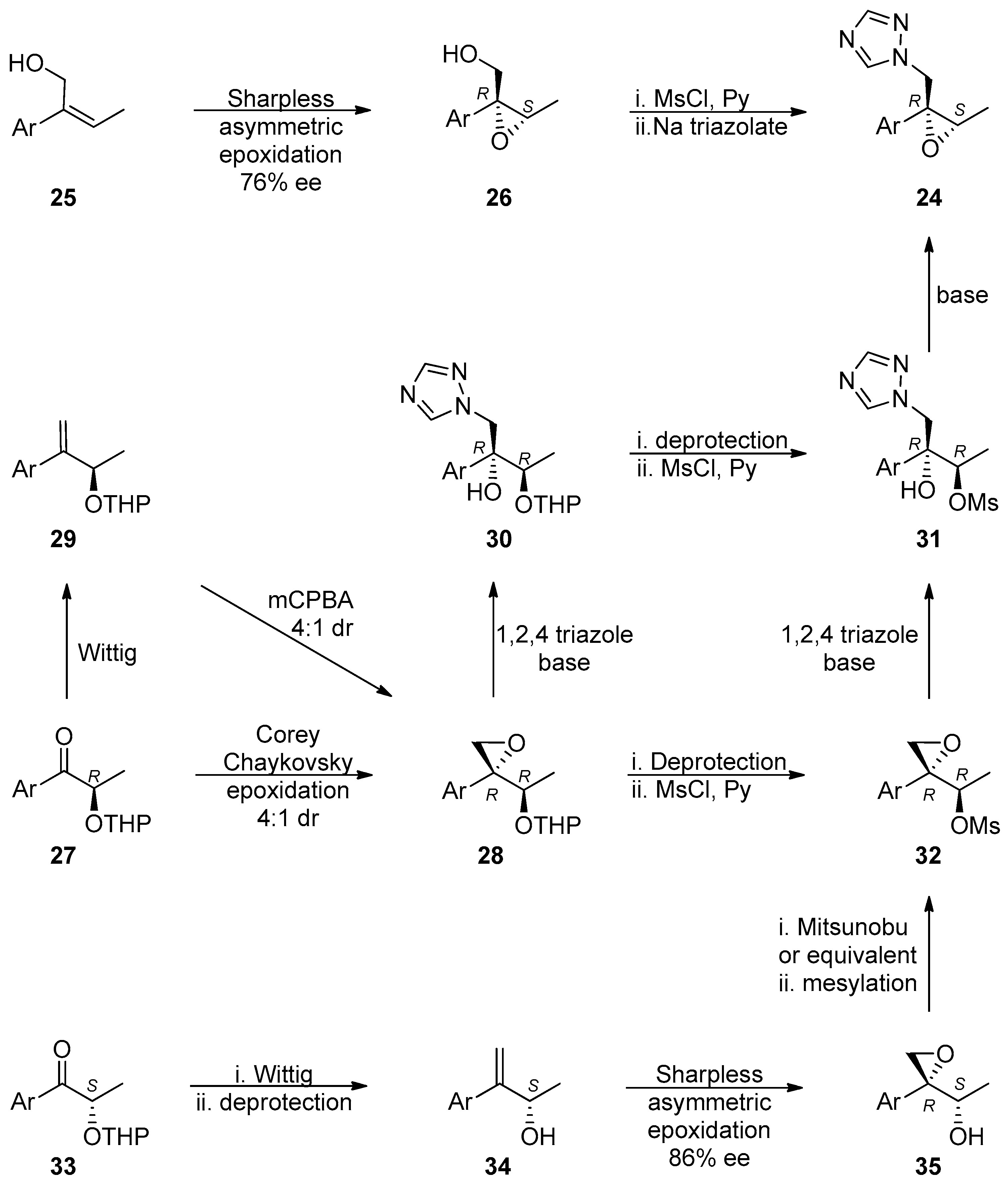{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
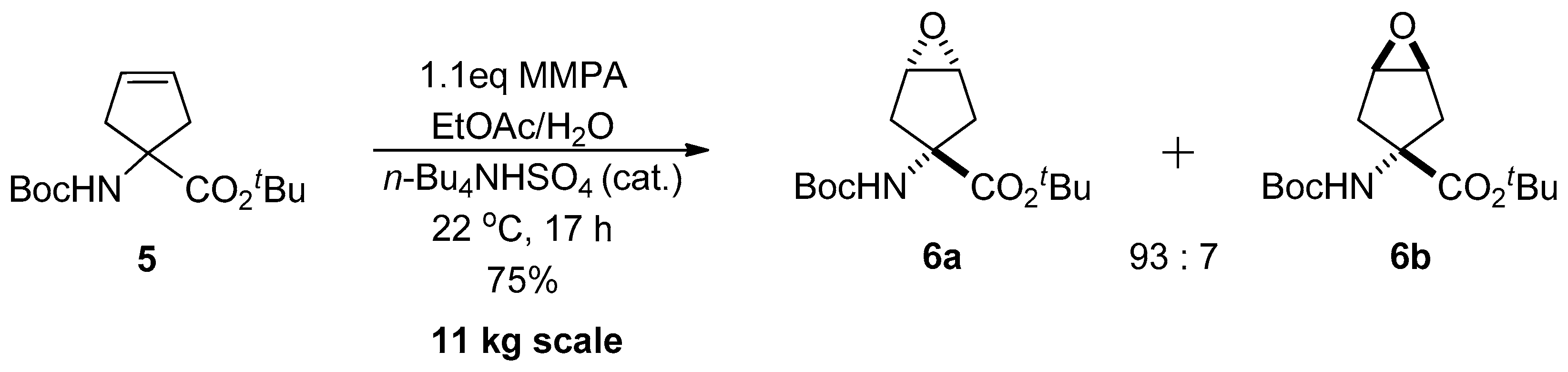
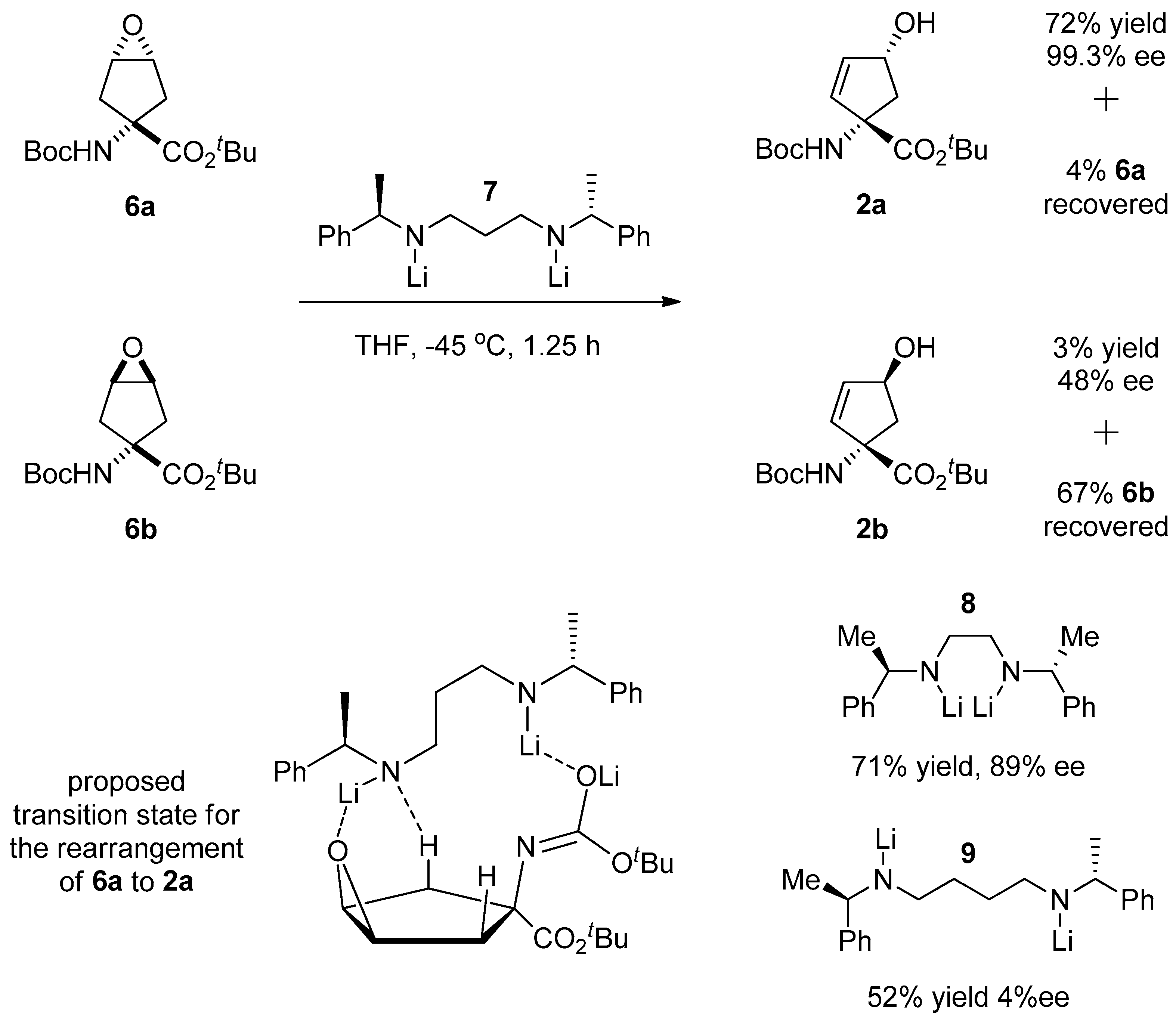
1.1. LY459477—Development of a Selective Route via a Desymmetrization Process of a Meso-Epoxide
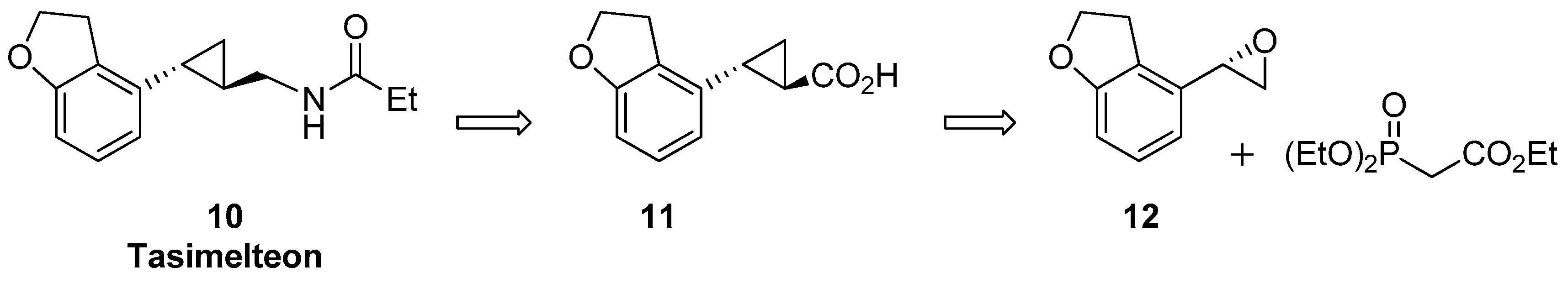
1.2. Tasimelteon—Comparison of Large-Scale Catalytic Asymmetric Epoxidation Processes Towards a Chiral Dihydrobenzofuran Epoxide
1.3. Efinaconazole, Ravuconazole, Isavuconazole, Albaconazole—Synthesis via Two Successive Chiral Epoxide Intermediates and Ring Opening Reactions
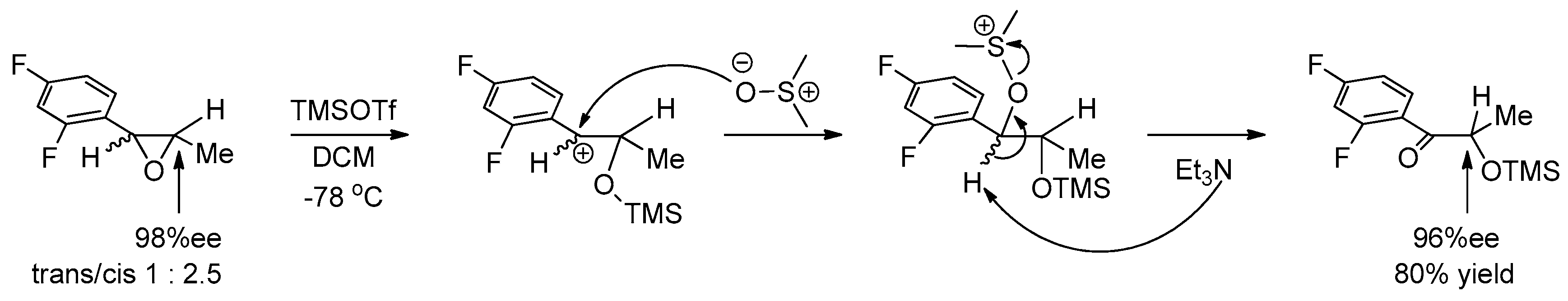
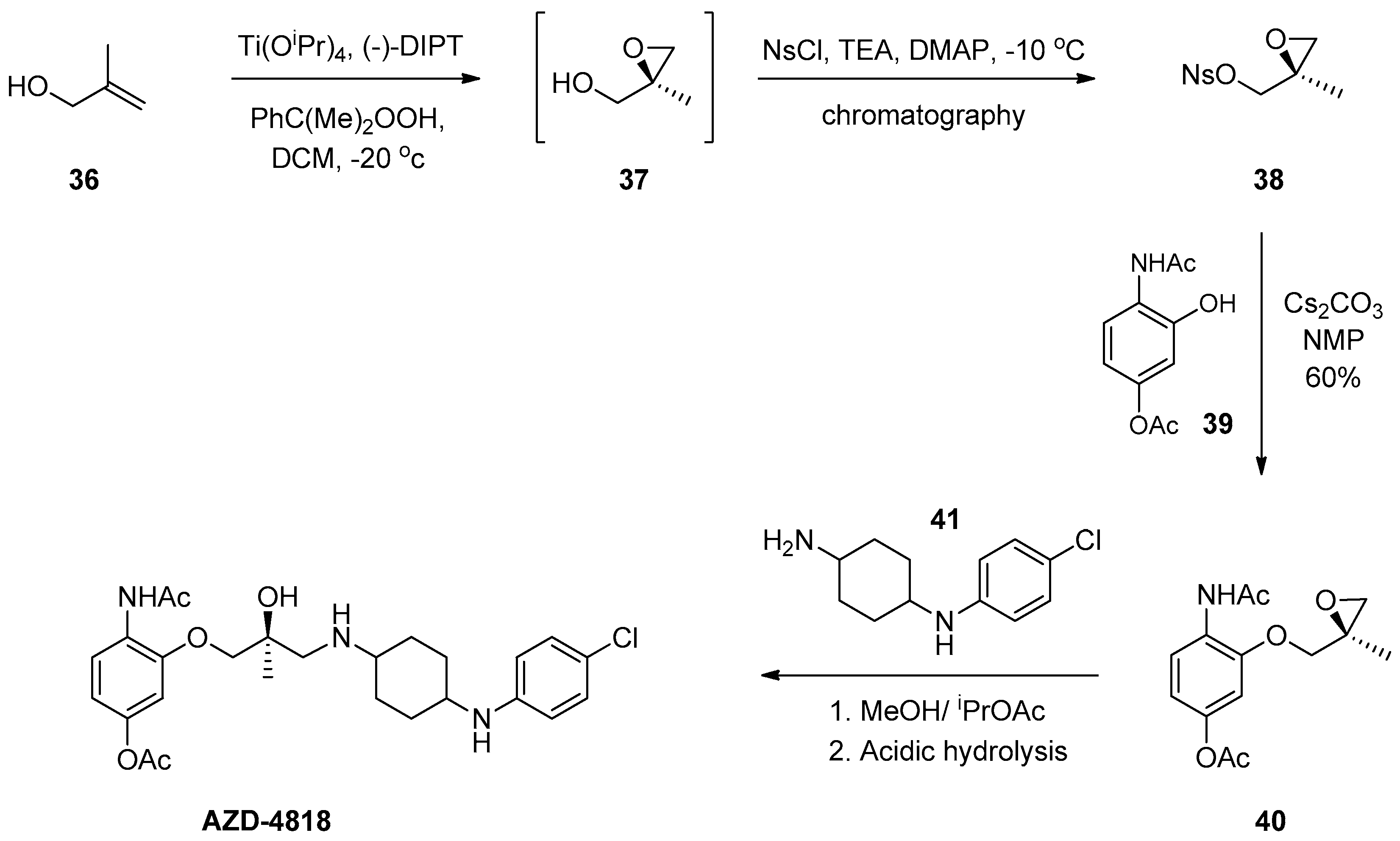
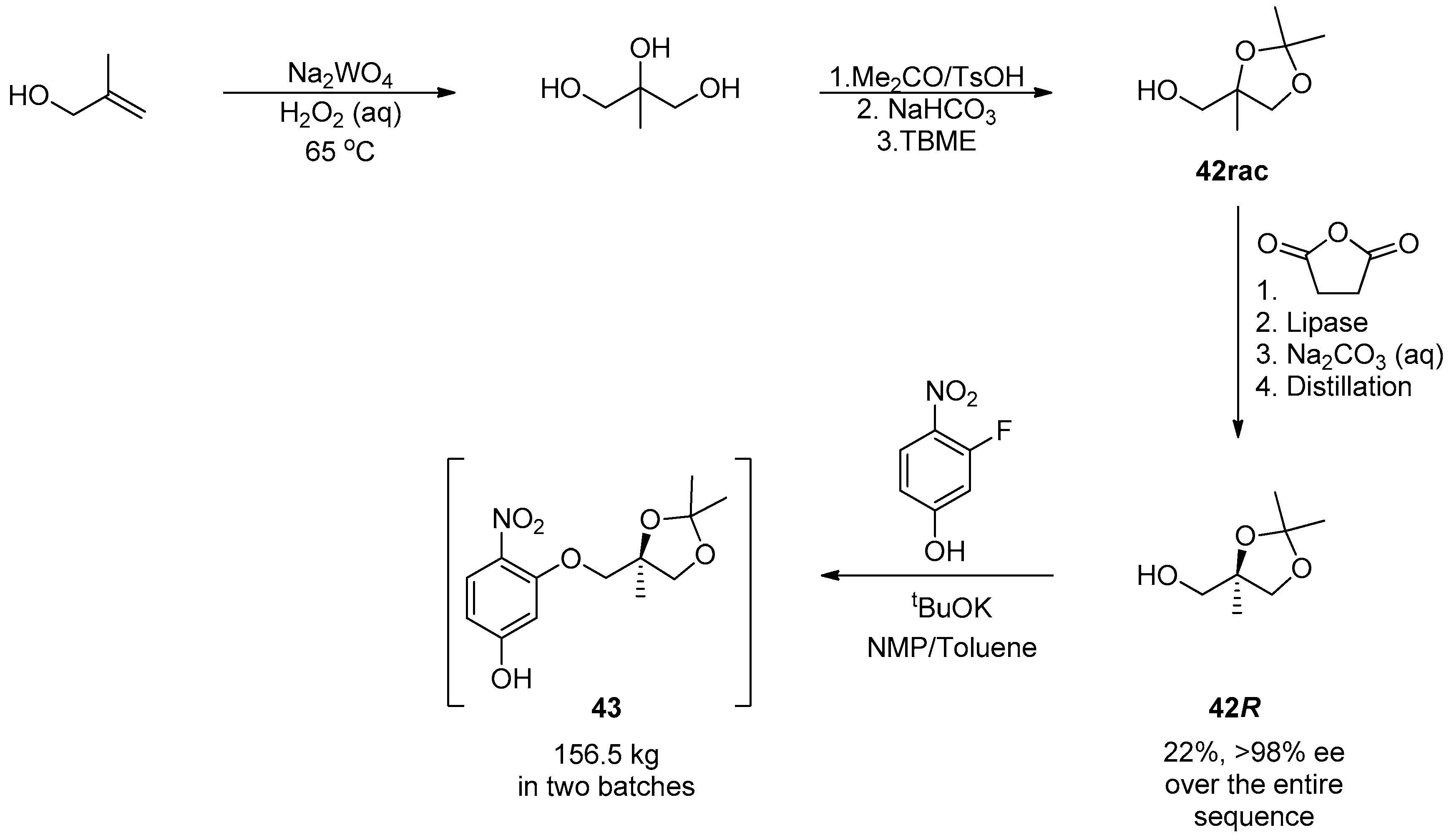
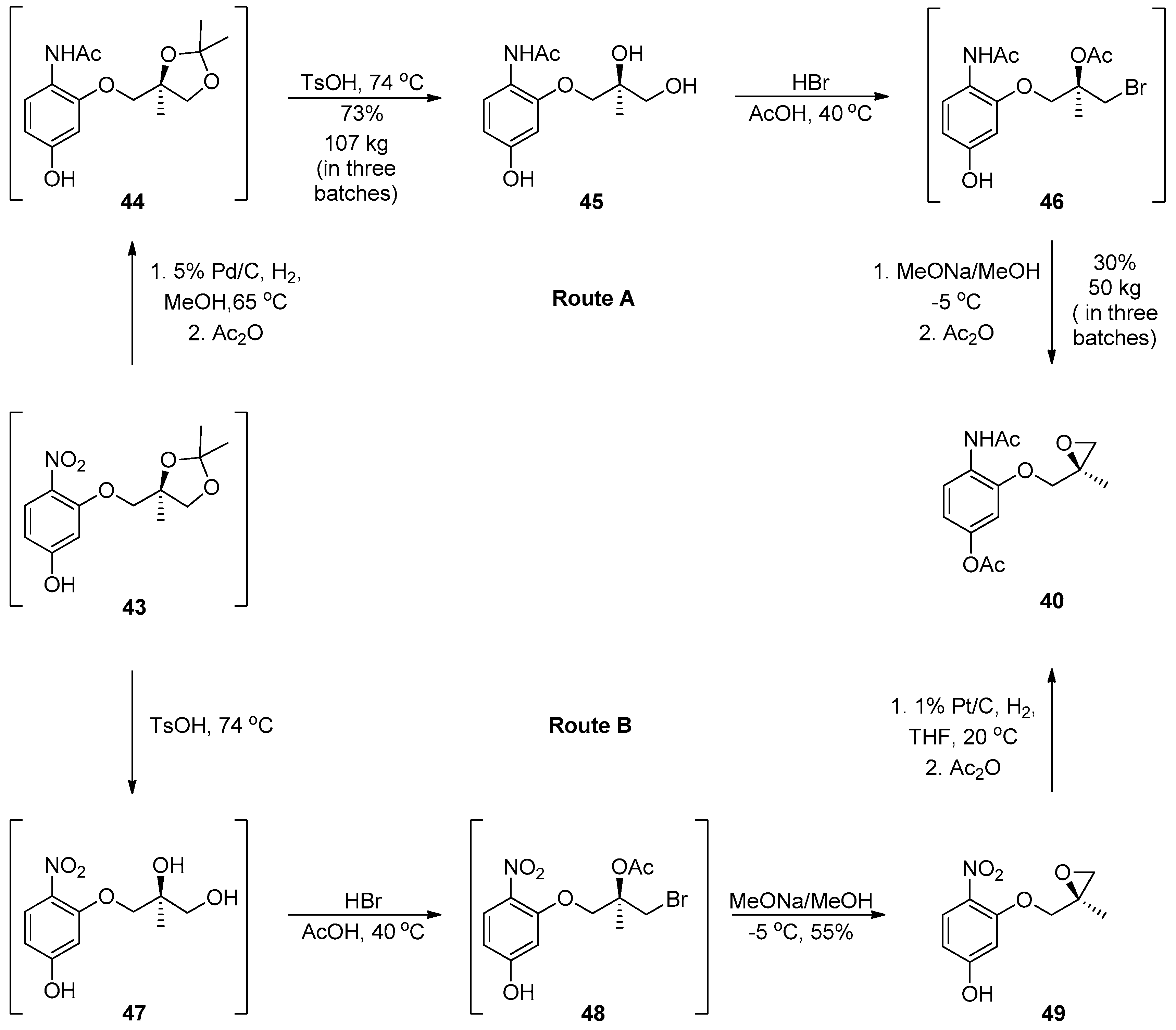
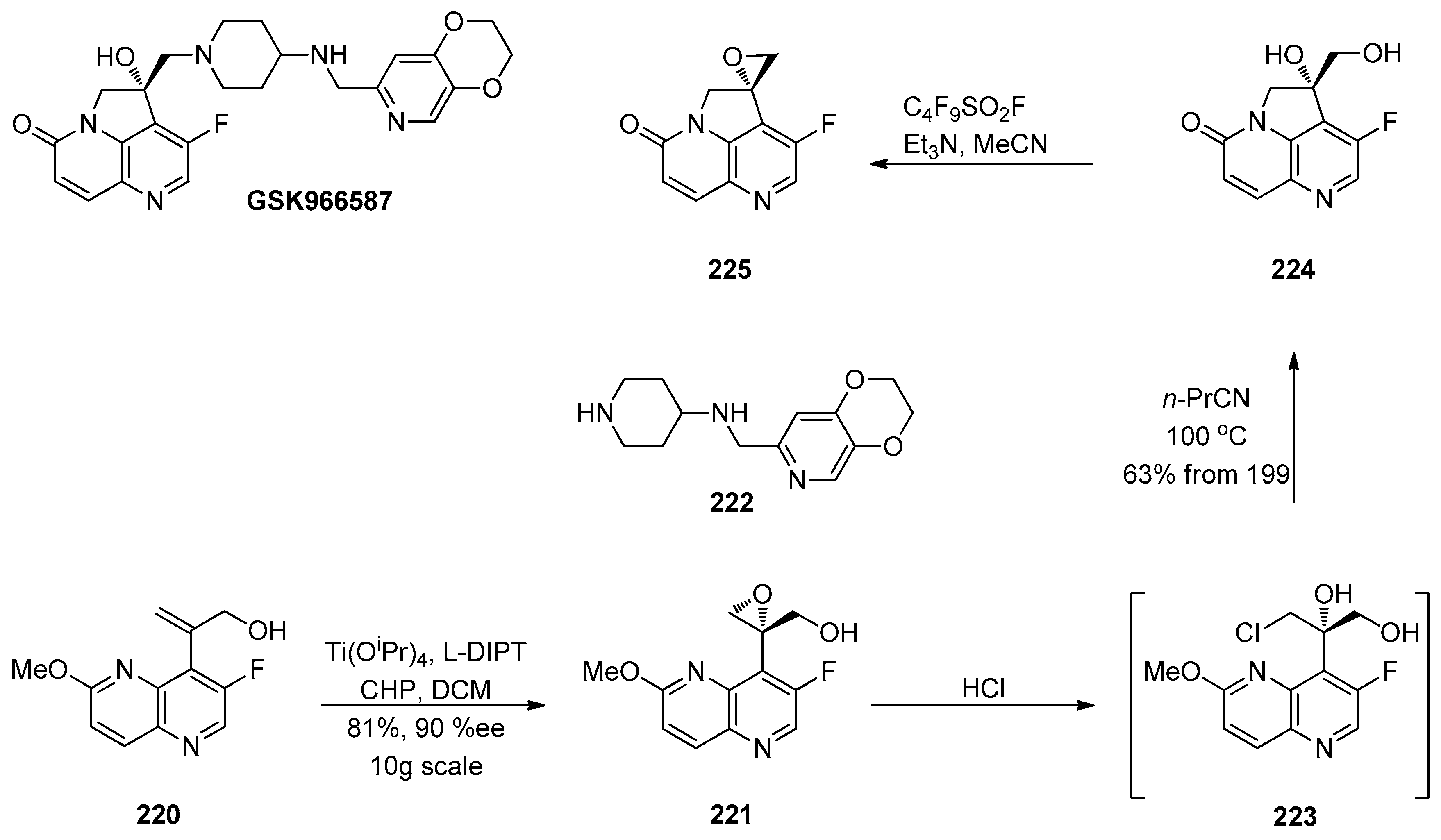
1.4. AZD-4818—Development of a Multikilogram Synthesis of a Chiral Epoxide Precursor to A CCR1 Antagonist
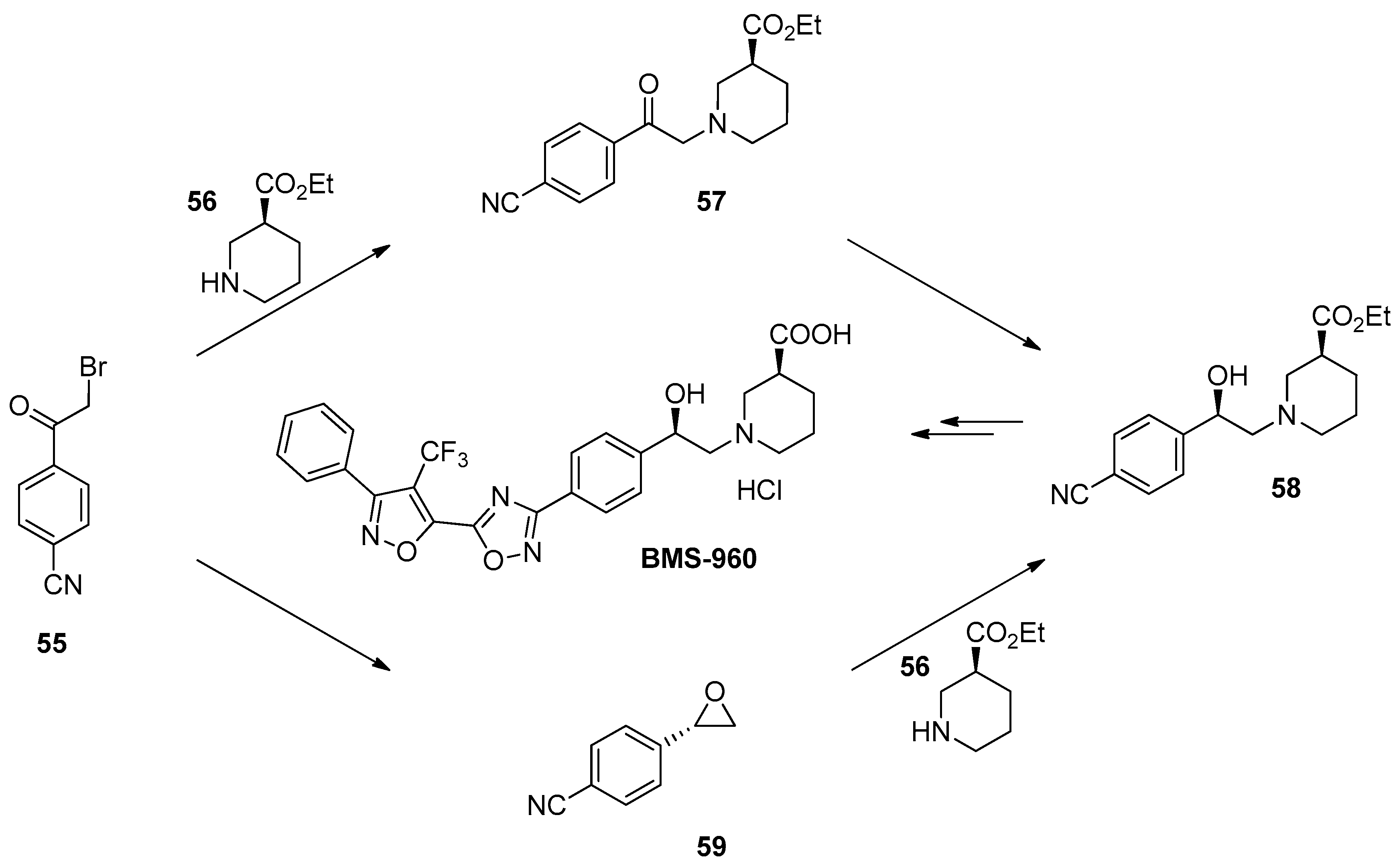
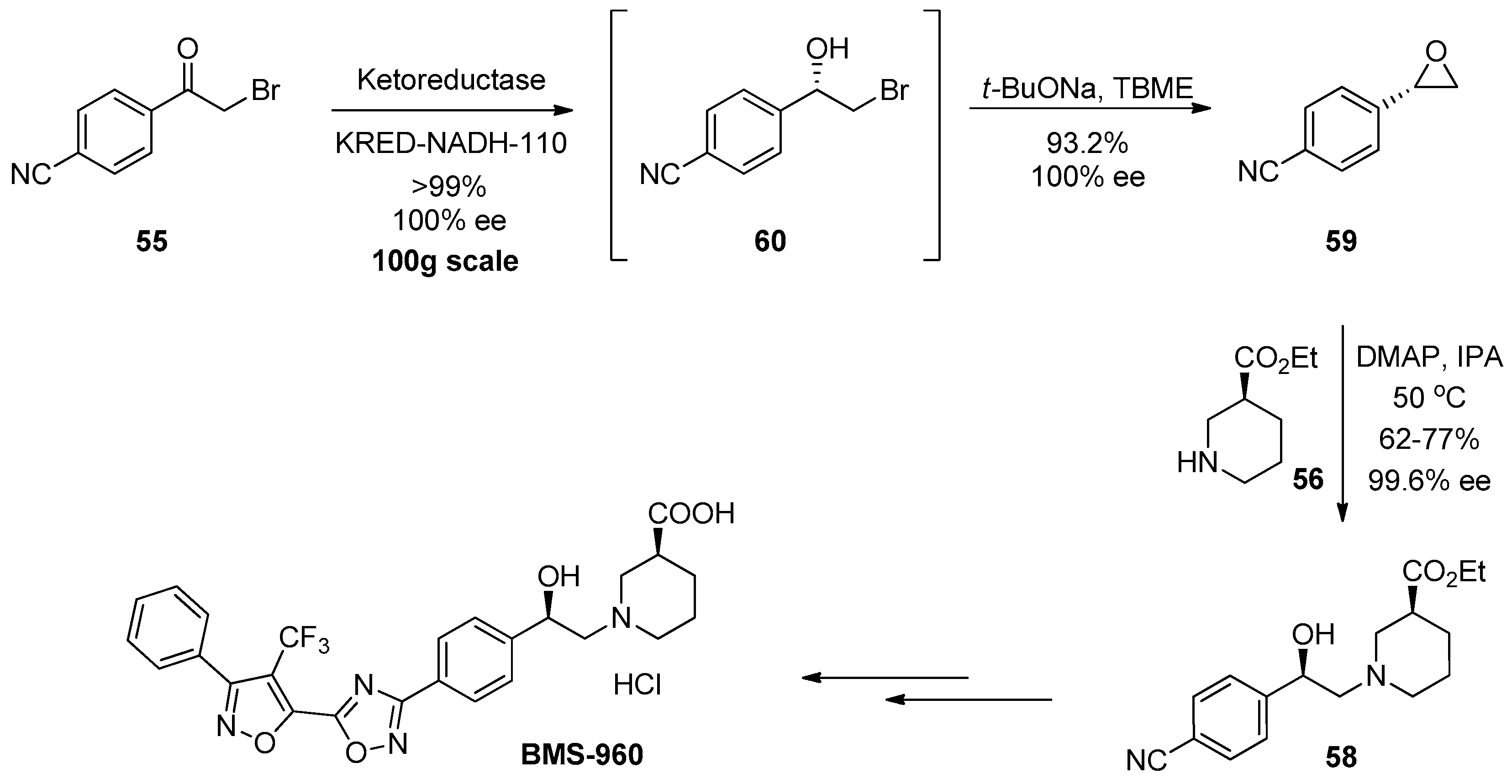
1.5. BMS-960—Development and Scale up of a Chiral Epoxide Route through an Enzymatic Reduction
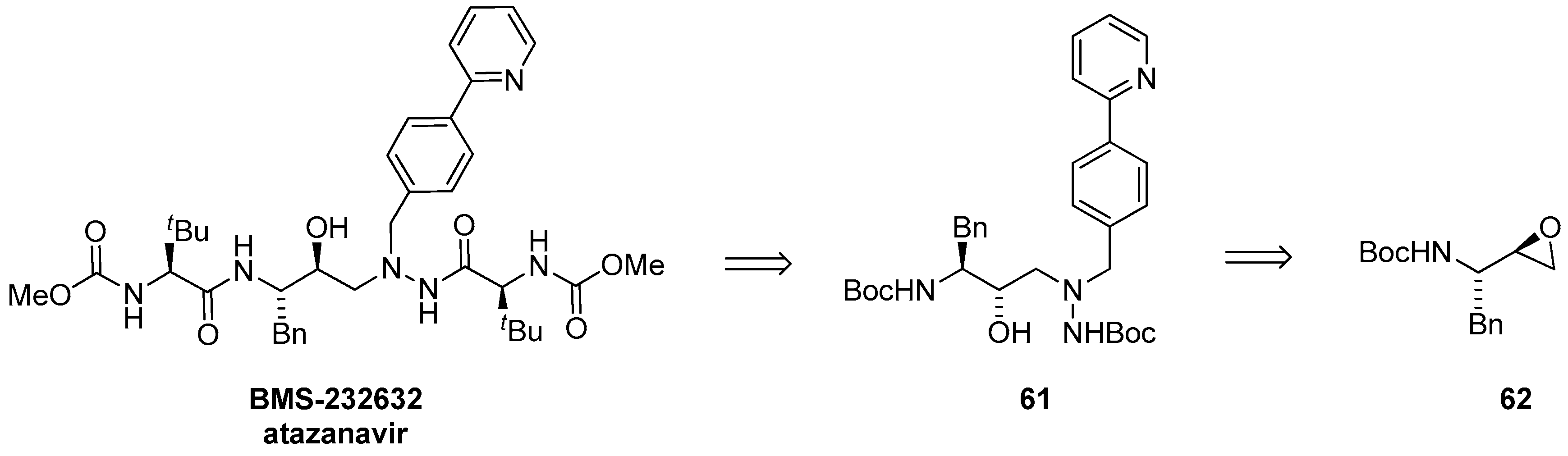
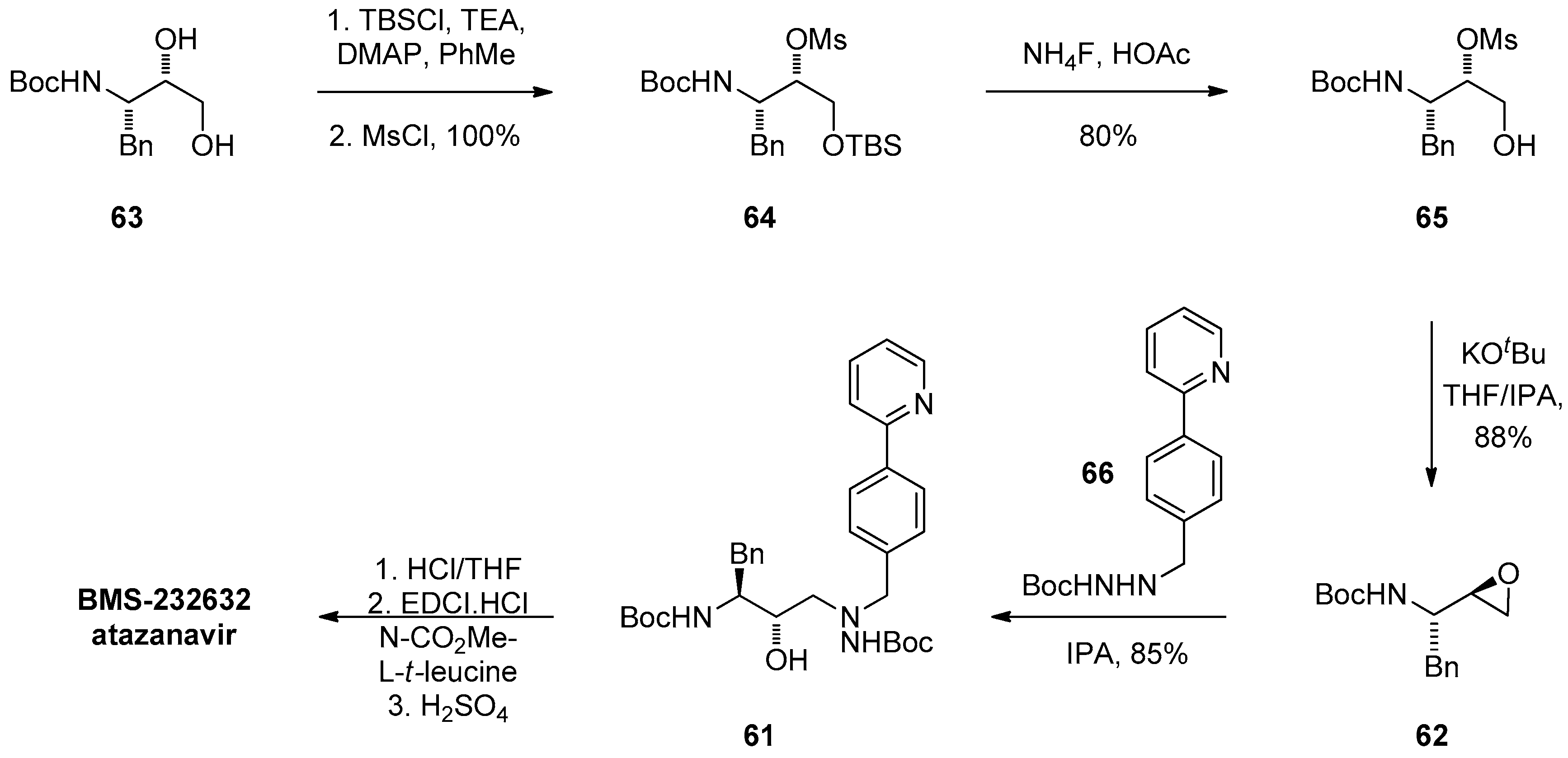
1.6. Atazanavir (BMS-232632)—Process Research and Development towards an Efficient Synthesis of an HIV Protease Inhibitor
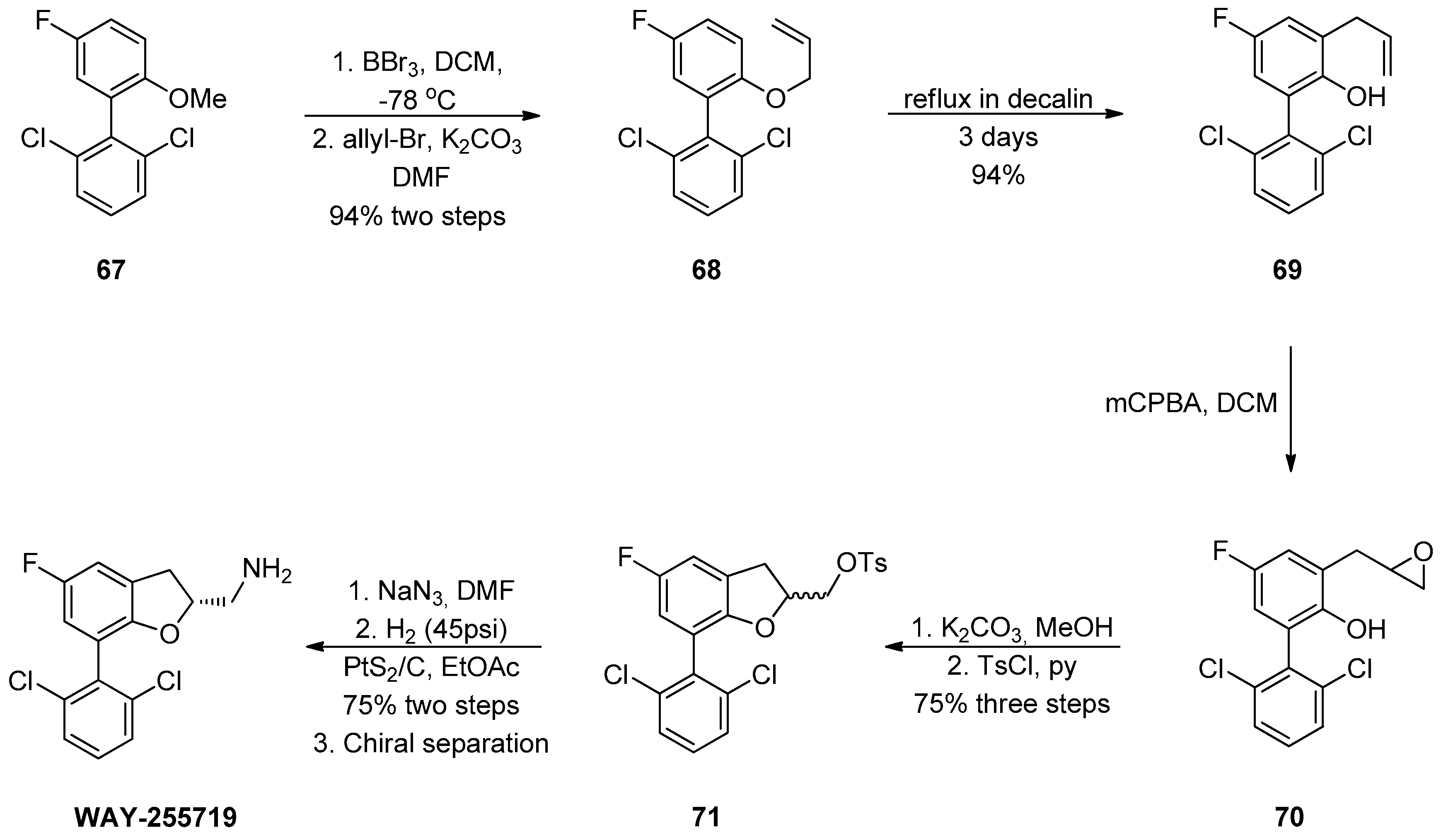
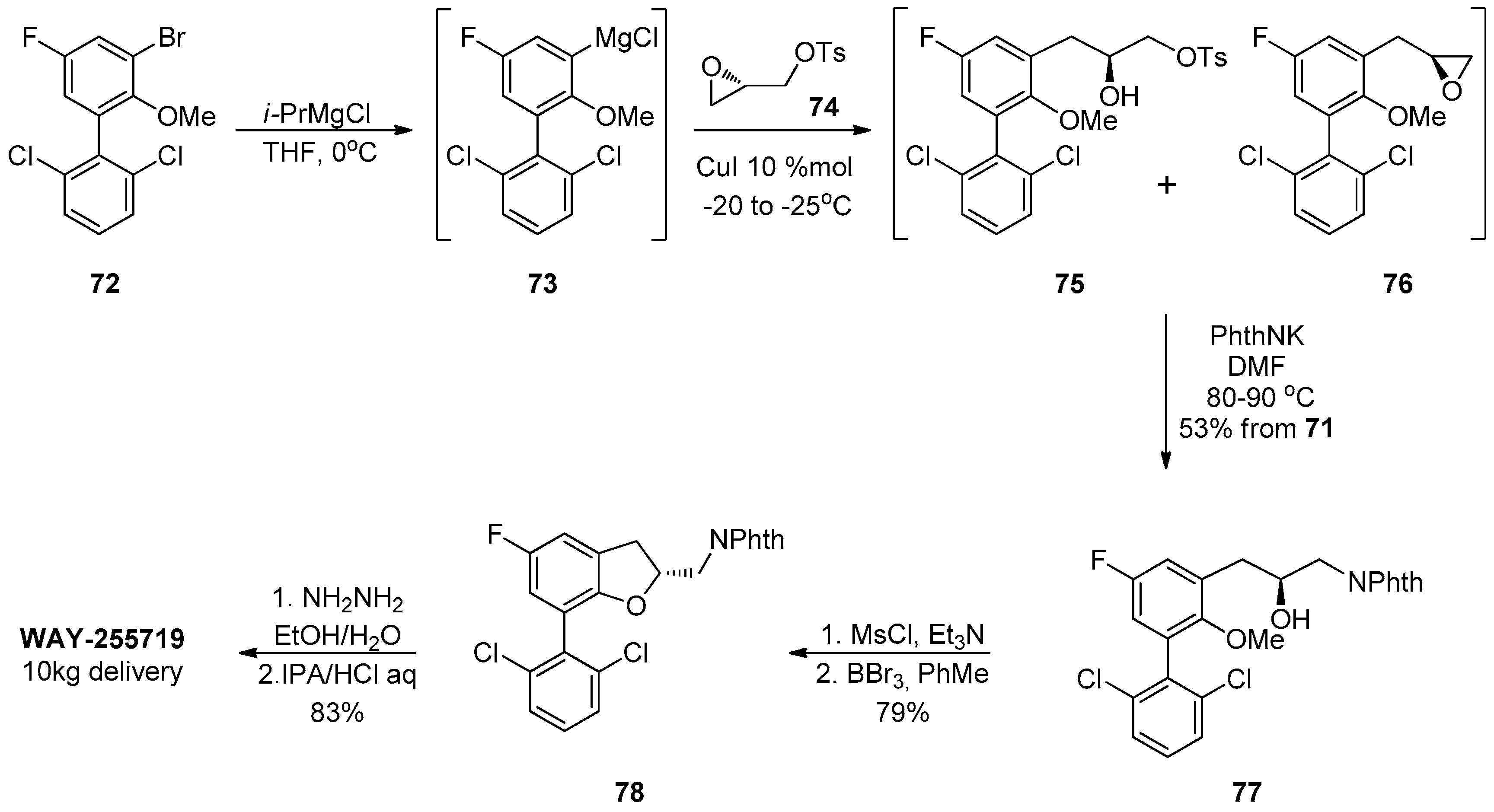
1.7. WAY-255719—Ring Opening of a Chiral Epoxide with Grignard Reagents
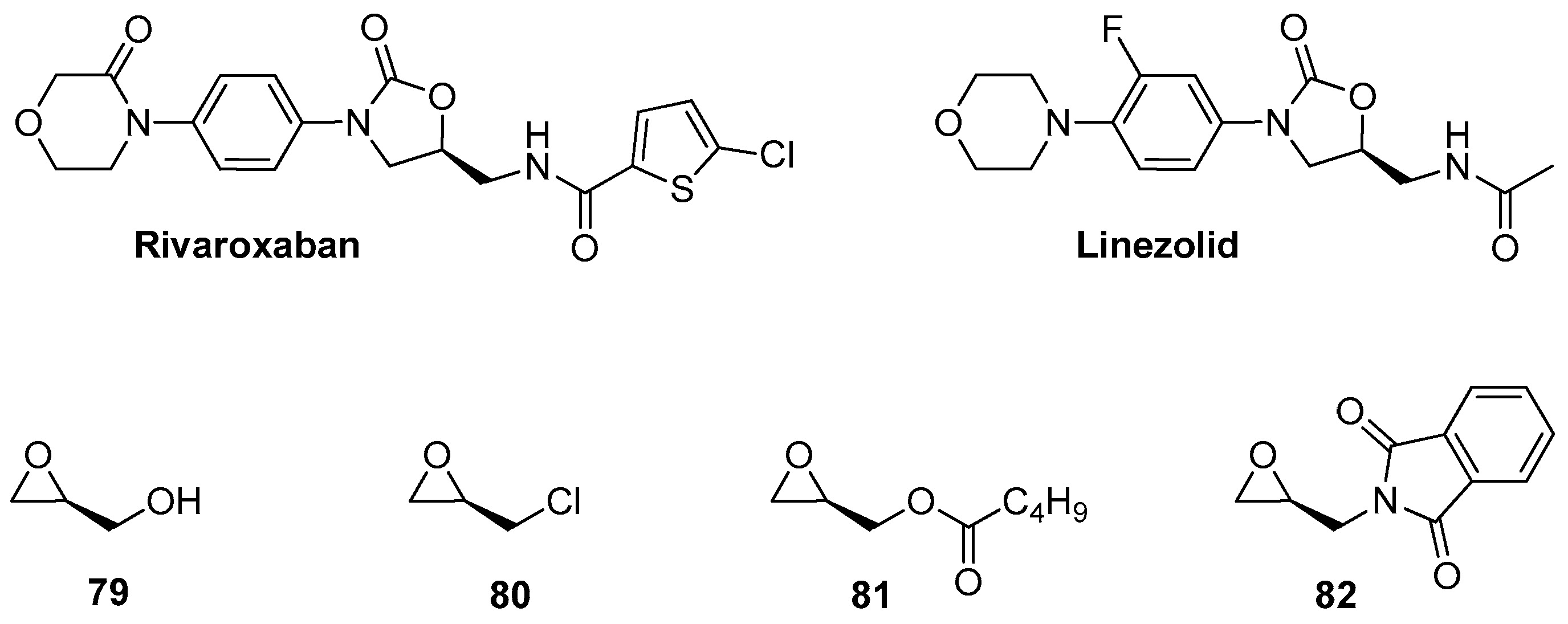
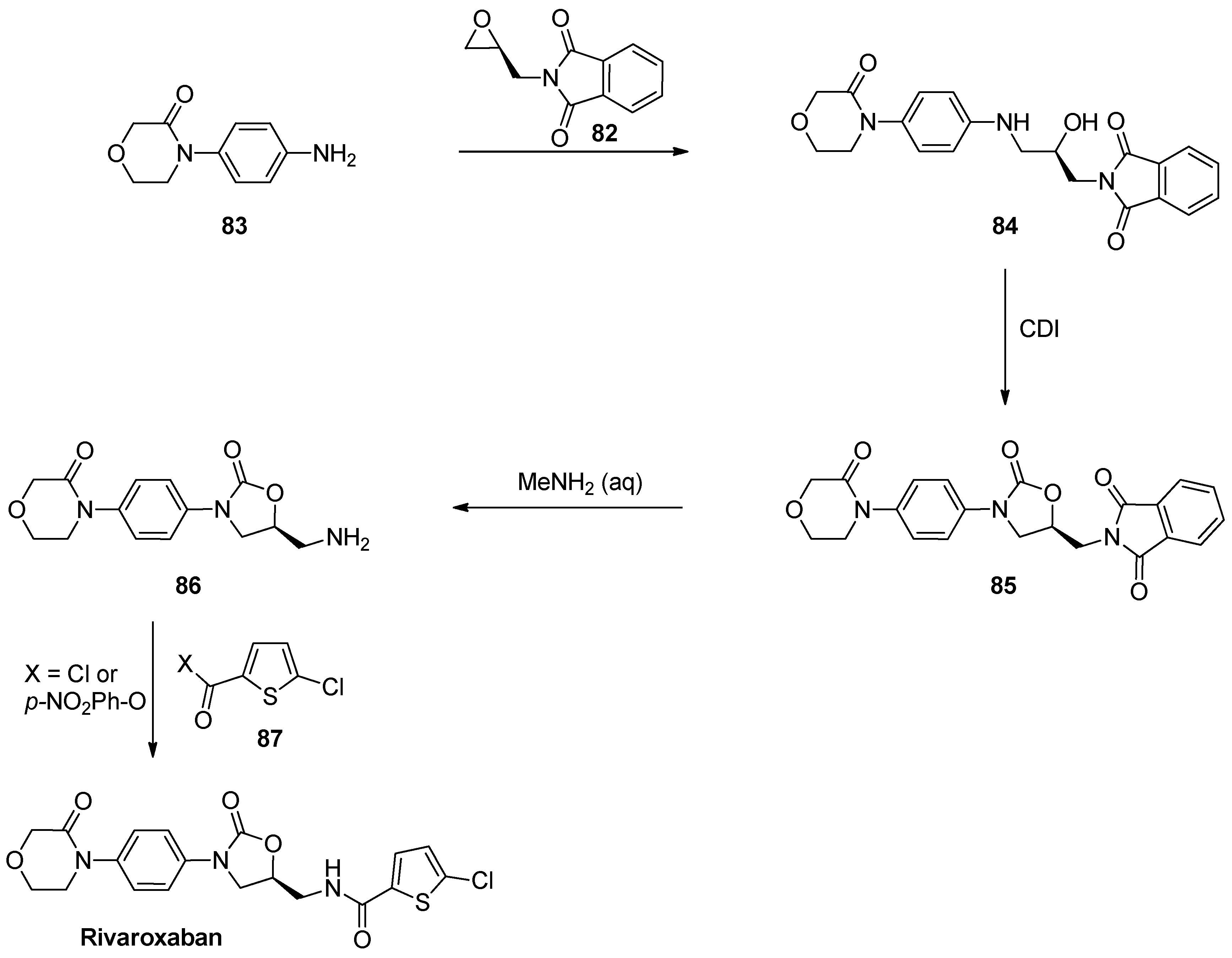
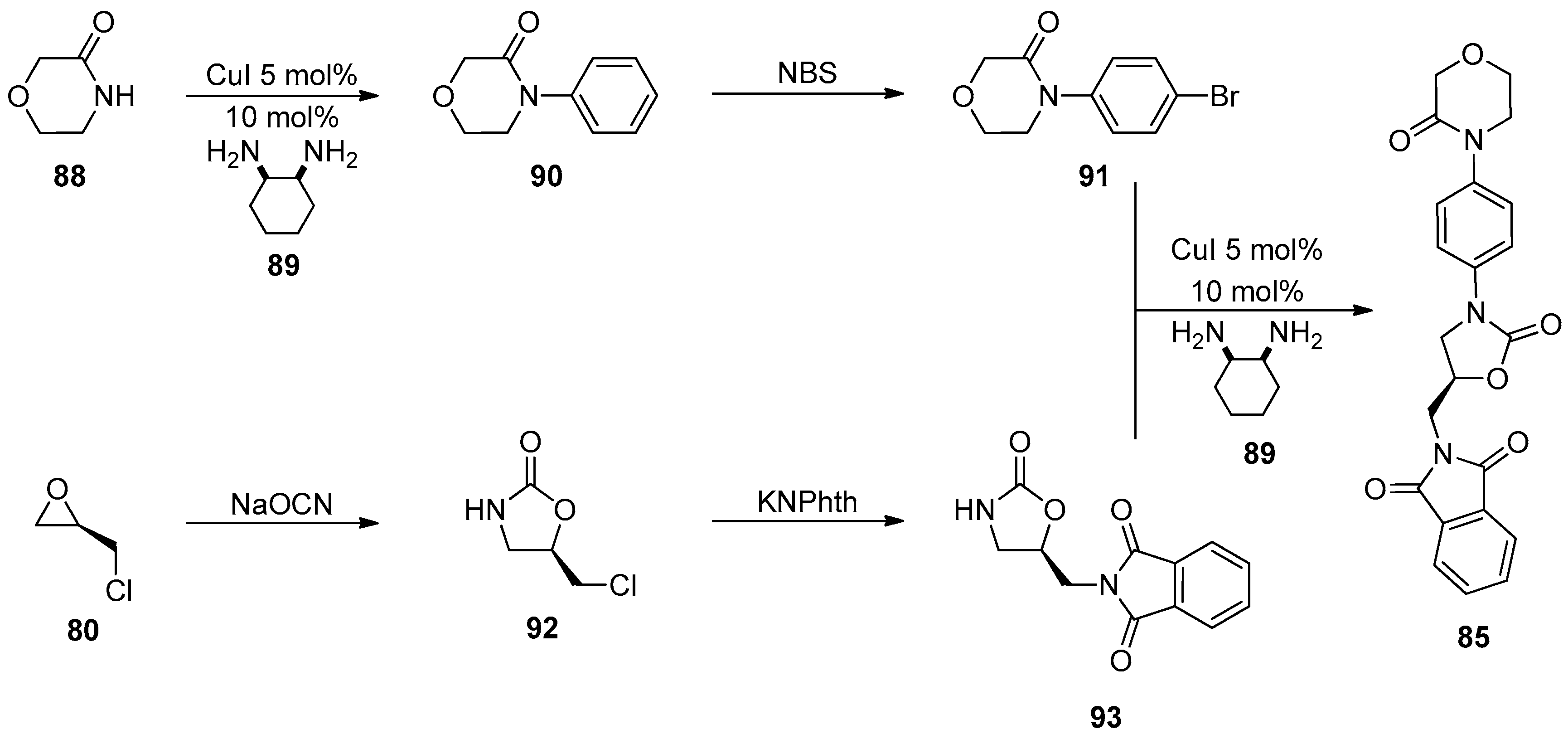
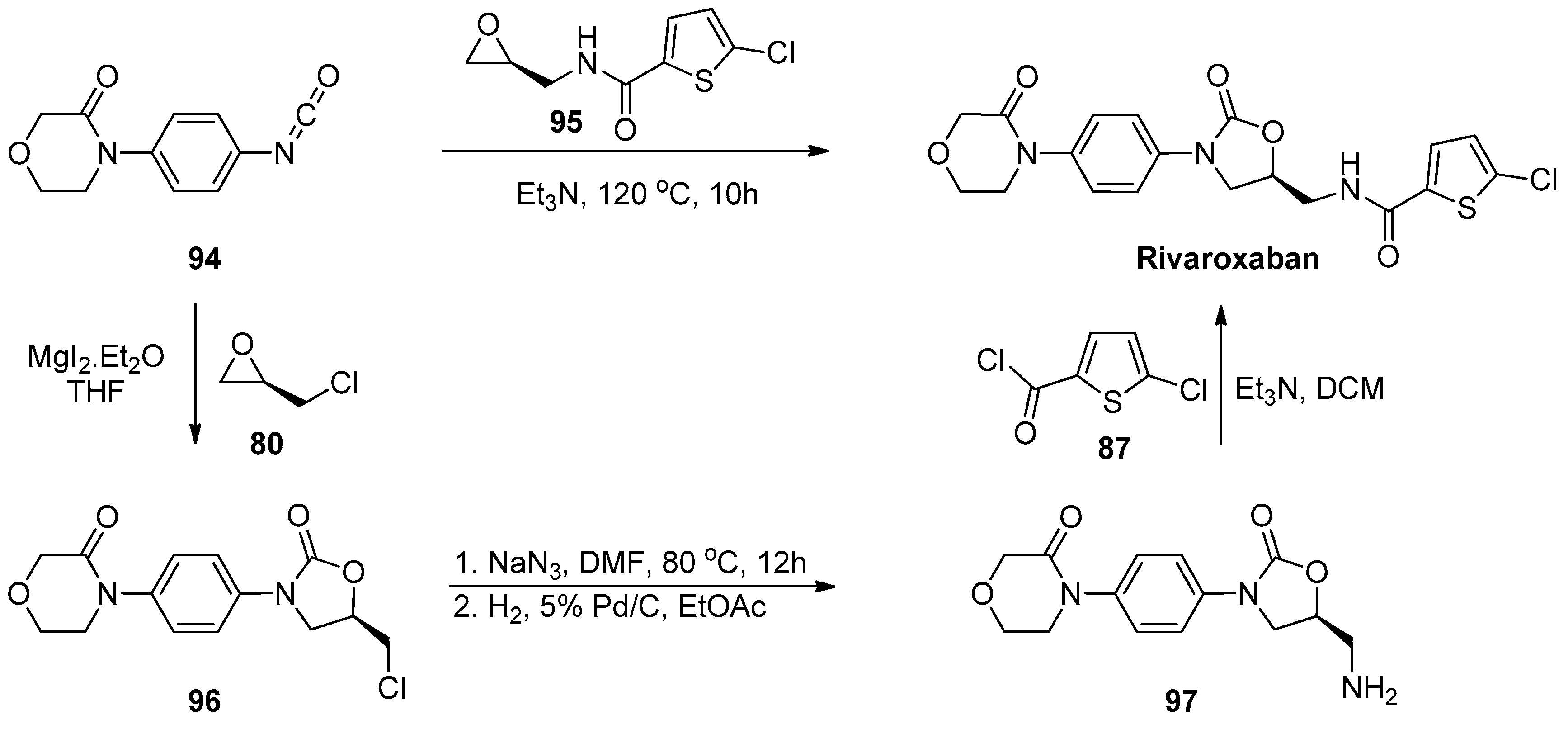
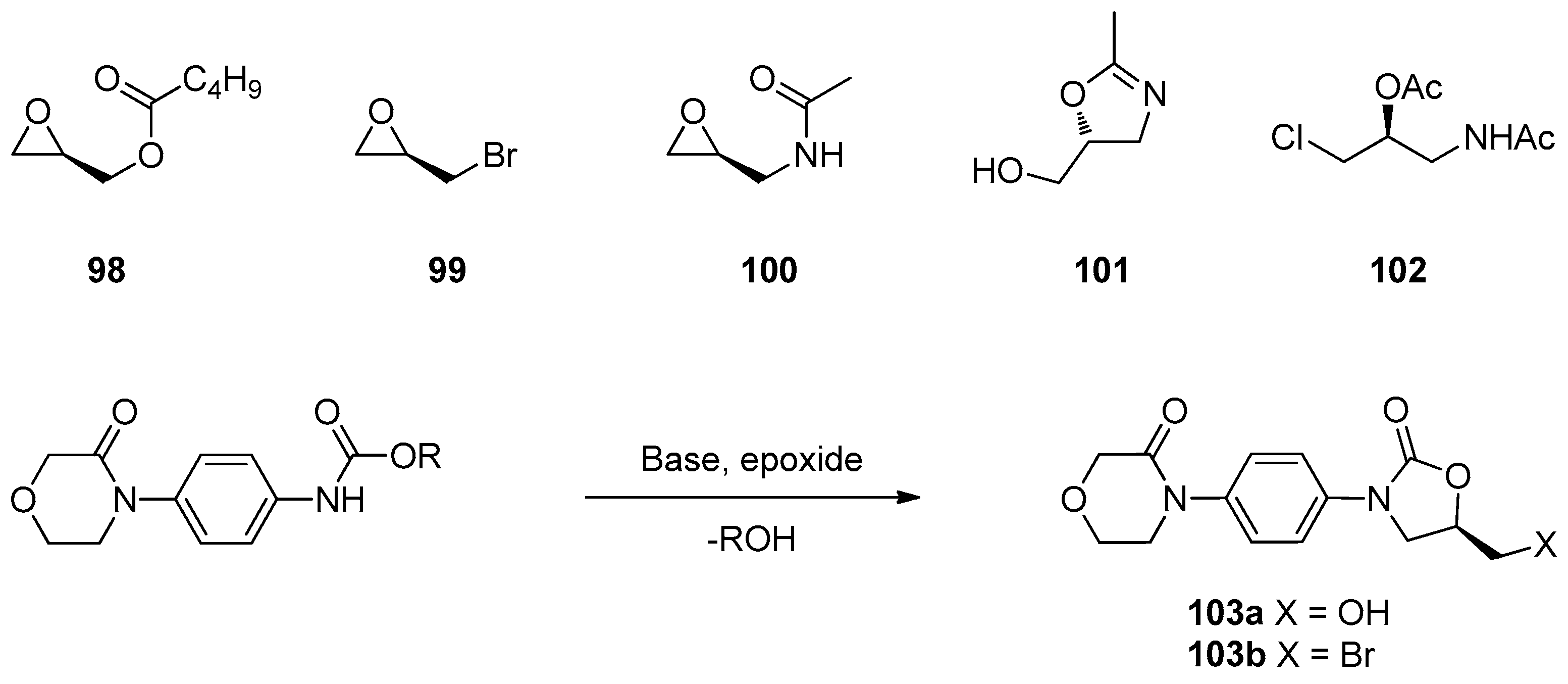
1.8. Linezolid and Rivaroxaban—Exploiting Glycerol Surrogates in Chiral Oxazolidinone Syntheses
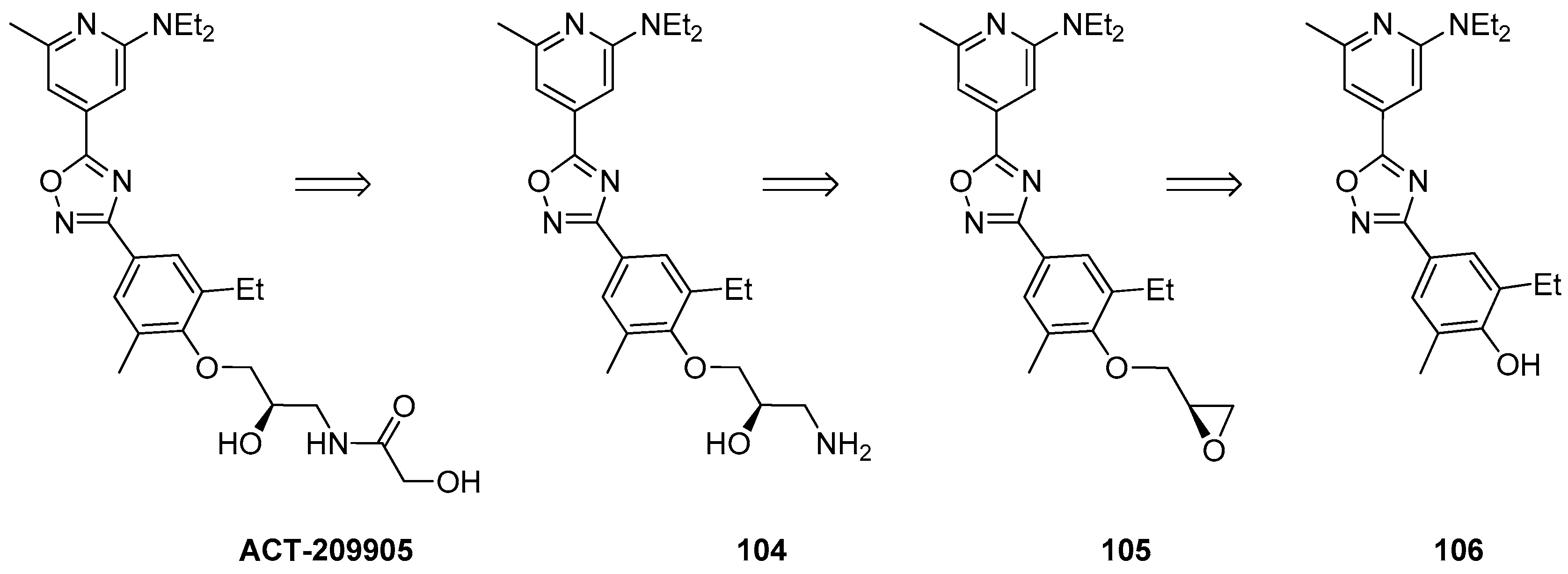
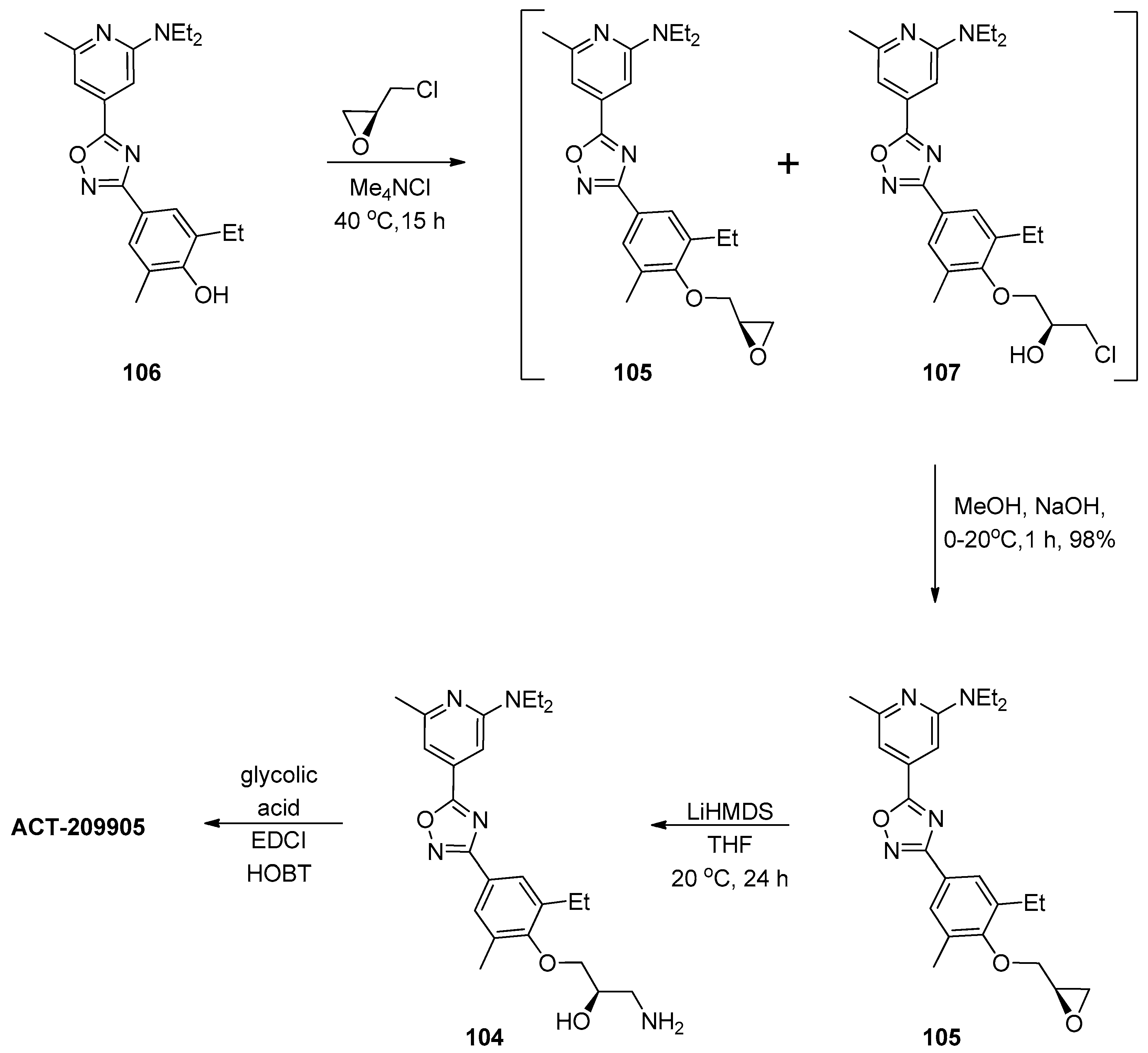
1.9. ACT-209905—Ring Opening of an Epoxide with Ammonia Surrogates
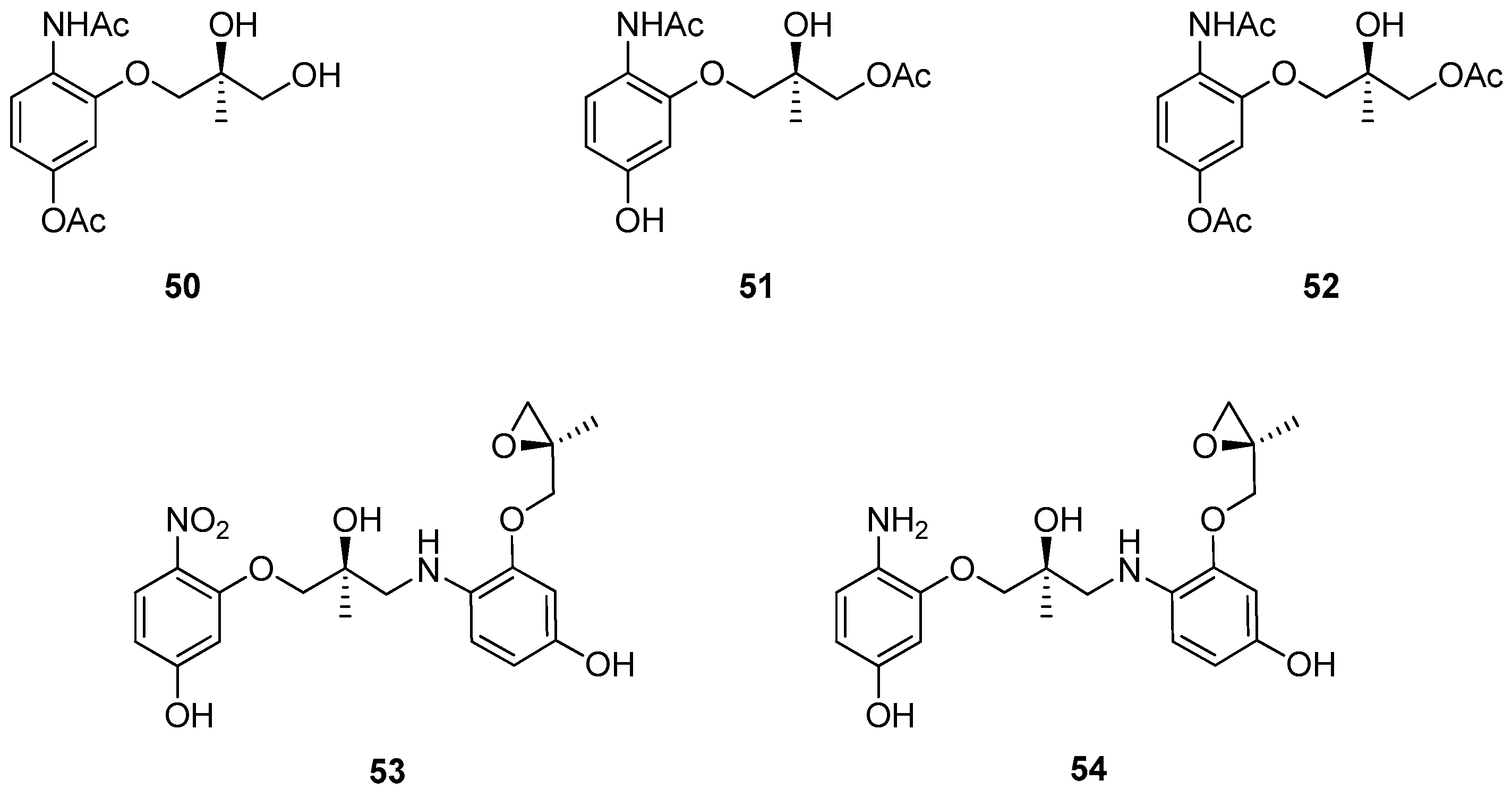
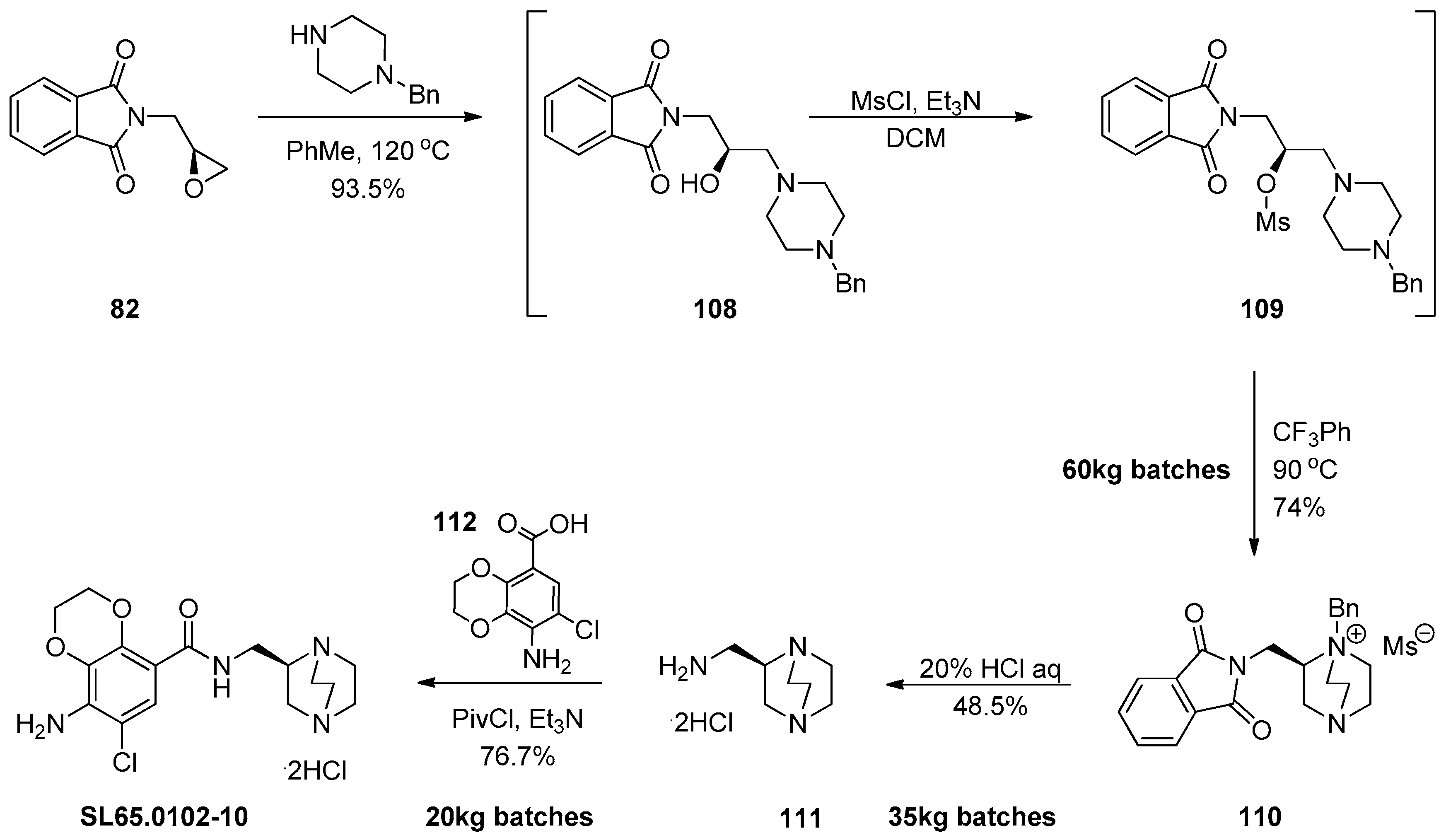
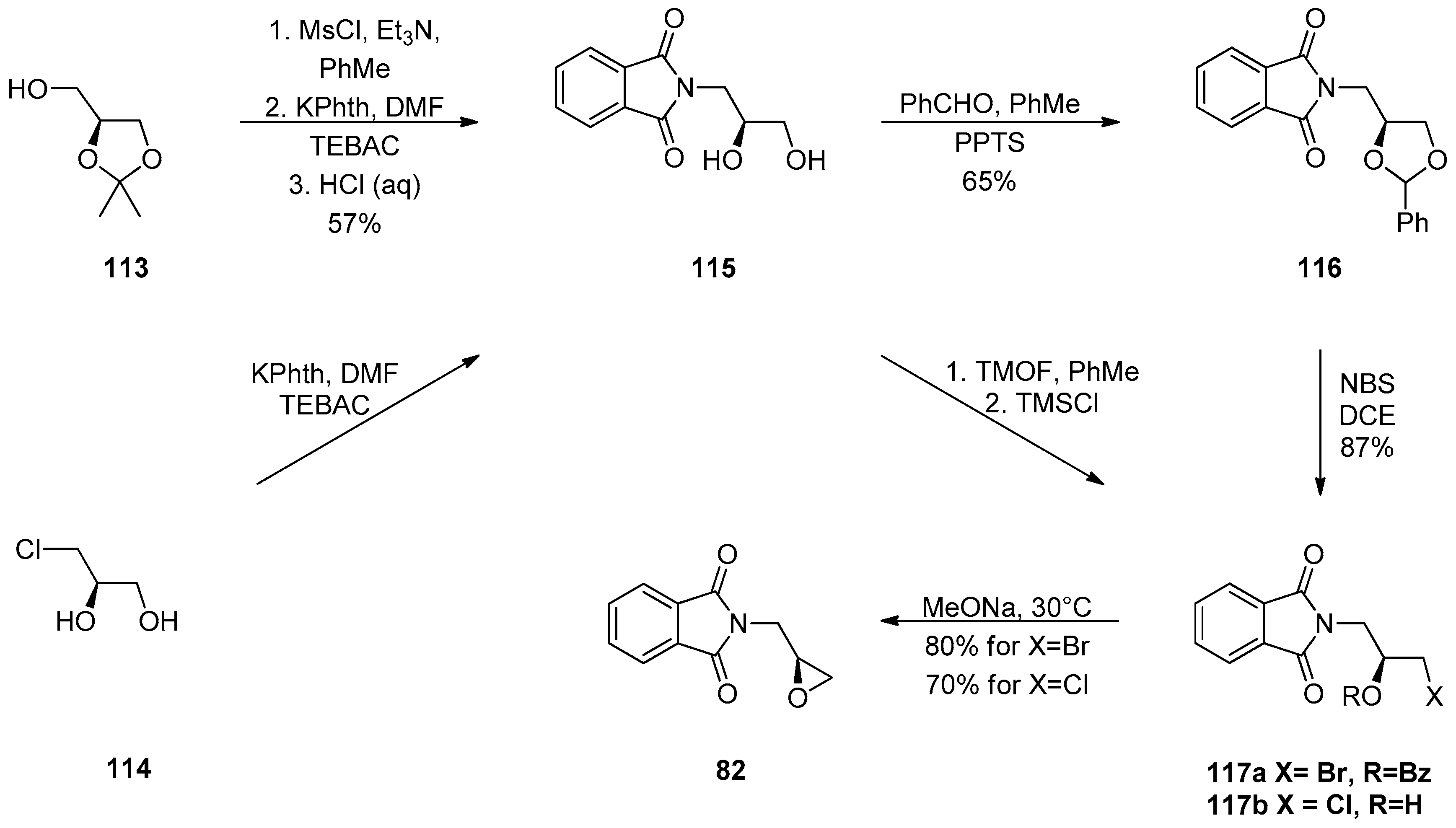
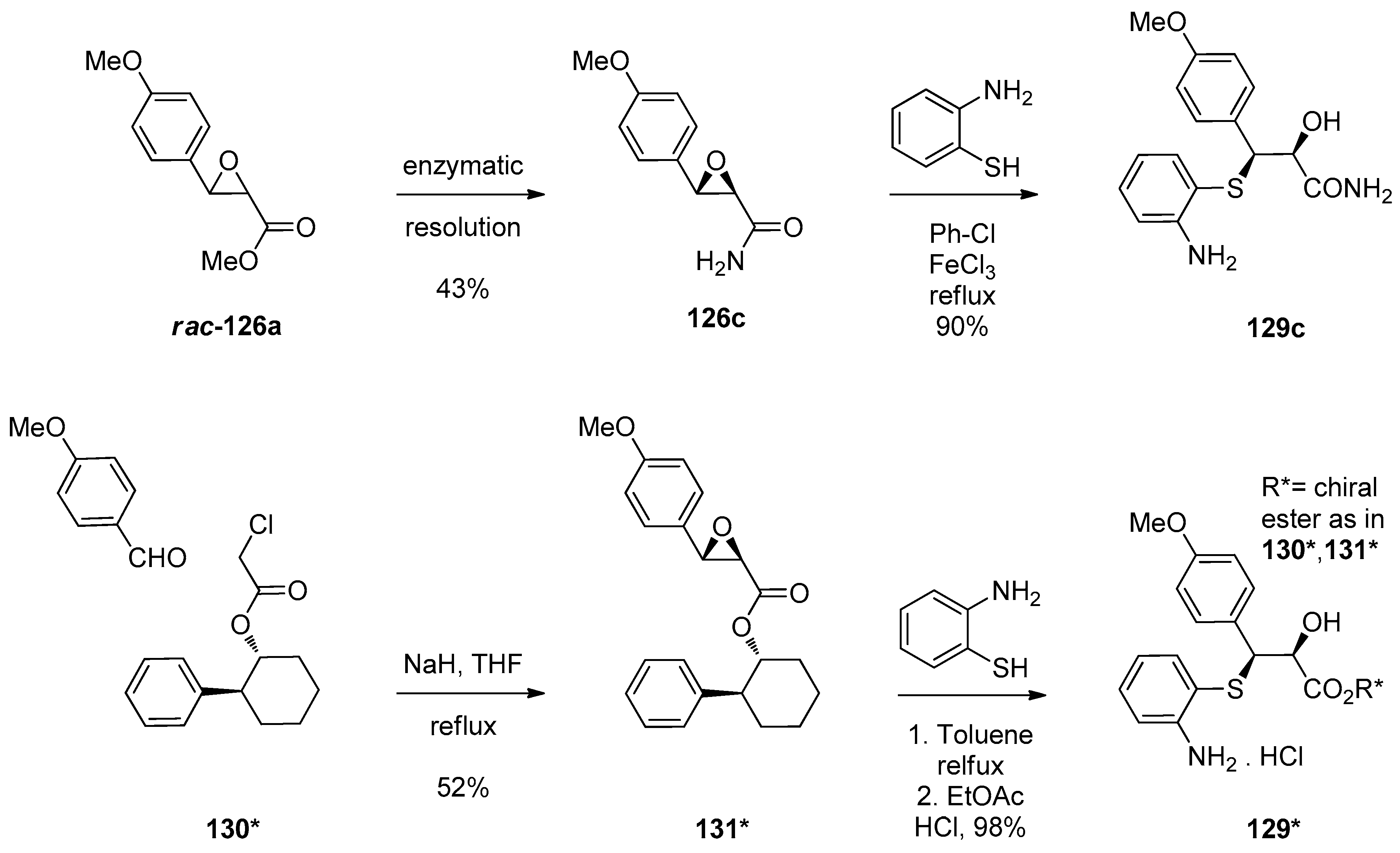
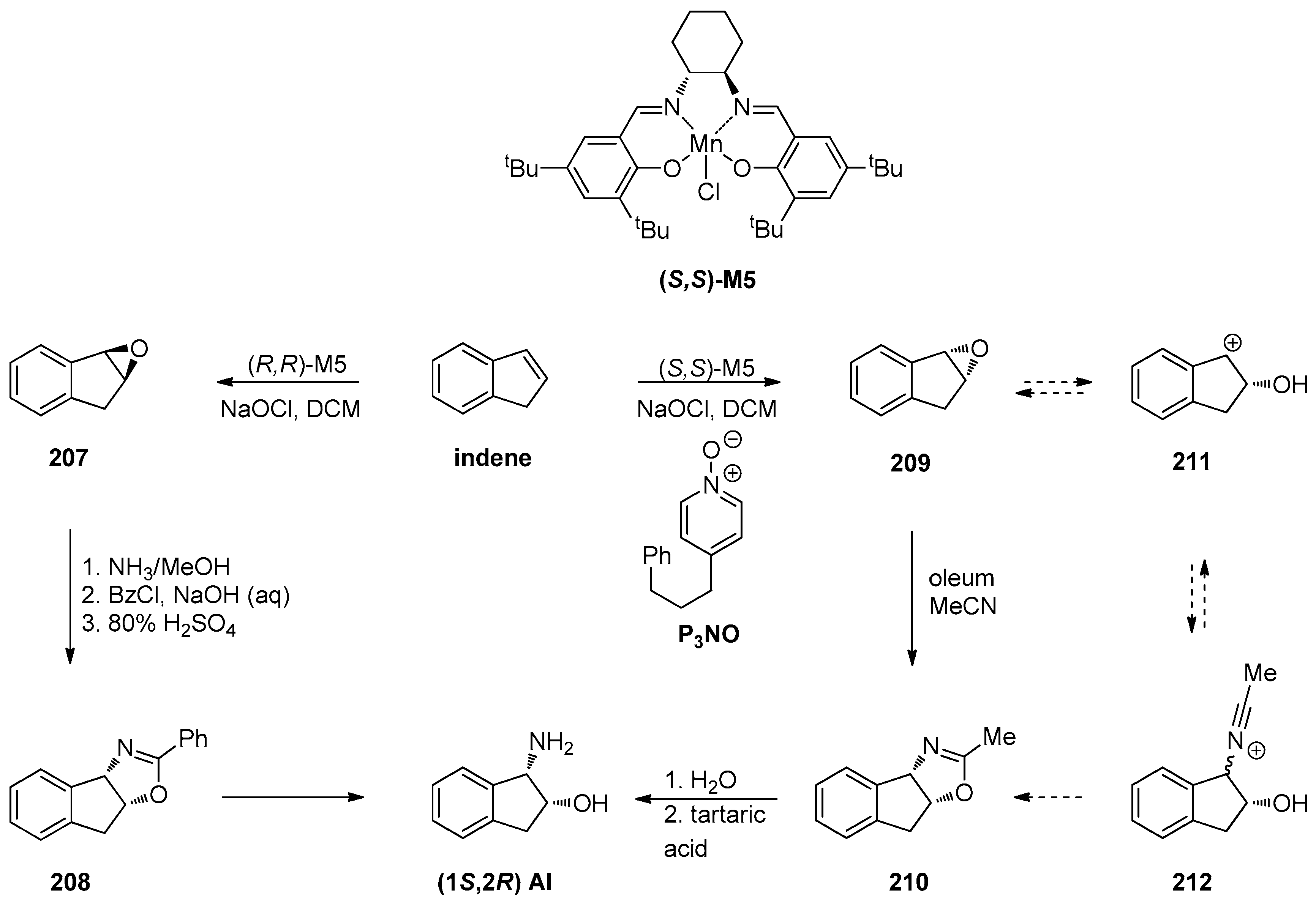
1.10. SL65.0102–10—Refocusing Attention from the Active Pharmaceutical Ingredient to the Chiral Epoxide Starting Material
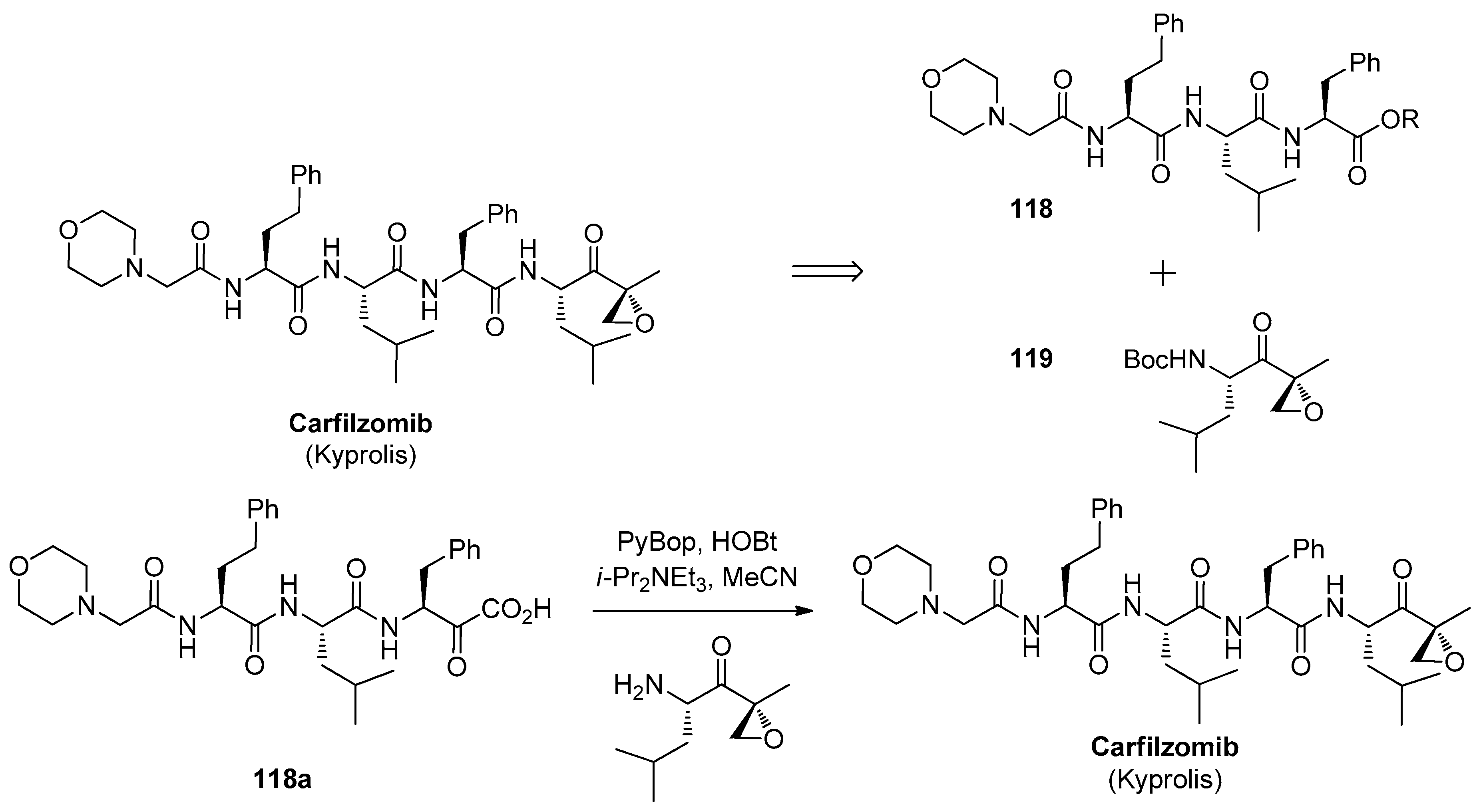
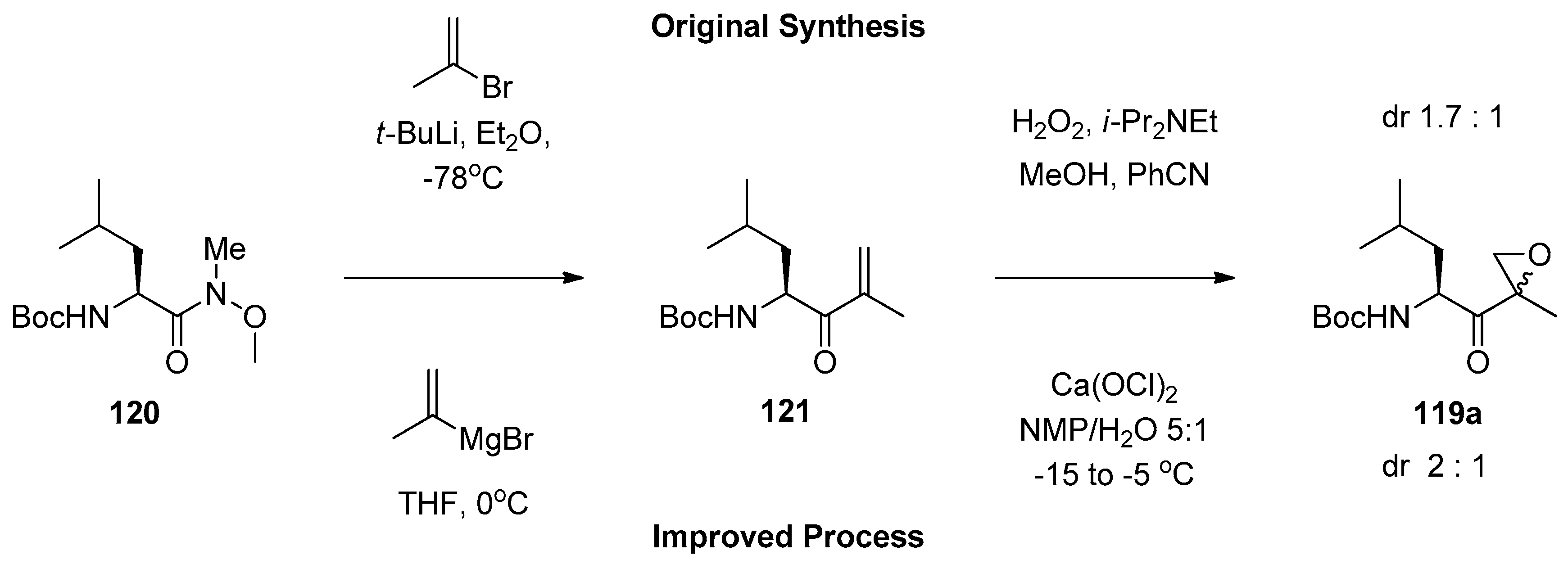
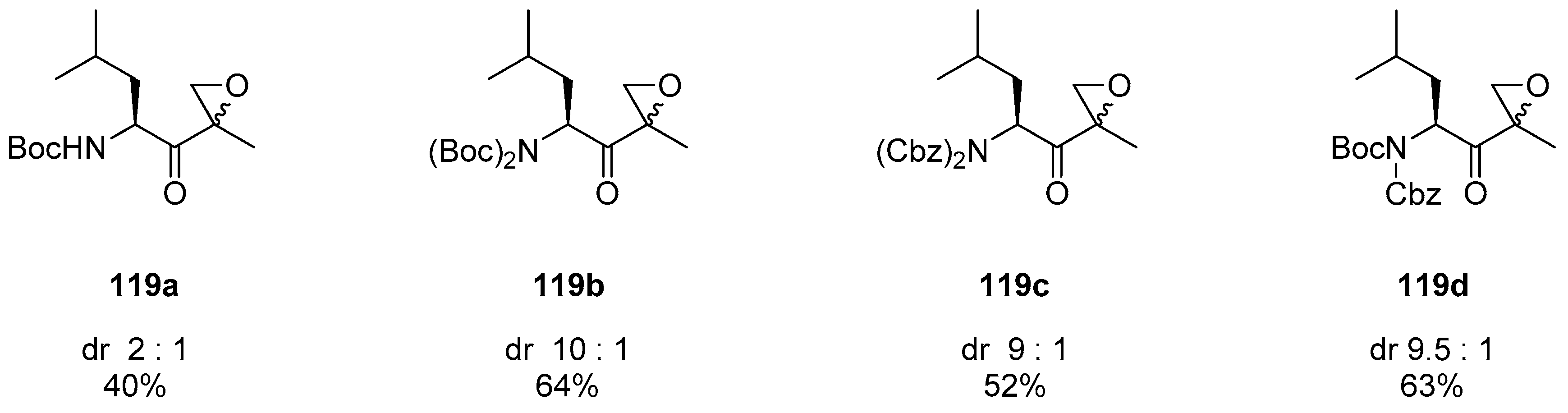
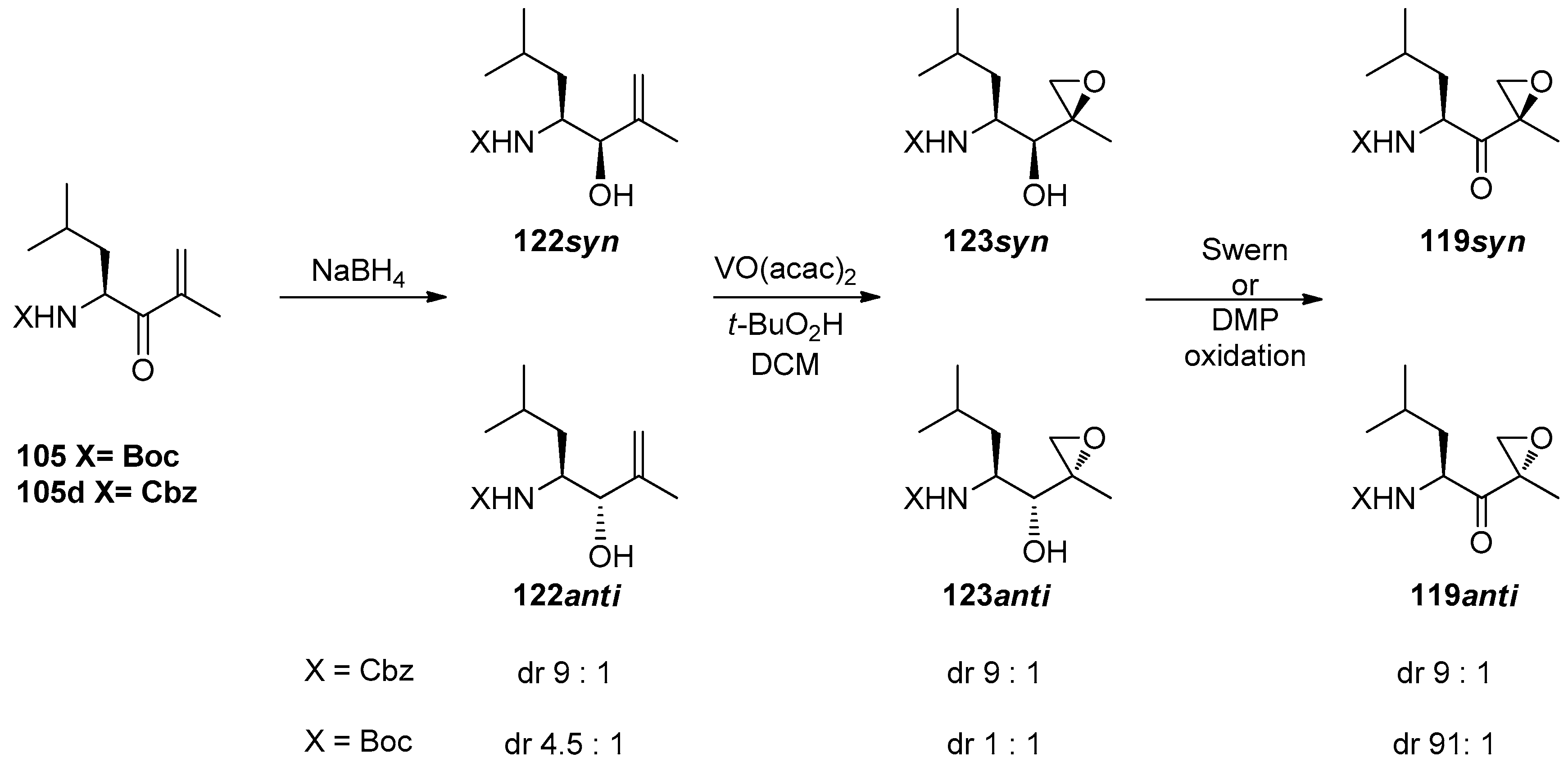
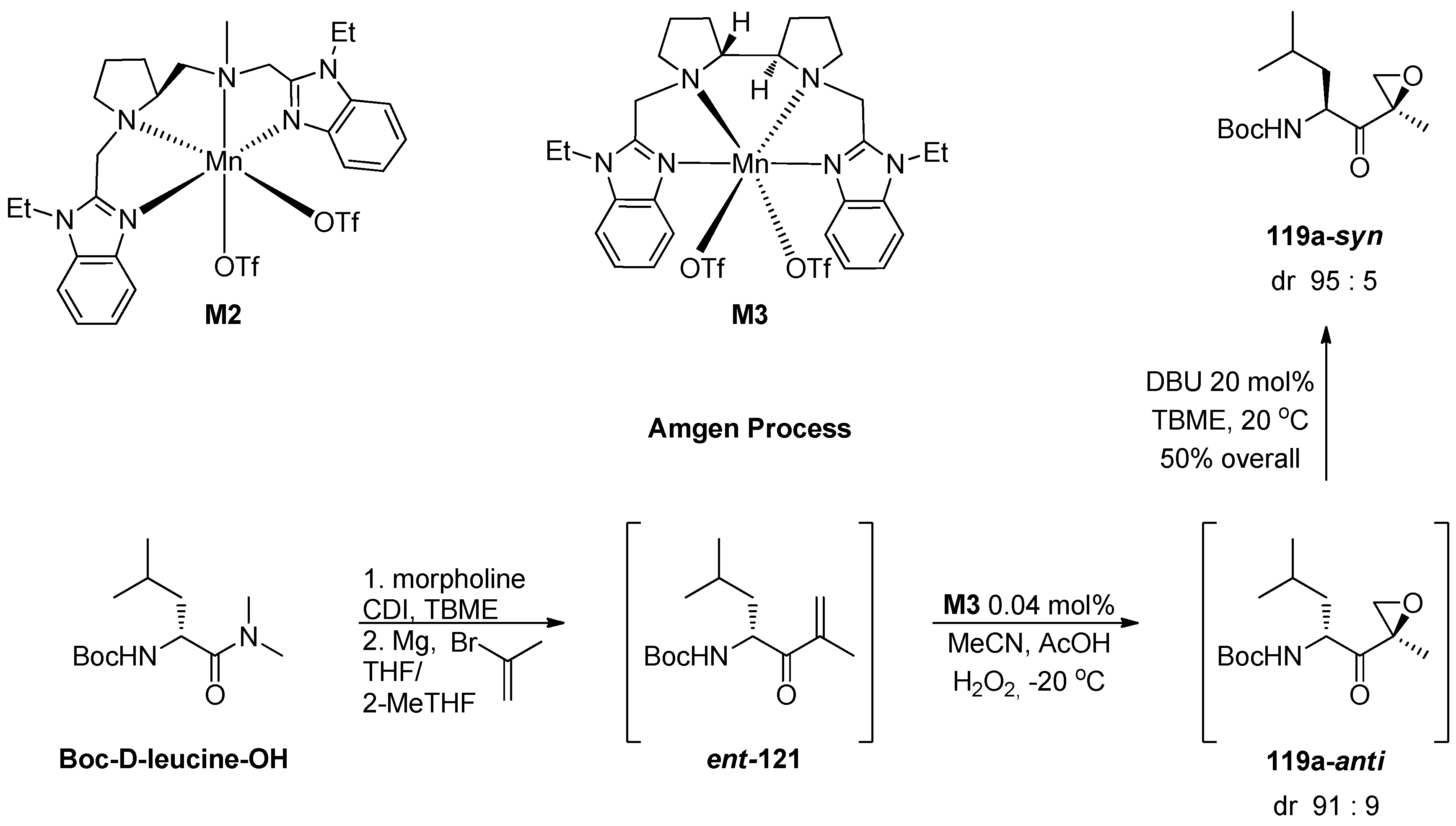
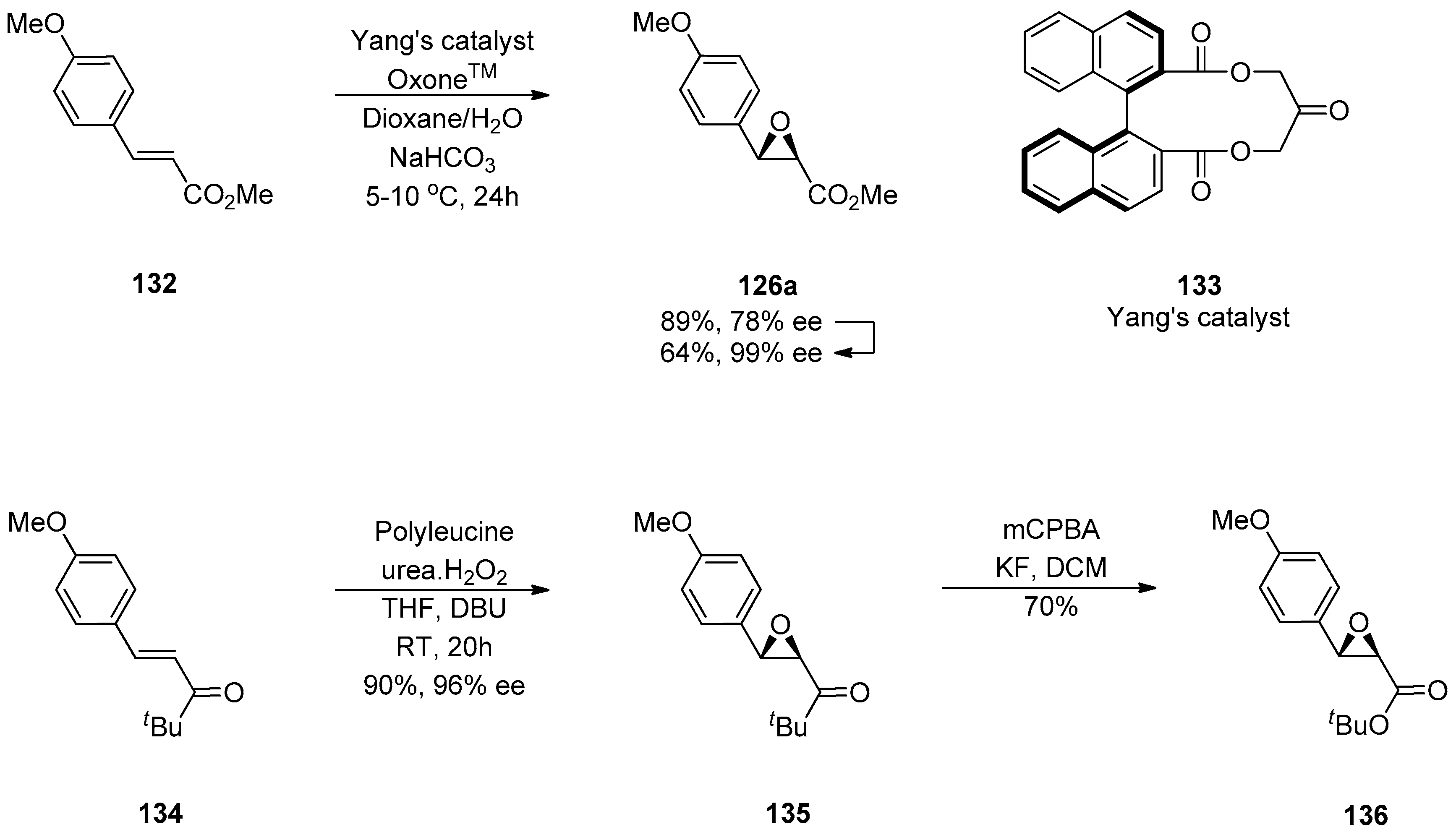
1.11. Carfilzomib—A Natural Epoxide Product Turned into a Drug and the Challenge of Its Diastereoselective Epoxidation
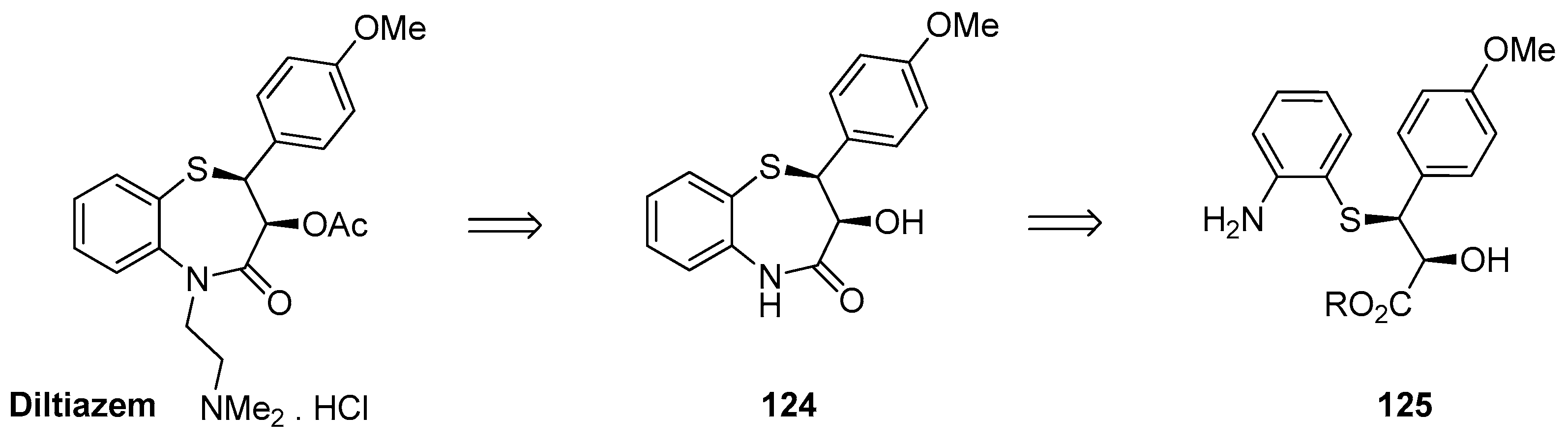
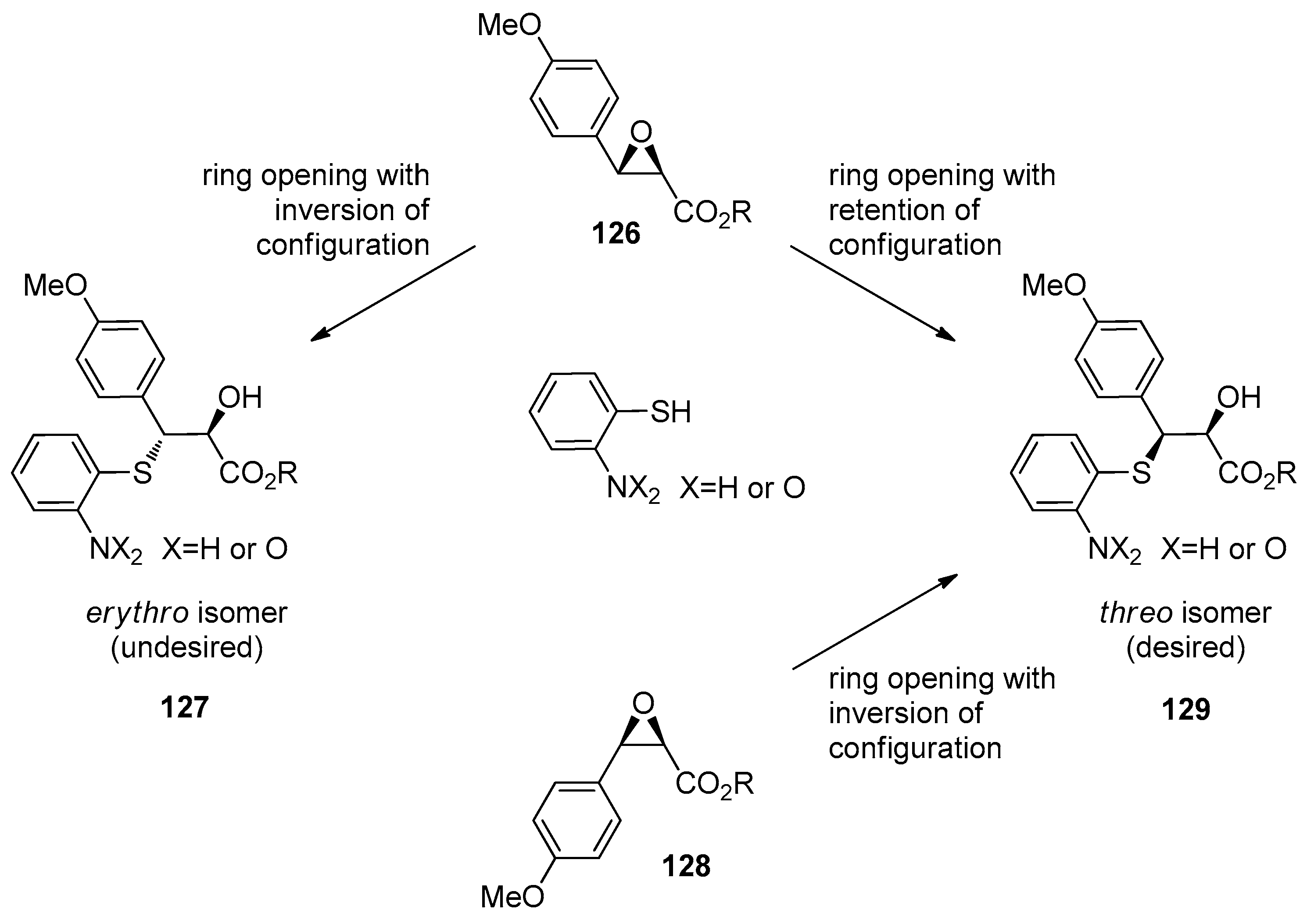
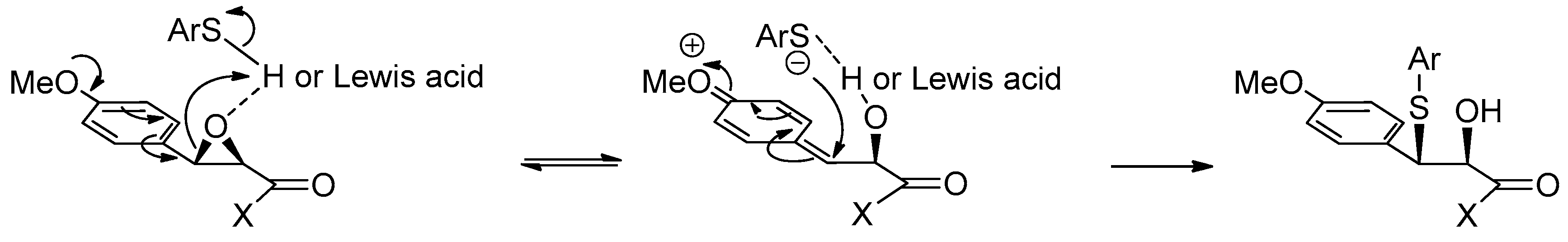
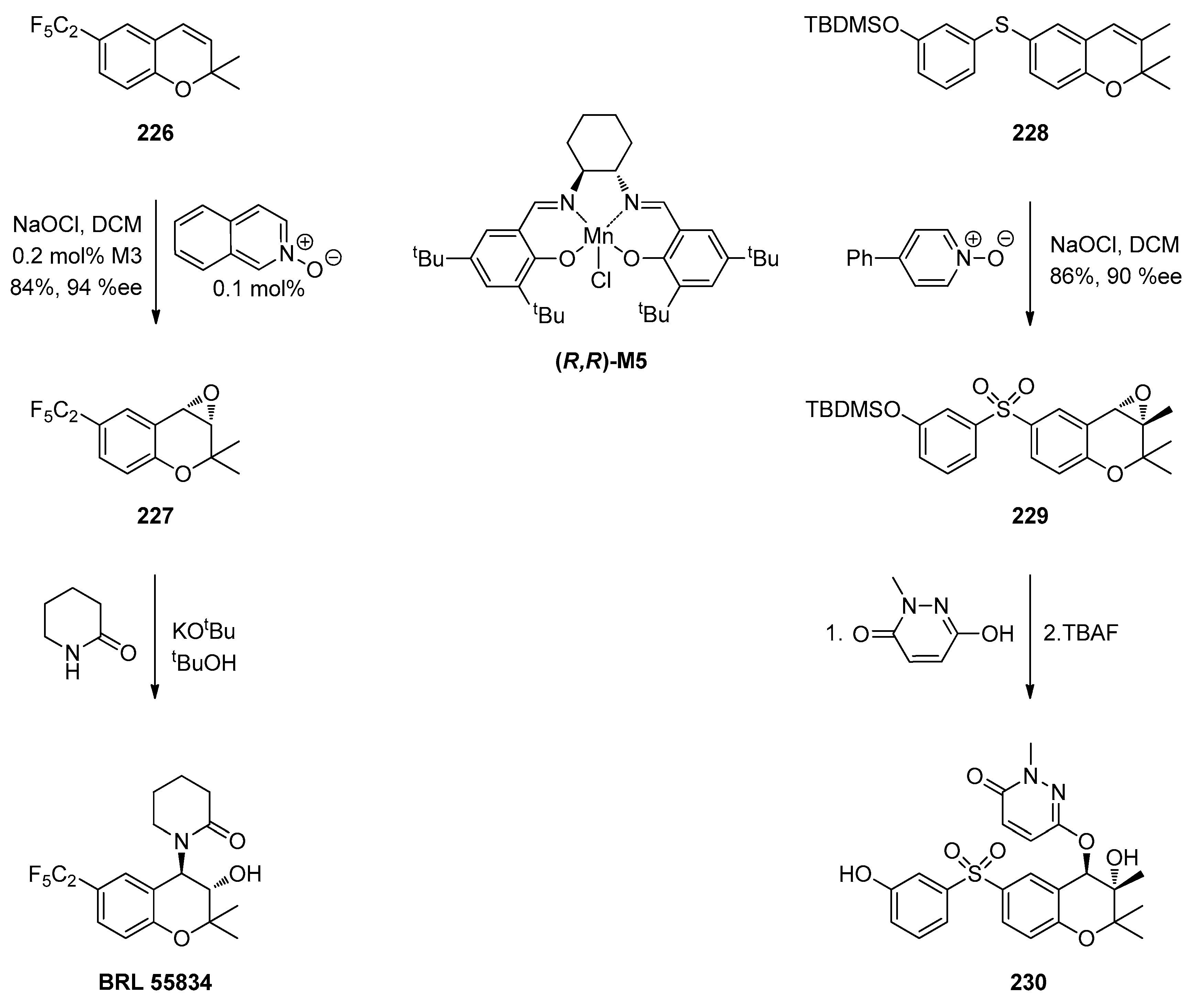
1.12. Diltiazem—A Rare Example of Chiral Epoxide Industrial Synthesis via Asymmetric Organocatalysis Followed by Ring Opening with Retention of Configuration
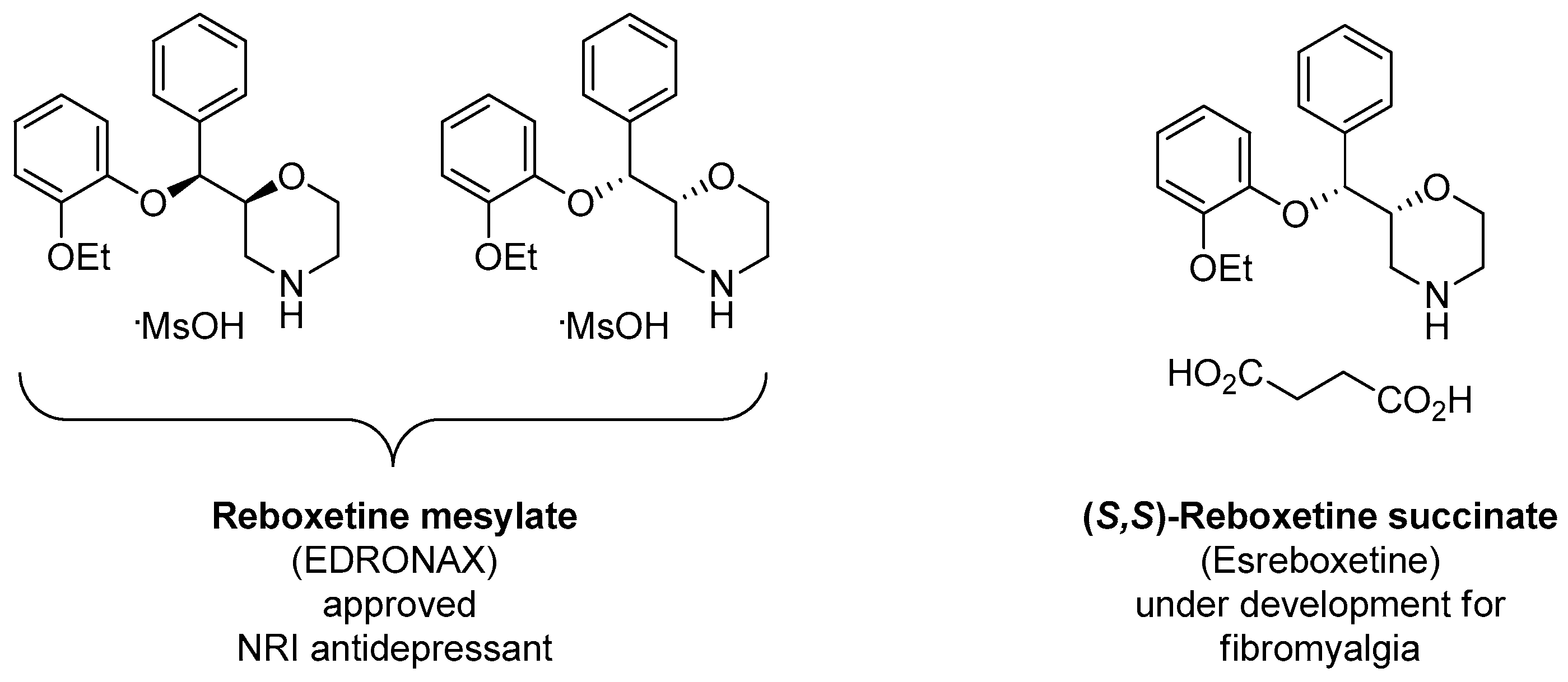
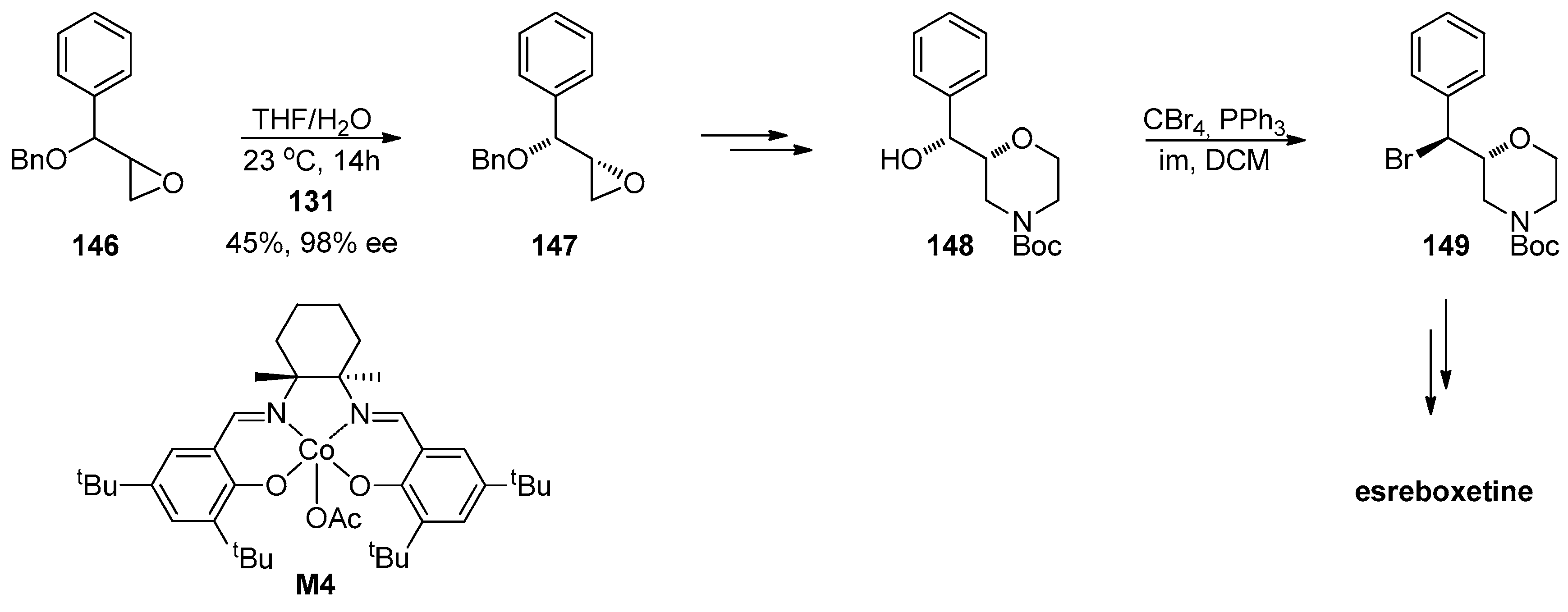
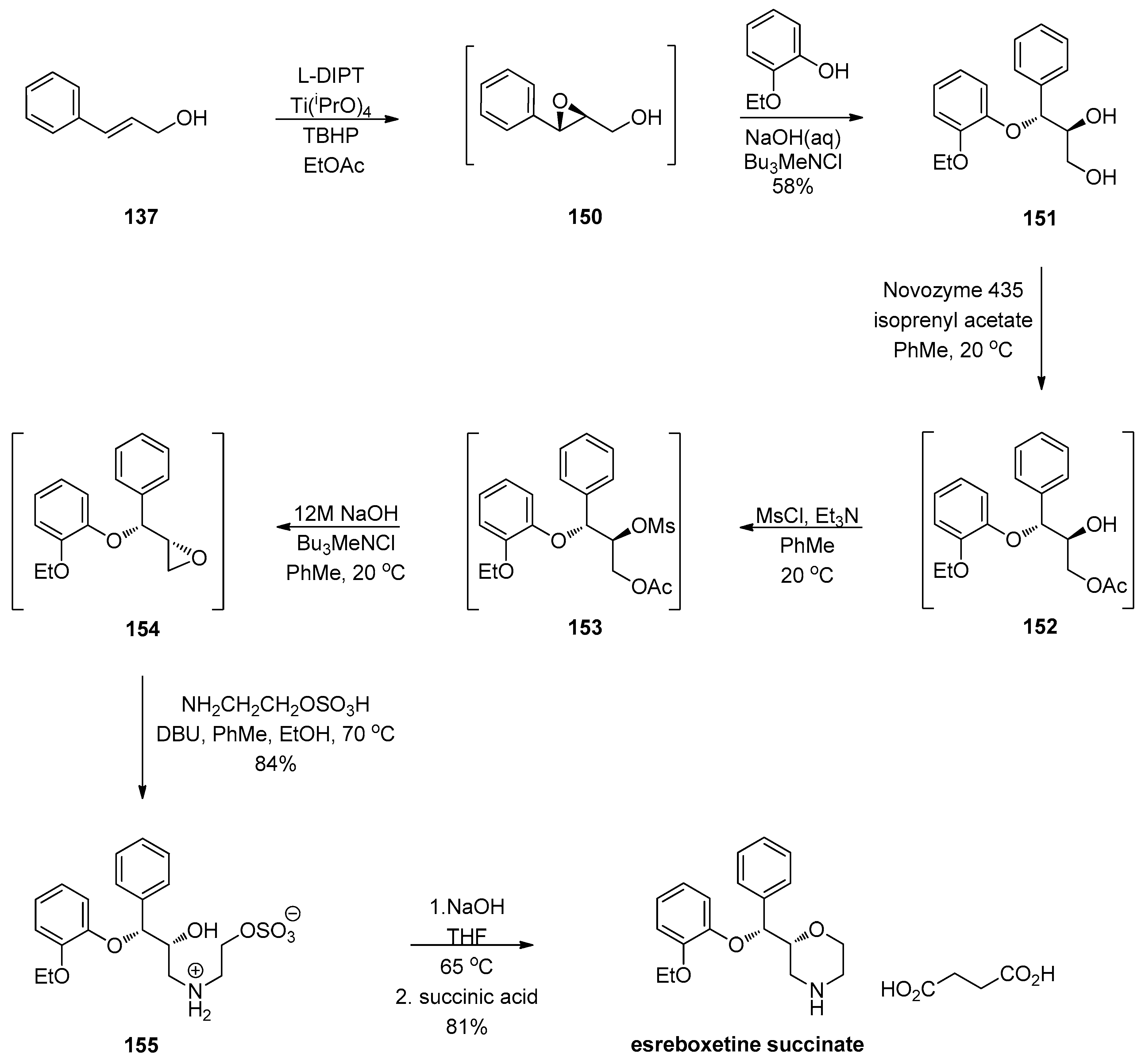
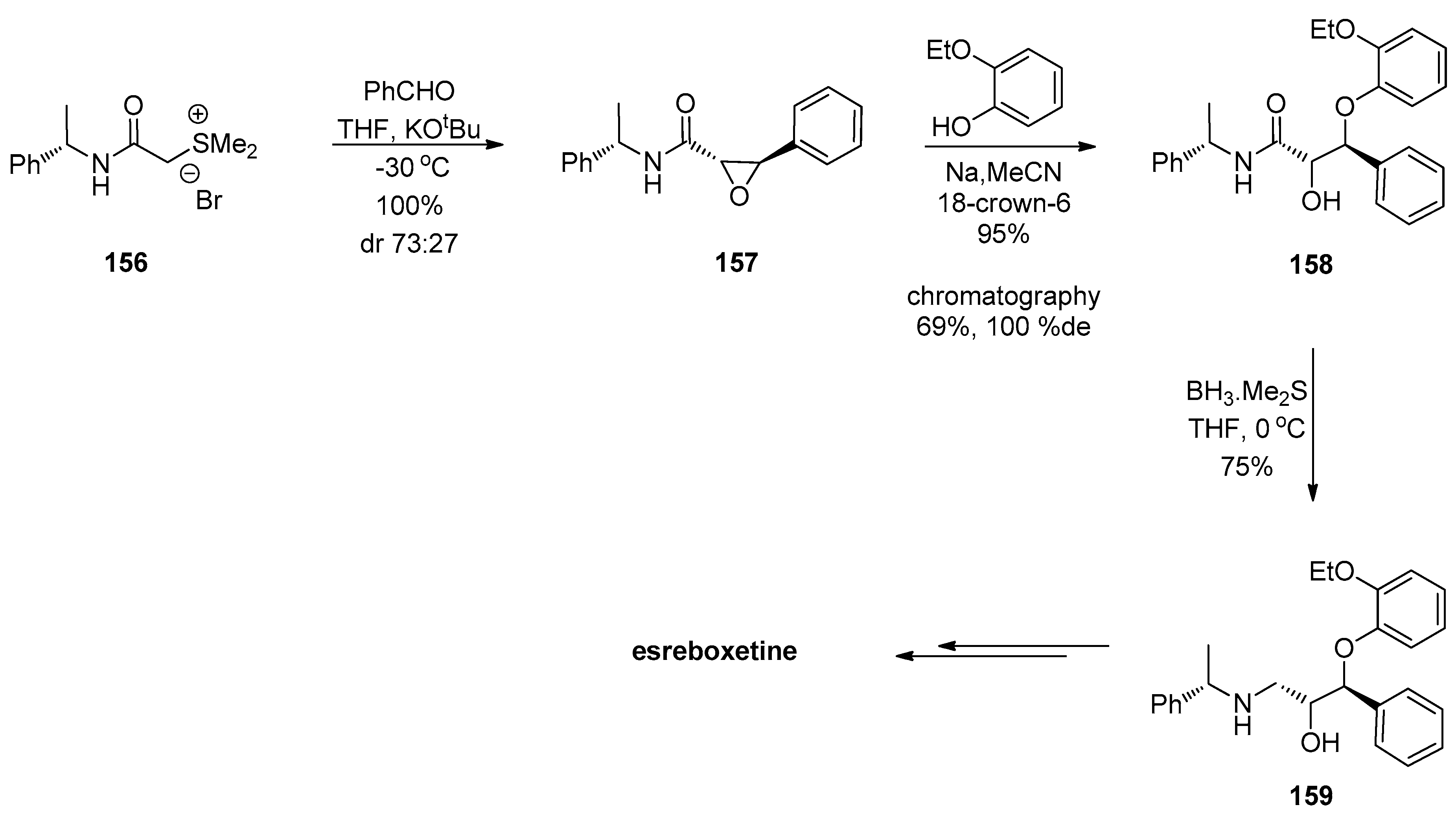
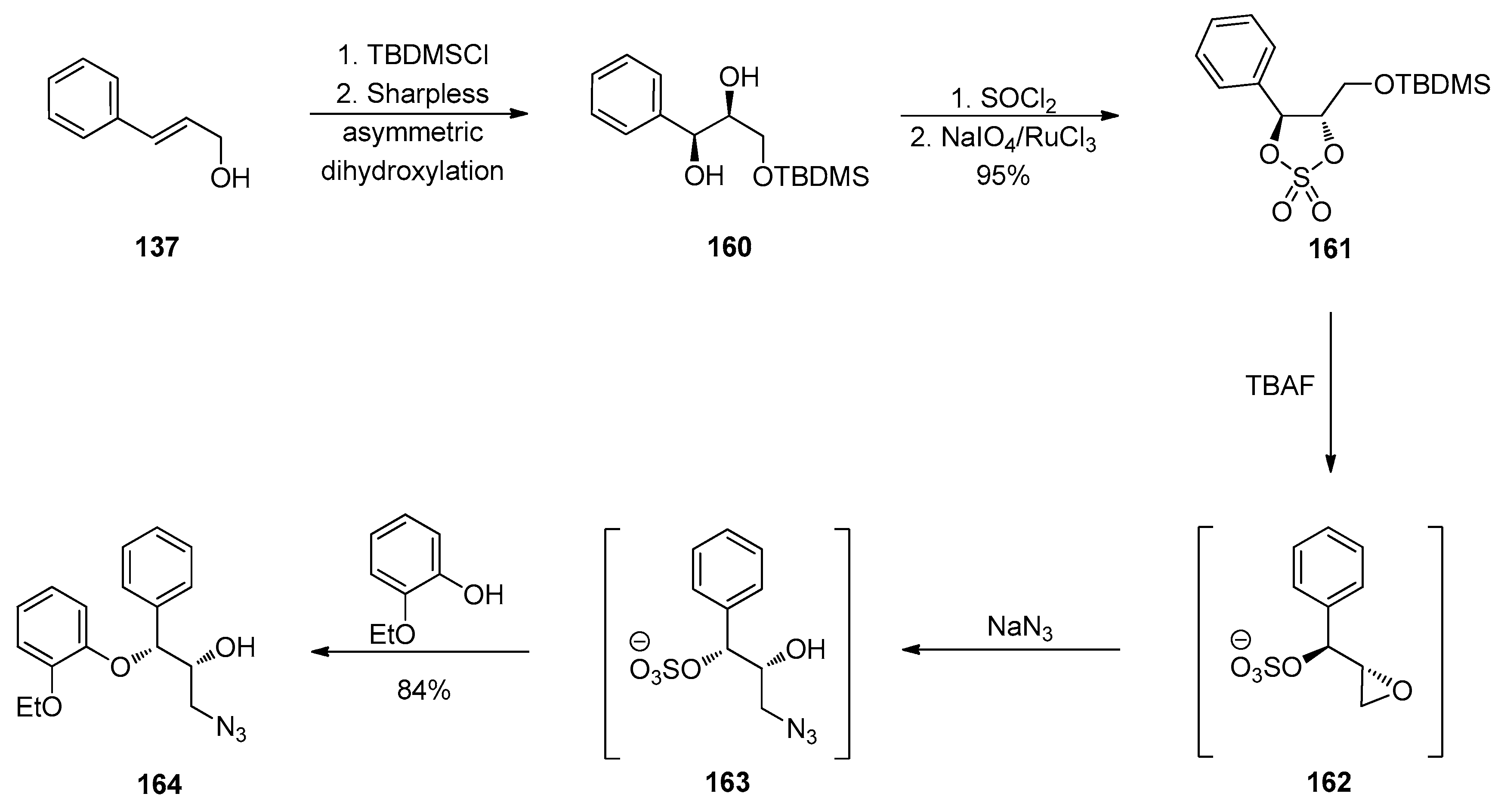
1.13. Reboxetine—Epoxide Intermediates Transcending from a Racemic to an Asymmetric Synthesis
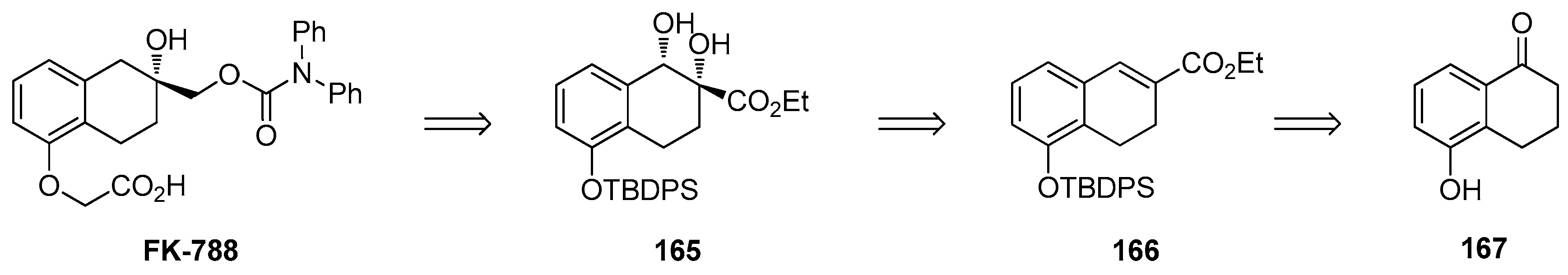
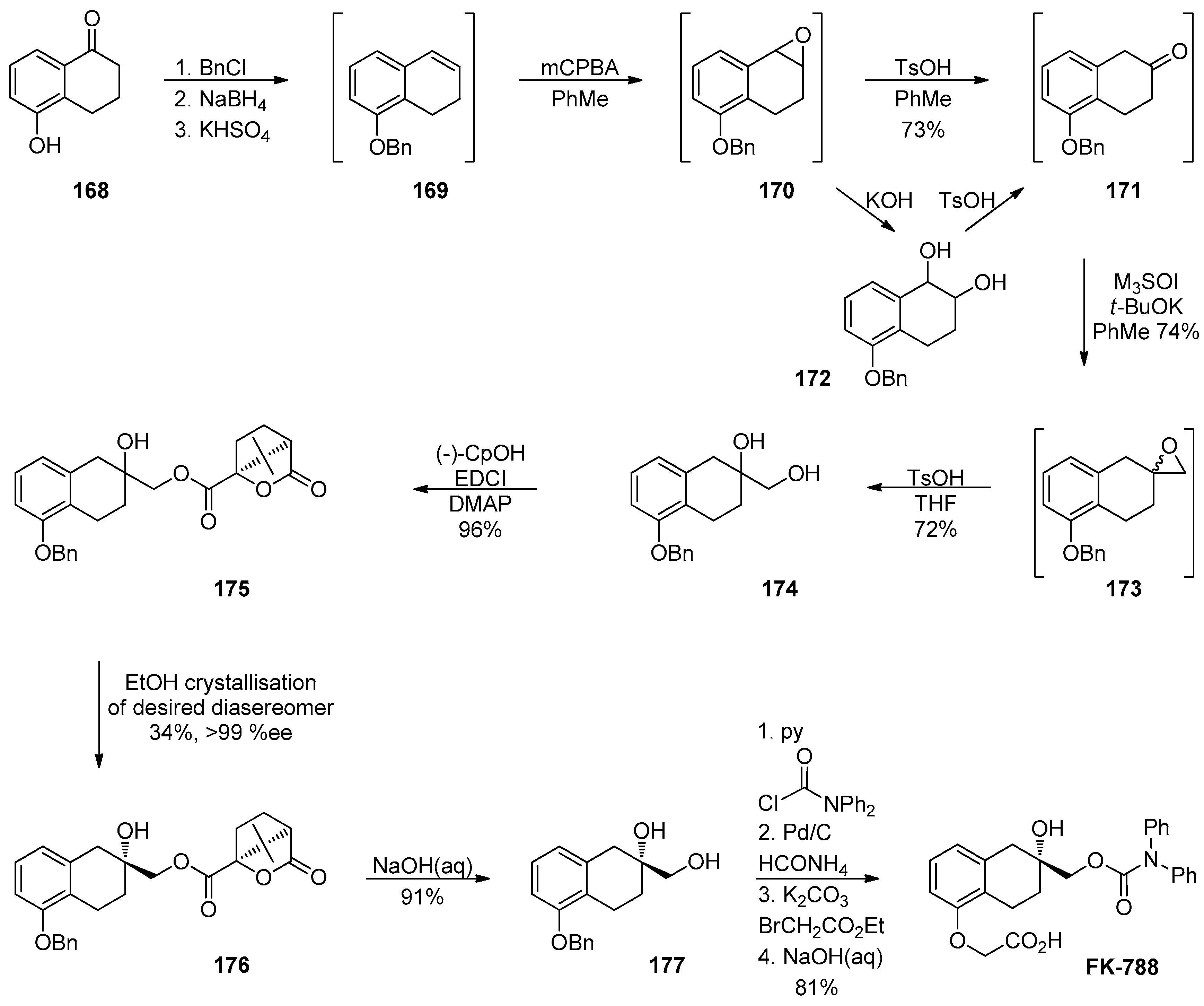
1.14. FK-788—Replacement of an Asymmetric Synthesis by a Resolution Process with Recycling of the Undesired Enantiomer; the Meinwald Rearrangement on Scale
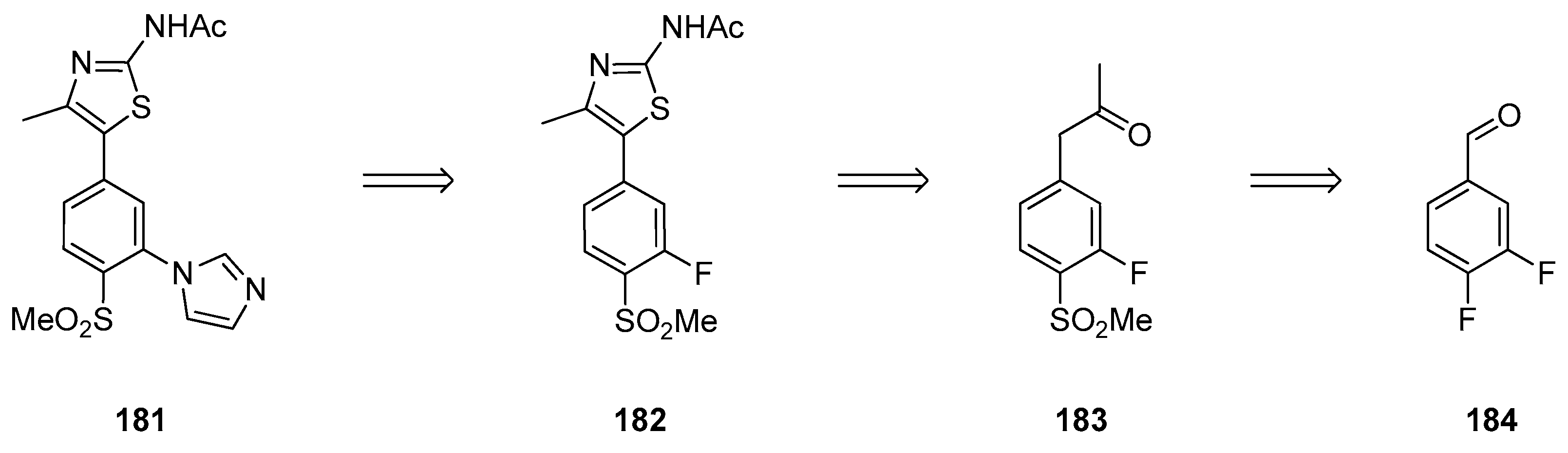
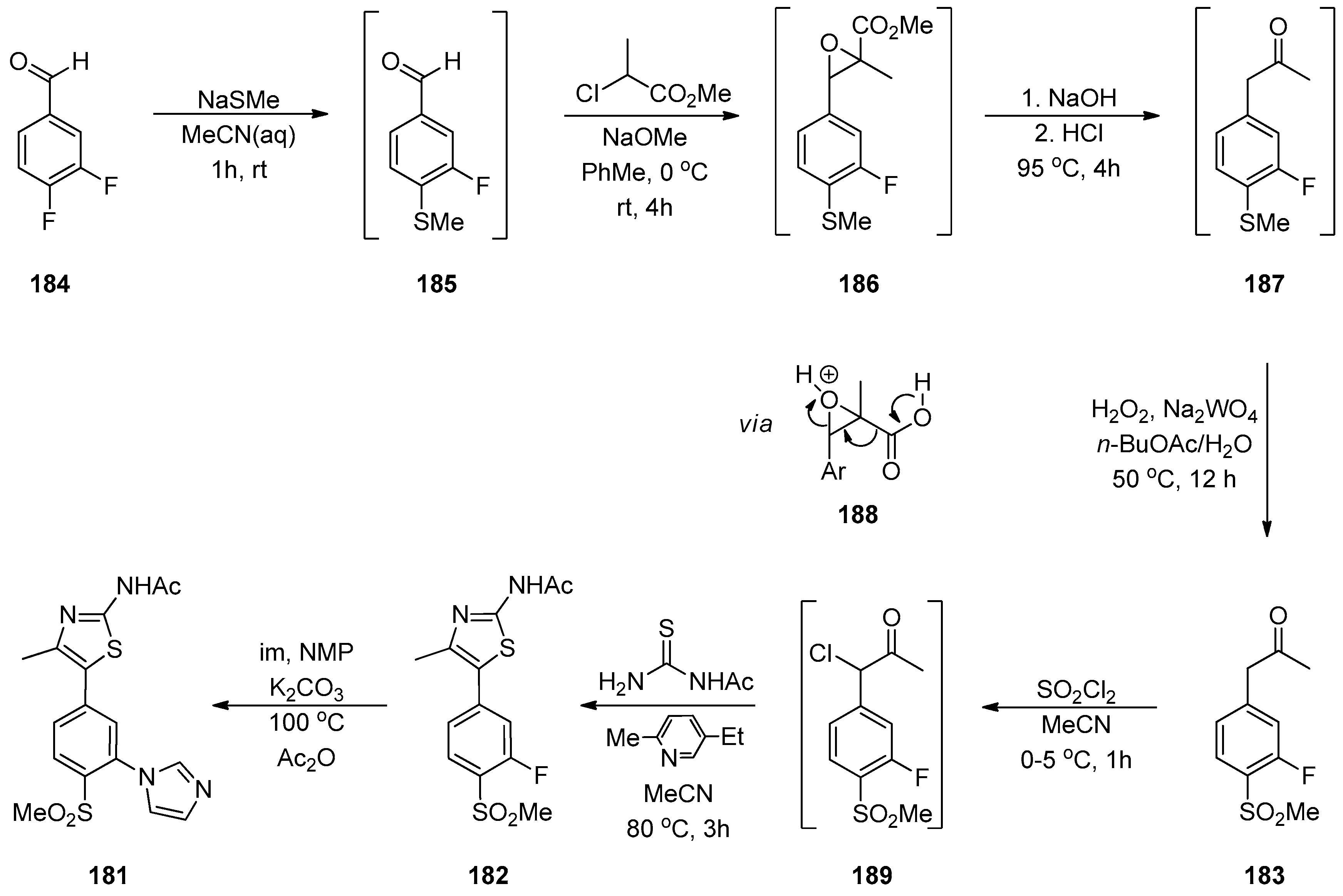
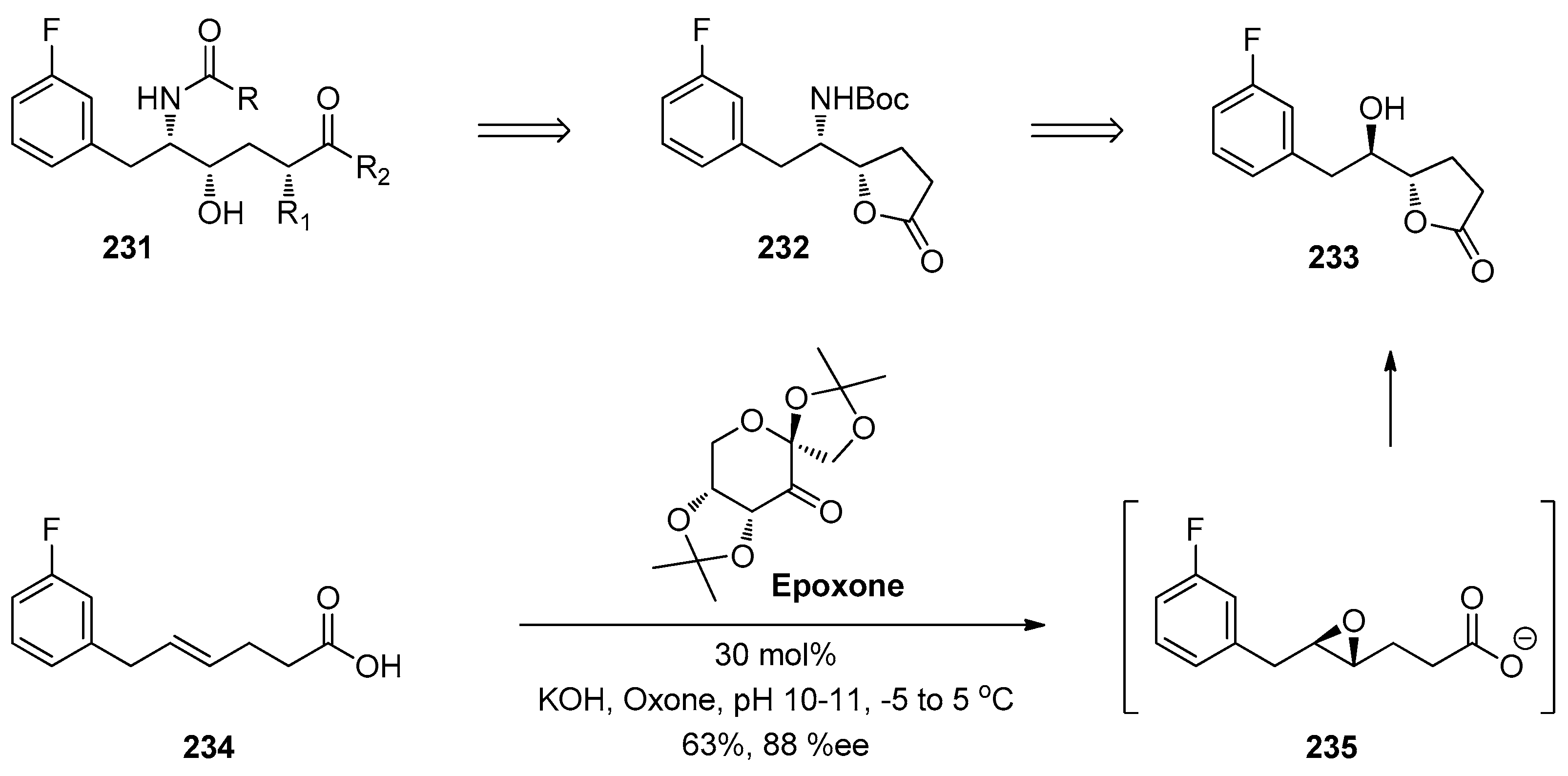
1.15. A Lead Candidate for Asthma—Improved Synthesis via a Decarboxylative Rearrangement of an α-Carboxyl Epoxide
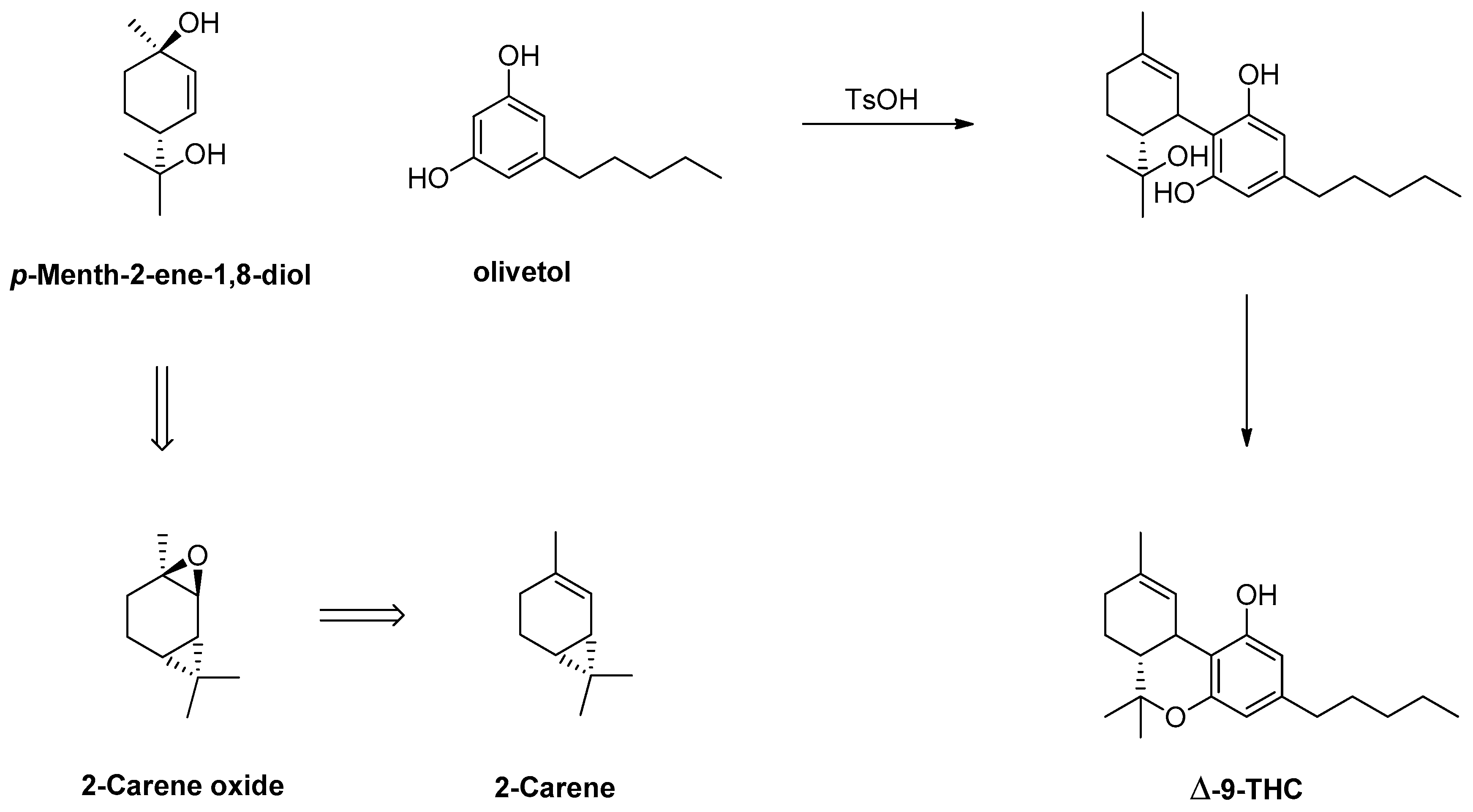
1.16. Δ-9-Tetrahydrocannabinol—From 3-Carene to 2-Carene Oxide and Δ-9-THC in Large Scale
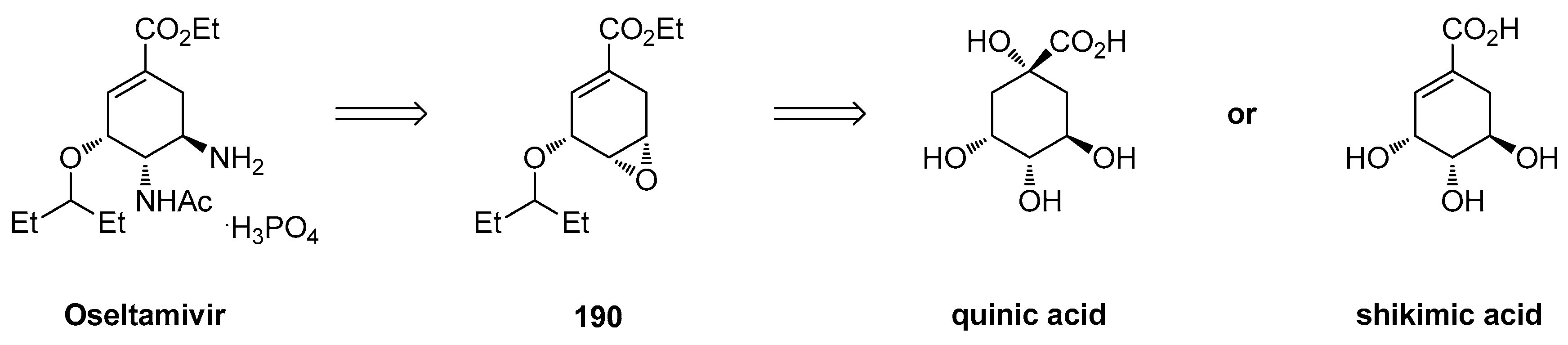
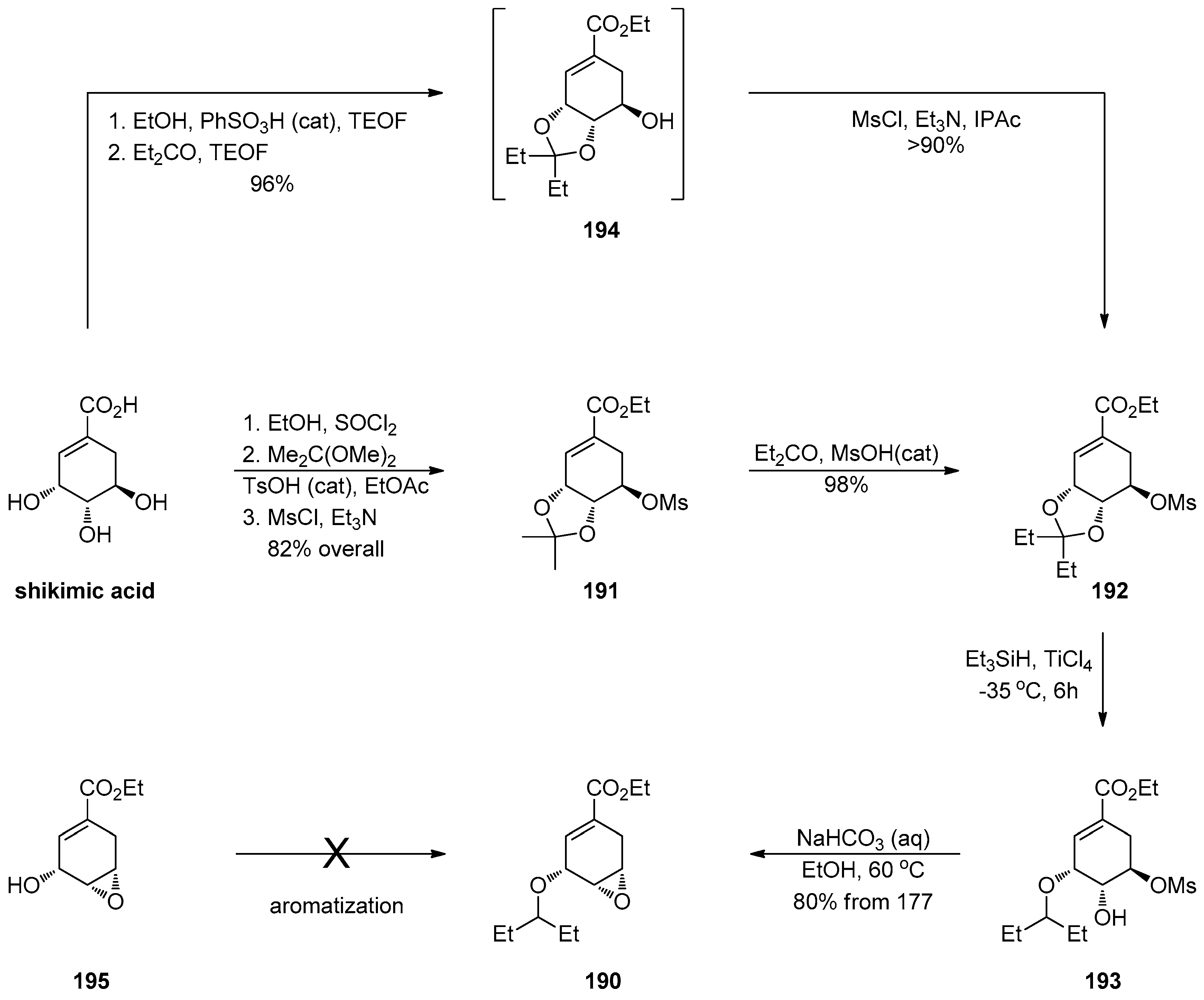
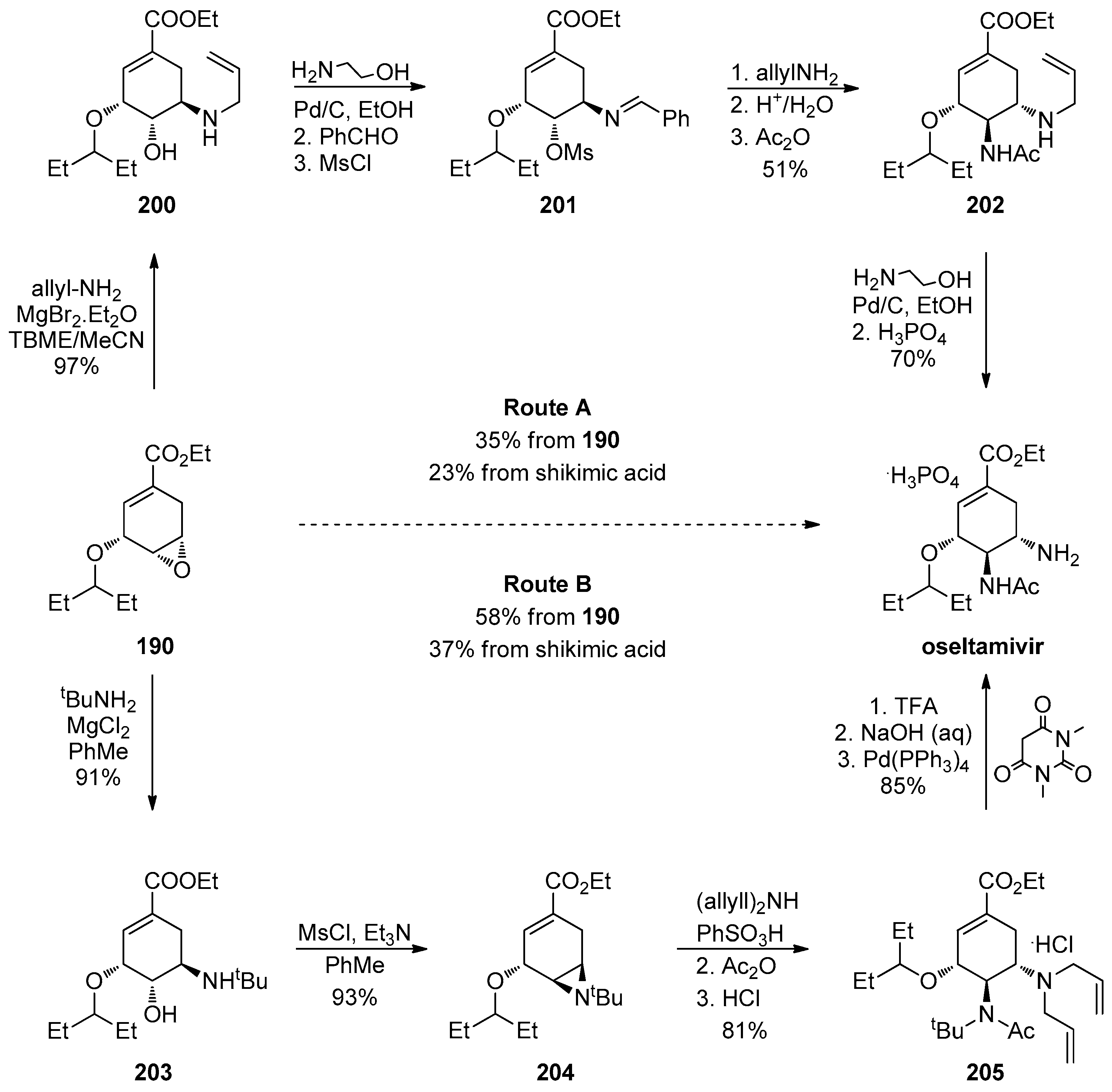
1.17. Oseltamivir—Development of Three Manufacturing Routes via the Same Key Epoxide Intermediate
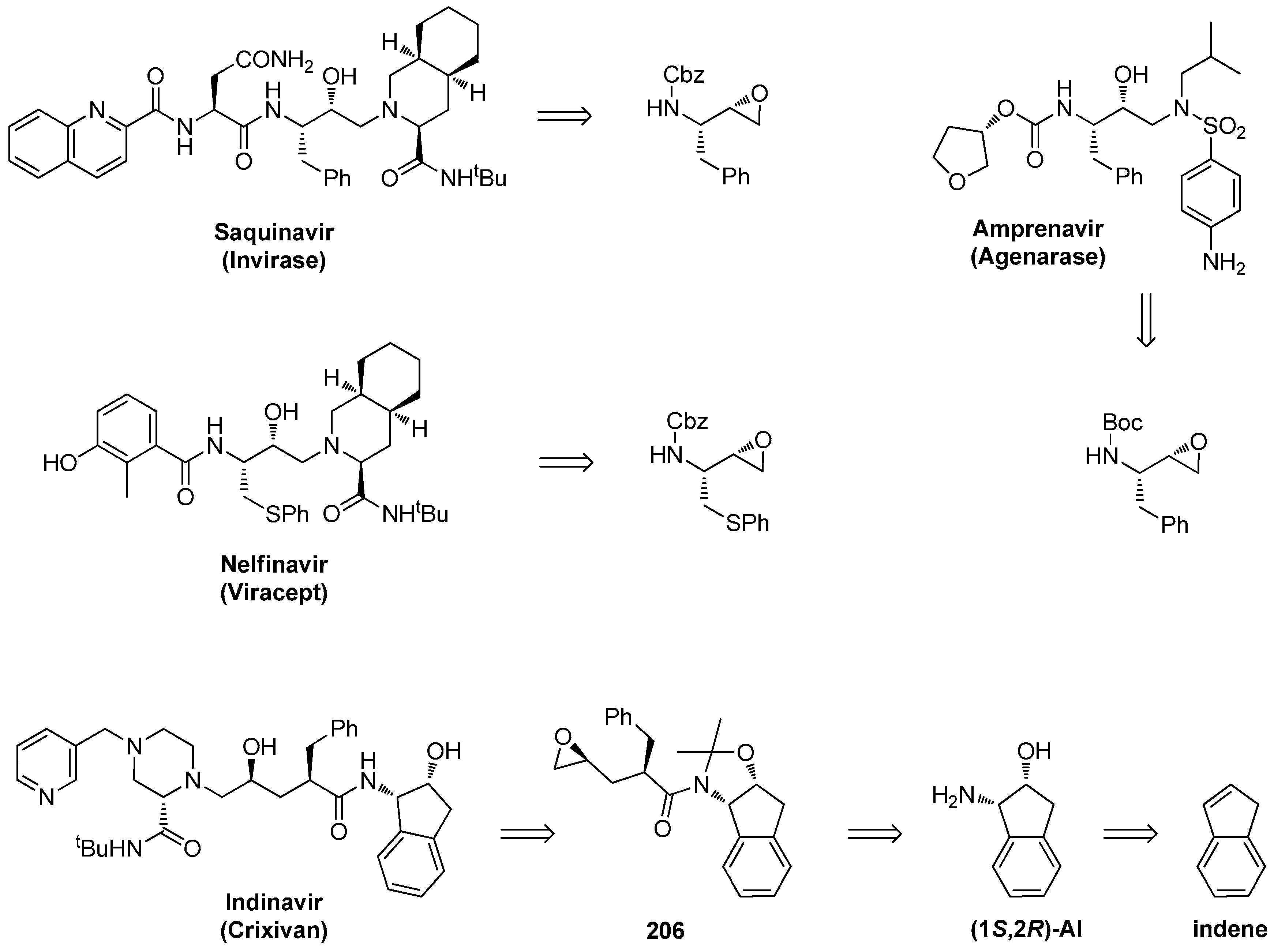
1.18. Indinavir—A Member of Approved HIV Protease Inhibitors Synthesized by Chiral Epoxide Intermediates
1.19. Metal-Catalyzed Asymmetric Epoxidations in Drug Development—Further Examples of the Sharpless and Jacobsen–Katsuki Methodologies
1.20. Organocatalytic Asymmetric Epoxidation in Drug Development—Chiral Ketones, Iminium Salts and Phase Transfer Catalysts
2. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gehringer, M.; Laufer, S.A. Emerging and re-emerging warheads for targeted covalent inhibitors: Applications in medicinal chemistry and chemical biology. J. Med. Chem. 2019, 62, 5673–5724. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hatcher, J.M.; Teng, M.; Gray, N.S.; Kostic, M. Recent advances in selective and irreversible covalent ligand development and validation. Cell Chem. Biol. 2019, 26, 1486–1500. [Google Scholar] [CrossRef] [PubMed]
- Kalgutkar, A.S. Designing around Structural Alerts in Drug Discovery. J. Med. Chem. 2020, 63, 6276–6302. [Google Scholar] [CrossRef]
- Coulup, S.K.; Huang, D.S.; Wong, H.L.; Georg, G.I. Identification of the metabolic profile of the alpha-tubulin-binding natural product (-)-pironetin. J. Med. Chem. 2019, 62, 1684–1689. [Google Scholar] [CrossRef] [PubMed]
- Marco-Contelles, J.; Molina, M.T.; Anjum, S. Naturally occurring cyclohexane epoxides: Sources, biological activities, and synthesis. Chem. Rev. 2004, 104, 2857–2899. [Google Scholar] [CrossRef]
- Delost, M.D.; Smith, D.T.; Anderson, B.J.; Njardarson, J.T. From oxiranes to oligomers: Architectures of U.S. FDA approved pharmaceuticals containing oxygen heterocycles. J. Med. Chem. 2018, 61, 10996–11020. [Google Scholar] [CrossRef] [PubMed]
- Yudin, K.A. Aziridines and Epoxides in Organic Synthesis; Wiley: Hoboken, NJ, USA, 2006. [Google Scholar]
- Meninno, S.; Lattanzi, A. Organocatalytic asymmetric reactions of epoxides: Recent progress. Chemistry 2016, 22, 3632–3642. [Google Scholar] [CrossRef]
- Delost, M.D.; Njardarson, J.T. Oxiranes and Oxirenes: Monocyclic; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Varie, D.L.; Beck, C.; Borders, S.K.; Brady, M.D.; Cronin, J.S.; Ditsworth, T.K.; Hay, D.A.; Hoard, D.H.; Hoying, R.C.; Linder, R.J.; et al. Design, development, and scale-up of a selective meso-epoxide desymmetrization process. Org. Process Res. Dev. 2007, 11, 546–559. [Google Scholar] [CrossRef]
- Célanire, S.; Duvey, G.; Poli, S.; Rocher, J.P. Chapter six-mGluR2 activators and mGluR5 blockers advancing in the clinic for major CNS disorders. Annu. Rep. Med. Chem. 2012, 47, 71–88. [Google Scholar] [CrossRef]
- Hodgson, D.M.; Thompson, A.J.; Wadman, S.; Clare, J.; Keats, C.J. On the possibility of carbamate-directed hydroboration. An approach to the asymmetric synthesis of 1-aminocy clopentane-1,3-dicarboxylic acid. Tetrahedron Lett. 1999, 55, 10815–10834. [Google Scholar] [CrossRef]
- Laudon, M.; Frydman-Marom, A. Therapeutic effects of melatonin receptor agonists on sleep and comorbid disorders. Int. J. Mol. Sci. 2014, 15, 15924–15950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, S.J.; Vu, T.; Totleben, M.J.; Crispino, G.A.; Kacsur, D.J.; Swaminathan, S.; Thornton, J.E.; Fritz, A.; Singh, A.K. Development of jacobsen asymmetric epoxidation and sharpless asymmetric dihydroxylation methods for the large-scale preparation of a chiral dihydrobenzofuran epoxide. Org. Process Res. Dev. 2003, 7, 821–827. [Google Scholar] [CrossRef]
- Brandes, B.D.; Jacobsen, E.N. Highly enantioselective, catalytic epoxidation of trisubstituted olefins. J. Org. Chem. 1994, 59, 4378–4380. [Google Scholar] [CrossRef]
- Zhang, W.; Loebach, J.L.; Wilson, S.R.; Jacobsen, E.N. Enantioselective epoxidation of unfunctionalized olefins catalyzed by (salen) manganese complexes. J. Am. Chem. Soc. 1990, 112, 2801–2803. [Google Scholar] [CrossRef]
- Jacobsen, E.N.; Zhang, W.; Muci, A.R.; Ecker, J.R.; Deng, L. Highly enantioselective epoxidation catalysts derived from 1,2-diaminocyclohexane. J. Am. Chem. Soc. 1991, 113, 7063–7064. [Google Scholar] [CrossRef]
- Ma, D.; Xia, C.; Tian, H. Oxidation of benzylic methylene compounds to ketones with m-chloroperoxybenzoic acid and oxygen. Tetrahedron Lett. 1999, 40, 8915–8917. [Google Scholar] [CrossRef]
- Konoike, T.; Araki, Y.; Kanda, Y. A novel allylic hydroxylation of sterically hindered olefins by Fe-porphyrin-catalyzed mCPBA oxidation. Tetrahedron Lett. 1999, 40, 6971–6974. [Google Scholar] [CrossRef]
- Sharpless, K.B.; Amberg, W.; Bennani, Y.L.; Crispino, G.A.; Hartung, J.; Jeong, K.S.; Kwong, H.L.; Morikawa, K.; Wang, Z.M. The osmium-catalyzed asymmetric dihydroxylation: A new ligand class and a process improvement. J. Org. Chem. 1992, 57, 2768–2771. [Google Scholar] [CrossRef]
- Kolb, H.C.; Sharpless, B.K. A simplified procedure for the stereospecific transformation of 1,2-diols into epoxides. Tetrahedron Lett. 1992, 48, 10515–10530. [Google Scholar] [CrossRef]
- Fisher, M.C.; Gurr, S.J.; Cuomo, C.A.; Blehert, D.S.; Jin, H.; Stukenbrock, E.H.; Stajich, J.E.; Kahmann, R.; Boone, C.; Denning, D.W.; et al. Threats posed by the fungal kingdom to humans, wildlife, and agriculture. mBio 2020, 11. [Google Scholar] [CrossRef]
- Brown, G.D.; Denning, D.W.; Gow, N.A.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci. Transl. Med. 2012, 4, 165rv113. [Google Scholar] [CrossRef] [Green Version]
- Ogura, H.; Kobayashi, H.; Nagai, K.; Nishida, T.; Naito, T.; Tatsumi, Y.; Yokoo, M.; Arika, T. Synthesis and antifungal activities of (2R,3R)-2-aryl-1-azolyl-3-(substituted amino)-2-butanol derivatives as topical antifungal agents. Chem. Pharm. Bull. (Tokyo) 1999, 47, 1417–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohwada, J.; Tsukazaki, M.; Hayase, T.; Oikawa, N.; Isshiki, Y.; Fukuda, H.; Mizuguchi, E.; Sakaitani, M.; Shiratori, Y.; Yamazaki, T.; et al. Design, synthesis and antifungal activity of a novel water soluble prodrug of antifungal triazole. Bioorg. Med. Chem. Lett. 2003, 13, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Hata, K.; Kimura, J.; Miki, H.; Toyosawa, T.; Nakamura, T.; Katsu, K. In vitro and in vivo antifungal activities of ER-30346, a novel oral triazole with a broad antifungal spectrum. Antimicrob. Agents Chemother. 1996, 40, 2237–2242. [Google Scholar] [CrossRef] [Green Version]
- Bartroli, J.; Turmo, E.; Alguero, M.; Boncompte, E.; Vericat, M.L.; Conte, L.; Ramis, J.; Merlos, M.; Garcia-Rafanell, J.; Forn, J. New azole antifungals. 3. Synthesis and antifungal activity of 3-substituted-4(3H)-quinazolinones. J. Med. Chem. 1998, 41, 1869–1882. [Google Scholar] [CrossRef]
- Bennett, F.; Ganguly, A.K.; Girijavallabhan, V.M.; Pinto, P.A. An enantioselective synthesis of the antifungal agent (2R, 3R)-2-(2,4-difluorophenyl)-3-(methylsulfonyl)-1-(1,2,4-triazol-1-yl)-2-butanol. Synlett 1995, 11, 1110–1112. [Google Scholar] [CrossRef]
- Method for Preparing Efinaconazole Intermediate. China Patent CN106608854A, 3 May 2017.
- Konosu, T.; Tajima, Y.; Takeda, N.; Miyaoka, T.; Kasahara, M.; Yasuda, H.; Oida, S. Triazole antifungals. II. Synthesis and antifungal activities of 3-acyl-4-methyloxazolidine derivatives. Chem. Pharm. Bull. (Tokyo) 1990, 38, 2476–2486. [Google Scholar] [CrossRef] [Green Version]
- Konosu, T.; Miyaoka, T.; Tajima, Y.; Oida, S. Triazole antifungals. III. Stereocontrolled synthesis of an optically active triazolylmethyloxirane precursor to antifungal oxazolidine derivatives. Chem. Pharm. Bull. (Tokyo) 1991, 3, 2241–2246. [Google Scholar] [CrossRef] [Green Version]
- Konosu, T.; Tajima, Y.; Takeda, N.; Miyaoka, T.; Kasahara, M.; Yasuda, H.; Oida, S. Triazole antifungals. IV. Synthesis and antifungal activities of 3-acylamino-2-aryl-2-butanol derivatives. Chem. Pharm. Bull. (Tokyo) 1991, 39, 2581–2589. [Google Scholar] [CrossRef] [Green Version]
- Tasaka, A.; Tamura, N.; Matsushita, Y.; Teranishi, K.; Hayashi, R.; Okonogi, K.; Itoh, K. Optically active antifungal azoles. I. Synthesis and antifungal activity of (2R,3R)-2-(2,4-difluorophenyl)-3-mercapto-1-(1H-1,2,4-triazol-1-yl)-2-b utanol and its stereoisomers. Chem. Pharm. Bull. (Tokyo) 1993, 41, 1035–1042. [Google Scholar] [CrossRef] [Green Version]
- Triazole Derivatives, Their Preparation and Their Use as Fungicides. European Patent Office EP0332387A1, 13 September 1989.
- Upadhayaya, R.S.; Sinha, N.; Jain, S.; Kishore, N.; Chandra, R.; Arora, S.K. Optically active antifungal azoles: Synthesis and antifungal activity of (2R,3S)-2-(2,4-difluorophenyl)-3-(5-[2-[4-aryl-piperazin-1-yl]-ethyl]-tetrazol-2-yl/1-yl)-1-[1,2,4]-triazol-1-yl-butan-2-ol. Bioorg. Med. Chem. 2004, 12, 2225–2238. [Google Scholar] [CrossRef] [PubMed]
- Process for the Manufacture of Epoxybutanol Intermediates. WIPO Patent WO2007062542A3, 7 June 2007.
- Mikailov, A.; Cohen, J.; Joyce, C.; Mostaghimi, A. Cost-effectiveness of confirmatory testing before treatment of onychomycosis. JAMA Derm. 2016, 152, 276–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gala, D.; Di Benedetto, D.J.; Mergelsberg, I.; Kugelman, M.; Research, S.-P. Total chiral synthesis of azole antifungals via α-hydroxylation of ketones. Tetrahedron Lett. 1996, 37, 8117–8120. [Google Scholar] [CrossRef]
- Bartroli, J.; Turmo, E.; Belloc, J.; Forn, J. Aldol condensation of evans chiral enolates with acetophenones. Its application to the stereoselective synthesis of homochiral antifungal agents. J. Org. Chem. 1995, 60, 3000–3012. [Google Scholar] [CrossRef]
- Tamura, K.; Kumagai, N.; Shibasaki, M. An enantioselective synthesis of the key intermediate for triazole antifungal agents; Application to the catalytic asymmetric synthesis of efinaconazole (Jublia). J. Org. Chem. 2014, 79, 3272–3278. [Google Scholar] [CrossRef]
- Karasawa, T.; Oriez, R.; Kumagai, N.; Shibasaki, M. Anti-selective catalytic asymmetric nitroaldol reaction of alpha-keto esters: Intriguing solvent effect, flow reaction, and synthesis of active pharmaceutical ingredients. J. Am. Chem. Soc. 2018, 140, 12290–12295. [Google Scholar] [CrossRef]
- Acetti, D.; Brenna, E.; Fuganti, C.; Gatti, F.G.; Serra, S. Enzyme-catalysed approach to the preparation of triazole antifungals: Synthesis of (−)-genaconazole. Tetrahedron Asymmetry 2009, 20, 2413–2420. [Google Scholar] [CrossRef]
- Gala, D.; Di Benedetto, D.J. A rational approach to chiral α-hydroxy aryl ketones from chiral aryl epoxides via regioselective, stereo retentive oxidative epoxide opening: Its application to the synthesis of antifungal Sch 42427/SM 9164. Tetrahedron Lett. 1994, 35, 8299–8302. [Google Scholar] [CrossRef]
- A Kind of Preparation Method of Ravuconazole Intermediate. China Patent CN106749202A, 31 May 2017.
- Process for the Synthesis of Efinaconazol. WIPO Patent WO2017114743A1, 6 July 2017.
- Antibacterial Compounds and Uses Thereof. WIPO Patent WO2017155909A1, 14 September 2018.
- Process for Preparing Intermediates Useful in the Synthesis of Antifungal Drugs. WIPO Patent WO2017178909, 19 October 2017.
- Zhu, F.; Xie, Y.; Zhang, J.; Tian, G.; Qin, H.; Yang, X.; Hu, T.; He, Y.; Aisa, H.A.; Shen, J. A facile epoxide aminolysis promoted by (t-BuO)2Mg and its application to the synthesis of efinaconazole. Org. Process Res. Dev. 2018, 22, 625–632. [Google Scholar] [CrossRef]
- Pesti, J.; Chen, C.-K.; Spangler, L.; DelMonte, A.J.; Benoit, S.; Berglund, D.; Bien, J.; Brodfuehrer, P.; Chan, Y.; Corbett, E.; et al. The process development of ravuconazole: An efficient multikilogram scale preparation of an antifungal agent. Org. Process Res. Dev. 2009, 13, 716–728. [Google Scholar] [CrossRef]
- Park, J.S.; Yu, K.A.; Kang, T.H.; Kim, S.; Suh, Y.G. Discovery of novel indazole-linked triazoles as antifungal agents. Bioorg. Med. Chem. Lett. 2007, 17, 3486–3490. [Google Scholar] [CrossRef]
- Guillon, R.; Pagniez, F.; Picot, C.; Hedou, D.; Tonnerre, A.; Chosson, E.; Duflos, M.; Besson, T.; Loge, C.; Le Pape, P. Discovery of a novel broad-spectrum antifungal agent derived from albaconazole. ACS Med. Chem. Lett. 2013, 4, 288–292. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Xu, Y.; Cao, Y.; Wang, R.; Zhou, R.; Chu, W.; Yang, Y. Design, synthesis, and structure-activity relationship studies of novel thienopyrrolidone derivatives with strong antifungal activity against Aspergillus fumigates. Eur. J. Med. Chem. 2015, 102, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Qian, A.; Ling, C.; Cao, X.; Cui, Y.; Yang, Y. Improved laboratory synthesis of YC-071, a potent azole antifungal agent. J. Chem. Res. 2017, 41, 241–245. [Google Scholar] [CrossRef]
- Ding, Z.; Ni, T.; Xie, F.; Hao, Y.; Yu, S.; Chai, X.; Jin, Y.; Wang, T.; Jiang, Y.; Zhang, D. Design, synthesis, and structure-activity relationship studies of novel triazole agents with strong antifungal activity against Aspergillus fumigatus. Bioorg. Med. Chem. Lett. 2020, 30, 126951. [Google Scholar] [CrossRef] [PubMed]
- Bachelerie, F.; Ben-Baruch, A.; Burkhardt, A.M.; Combadiere, C.; Farber, J.M.; Graham, G.J.; Horuk, R.; Sparre-Ulrich, A.H.; Locati, M.; Luster, A.D.; et al. International union of basic and clinical pharmacology. [corrected]. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharm. Rev. 2014, 66, 1–79. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.F.; Jack, R. CCR1 antagonists. Mol. Divers. 2008, 12, 17–23. [Google Scholar] [CrossRef]
- Karash, A.R.; Gilchrist, A. Therapeutic potential of CCR1 antagonists for multiple myeloma. Future Med. Chem. 2011, 3, 1889–1908. [Google Scholar] [CrossRef]
- Gladue, R.P.; Brown, M.F.; Zwillich, S.H. CCR1 antagonists: What have we learned from clinical trials. Curr. Top. Med. Chem. 2010, 10, 1268–1277. [Google Scholar] [CrossRef]
- Ainge, D.; Booker, J.E.M.; Pedge, N.; Sinclair, R.; Sleigh, C.; Stefinovic, M.; Vaz, L.-M.; Way, E. Development of a multikilogram synthesis of a chiral epoxide precursor to a CCR1 antagonist. Use of in situ monitoring for informed optimisation via fragile intermediates. Org. Process Res. Dev. 2010, 14, 72–84. [Google Scholar] [CrossRef]
- Golding, B.T.; Hall, D.R.; Sakrikar, S. Reaction between vicinal diols and hydrogen bromide in synthesis of chiral propylene oxide. J. Chem. Soc. Perkin Trans. 1 1973, 1214–1220. [Google Scholar] [CrossRef]
- Di Pardo, A.; Maglione, V. The S1P axis: New exciting route for treating huntington’s disease. Trends Pharm. Sci. 2018, 39, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, A.; Camp, S.M.; Garcia, J.G.N.; Polt, R. An update on sphingosine-1-phosphate receptor 1 modulators. Bioorg. Med. Chem. Lett. 2018, 28, 3585–3591. [Google Scholar] [CrossRef]
- Stepanovska, B.; Huwiler, A. Targeting the S1P receptor signaling pathways as a promising approach for treatment of autoimmune and inflammatory diseases. Pharm. Res. 2020, 154, 104170. [Google Scholar] [CrossRef] [PubMed]
- Chiba, K. Discovery of fingolimod based on the chemical modification of a natural product from the fungus, Isaria sinclairii. J. Antibiot. (Tokyo) 2020. [Google Scholar] [CrossRef]
- Lamb, Y.N. Ozanimod: First approval. Drugs 2020, 80, 841–848. [Google Scholar] [CrossRef]
- Huwiler, A.; Zangemeister-Wittke, U. The sphingosine 1-phosphate receptor modulator fingolimod as a therapeutic agent: Recent findings and new perspectives. Pharm. Ther. 2018, 185, 34–49. [Google Scholar] [CrossRef]
- Hou, X.; Zhang, H.; Chen, B.-G.; Guo, Z.; Singh, A.; Goswami, A.; Gilmore, J.L.; Sheppeck, J.E.; Dyckman, A.J.; Carter, P.H.; et al. Regioselective epoxide ring opening for the stereospecific scale-up synthesis of BMS-960, a potent and selective isoxazole-containing S1P1 receptor agonist. Org. Process Res. Dev. 2017, 21, 200–207. [Google Scholar] [CrossRef]
- Bentley, W.T.; Jones, R.V.H.; Larder, A.H.; Lock, S.J. Solvents as phase transfer catalysts. Reaction of trimethylsulfonium iodide and solid potassium hydroxide in acetonitrile leading to an epoxide of benzophenone. J. Chem. Soc. Perkin Trans. 2 1996, 6, 1407–1412. [Google Scholar] [CrossRef]
- Choi, J.; Horner, K.A.; Carnevale, K. Atazanavir; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Antunes, F. Atazanavir sulfate + cobicistat for the treatment of HIV infection. Expert Rev. Anti Infect. Ther. 2017, 15, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Eckhardt, B.J.; Gulick, R.M. Infectious Diseases; Elsevier: Amsterdam, The Netherlands, 2017; Volume 2, pp. 1293.e1292–1308.e1292. [Google Scholar]
- Atazanavir (Reyataz). Available online: https://www.aidsmap.com/about-hiv/arv-background-information/atazanavir-reyataz (accessed on 20 August 2020).
- Bold, B.; Fässler, A.; Capraro, H.-G.; Cozens, R.; Klimkait, T.; Lazdins, J.; Mestan, J.; Poncioni, B.; Rösel, J.; Stover, D.; et al. New aza-dipeptide analogues as potent and orally absorbed HIV-1 protease inhibitors: Candidates for clinical development. J. Med. Chem. 1998, 41, 3387–3401. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Singh, J.; Schwinden, M.D.; Zheng, B.; Kissick, T.P.; Patel, B.; Humora, M.J.; Quiroz, F.; Dong, L.; Hsieh, D.-M.; et al. Process research and development for an efficient synthesis of the HIV protease inhibitor BMS-232632. Org. Proc. Res. Dev. 2002, 6, 323–328. [Google Scholar] [CrossRef]
- Castejón, P.; Pastó, M.; Moyano, A.; Pericàs, M.; Riera, A. A convenient, stereodivergent approach to the enantioselective synthesis of N-Boc-aminoalkyl epoxides. Tetrahedron Lett. 1995, 36, 3019–3022. [Google Scholar] [CrossRef]
- Wold, E.A.; Wild, C.T.; Cunningham, K.A.; Zhou, J. Targeting the 5-HT2C receptor in biological context and the current state of 5-HT2C receptor ligand development. Curr. Top. Med. Chem. 2019, 19, 1381–1398. [Google Scholar] [CrossRef] [PubMed]
- Dihydrobenzofuranyl Alkanamine Derivatives and Methods for Using Same. U.S. Patent 7435837B2, 14 October 2008.
- Gontcharov, A.; Cheng Shaw, C.-C.; Yu, Q.; Tadayon, S.; Bernatchez, M.; Lankau, M.; Cantin, M.; Potoski, J.; Khafizov, G.; Stac, G.; et al. Development of a new practical synthesis of a 5-HT2C receptor agonist. Org. Process Res. Dev. 2010, 14, 1438–1447. [Google Scholar] [CrossRef]
- Roger, C.; Roberts, J.A.; Muller, L. Clinical pharmacokinetics and pharmacodynamics of oxazolidinones. Clin. Pharm. 2018, 57, 559–575. [Google Scholar] [CrossRef]
- Wright, A.; Deane-Alder, K.; Marschall, E.; Bamert, R.; Venugopal, H.; Lithgow, T.; Lupton, D.W.; Belousoff, M.J. Characterization of the core ribosomal binding region for the oxazolidone family of antibiotics using cryo-EM. ACS Pharm. Trans. Sci. 2020, 3, 425–432. [Google Scholar] [CrossRef]
- Saini, J.S.; Homeyer, N.; Fulle, S.; Gohlke, H. Determinants of the species selectivity of oxazolidinone antibiotics targeting the large ribosomal subunit. Biol. Chem. 2013, 394, 1529–1541. [Google Scholar] [CrossRef]
- Warner, K.D.; Hajdin, C.E.; Weeks, K.M. Principles for targeting RNA with drug-like small molecules. Nat. Rev. Drug Discov. 2018, 17, 547–558. [Google Scholar] [CrossRef]
- Perzborn, E.; Roehrig, S.; Straub, A.; Kubitza, D.; Misselwitz, F. The discovery and development of rivaroxaban, an oral, direct factor Xa inhibitor. Nat. Rev. Drug Discov. 2011, 10, 61–75. [Google Scholar] [CrossRef]
- Roehrig, S.; Straub, A.; Pohlmann, J.; Lampe, T.; Pernerstorfer, J.; Schlemmer, K.-H.; Reinemer, P.; Perzborn, E. Discovery of the novel antithrombotic agent 5-Chloro-N-({(5S)-2-oxo-3-[4-(3-oxomorpholin-4-yl)phenyl]-1,3-oxazolidin-5-yl}methyl)thiophene-2-carboxamide (BAY 59–7939): An oral, direct factor Xa inhibitor. J. Med. Chem. 2005, 48, 5900–5908. [Google Scholar] [CrossRef] [PubMed]
- Substituted Oxazolidinones and Their Use in the Field of Blood Coagulation. U.S. Patent US7576111B2, 18 August 2009.
- Method for the Preparation of Rivaroxaban. WIPO (PCT) Patent WO2011098501A1, 18 August 2011.
- Processes for the Preparation of Rivaroxaban and Intermediates Thereof. WIPO (PCT) Patent WO2012051692, 26 April 2012.
- A Process for the Preparation of Rivaroxaban Based on Saving of 1,1’-Carbonyl Diimidazole. WIPO (PCT) Patent WO2013120464A1, 22 August 2013.
- Process for the Synthesis of Rivaroxaban and Intermediate for the Production Thereof. WIPO (PCT) Patent WO2015198259A1, 30 December 2015.
- Mali, A.C.; Deshmukh, D.G.; Joshi, D.R.; Lad, H.D.; Patel, P.I.; Medhane, V.J.; Mathad, V.T. Facile approach for the synthesis of rivaroxaban using alternate synthon: Reaction, crystallization and isolation in single pot to achieve desired yield, quality and crystal form. Sustain. Chem. Process 2015, 3. [Google Scholar] [CrossRef]
- Fattah, T.A.; Saeed, A. A review on the synthetic approaches of rivaroxaban: An anticoagulant drug. Tetrahedron Asymmetry 2017, 28, 485–504. [Google Scholar] [CrossRef]
- Yuan, J.; Liu, K.; Li, L.; Yuan, Y.; Liu, X.; Li, Y. A novel synthesis of the oxazolidinone antithrombotic agent rivaroxaban. Molecules 2014, 19, 14999–15004. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Liu, Y.; Zhang, Y.; Zhang, X. An approach to the anticoagulant agent rivaroxaban via an isocyanate-oxirane cycloaddition promoted by MgI2.etherate. J. Chem. Res. 2011, 35, 400–401. [Google Scholar] [CrossRef]
- Oxazolidinones Substituees et Leur Utilisation Dans le Domaine de la Coagulation Sanguine. WIPO (PCT) Patent WO2001047919A9, 7 May 2001.
- Process for the Preparation of Rivaroxaban and Intermediates Thereof. WIPO (PCT) Patent WO2011080341A1, 7 July 2011.
- Siddaraj, R.; Govindaiah, S.; Ningegowda, R.; Swamy Shivananju, N.; Shubha Priya, B. A novel and expeditious synthesis of oxazolidinone drugs linezolid and eperezolid. Eur. J. Chem. 2018, 9, 353–359. [Google Scholar] [CrossRef]
- Perrault, W.R.; Pearlman, B.A.; Godrej, D.B.; Jeganathan, A.; Yamagata, K.; Chen, J.J.; Lu, C.V.; Herrinton, P.M.; Gadwood, R.C.; Chan, L.; et al. The synthesis of N-Aryl-5(S)-aminomethyl-2-oxazolidinone antibacterials and derivatives in one step from aryl carbamates. Org. Proc. Res. Dev. 2003, 7, 533–546. [Google Scholar] [CrossRef]
- Halama, A.; Kruliš, R.; Rymeš, J. A convenient synthesis of rivaroxaban from (S)-epichlorohydrin. Org. Prep. Proced. Int. 2020, 52, 201–211. [Google Scholar] [CrossRef]
- Schmidt, G.; Reber, S.; Bolli, M.H.; Abele, S. Practical and scalable synthesis of S1P1 receptor agonist ACT-209905. Org. Process Res. Dev. 2012, 16, 595–604. [Google Scholar] [CrossRef]
- Meneses, A. The Role of 5-HT Systems on Memory and Dysfunctional Memory; Elsevier Inc.: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Mohler, E.G.; Shacham, S.; Noiman, S.; Lezoualc’h, F.; Robert, S.; Gastineau, M.; Rutkowski, J.; Marantz, Y.; Dumuis, A.; Bockaert, J.; et al. VRX-03011, a novel 5-HT4 agonist, enhances memory and hippocampal acetylcholine efflux. Neuropharmacology 2007, 53, 563–573. [Google Scholar] [CrossRef]
- Shen, F.; Smith, J.A.; Chang, R.; Bourdet, D.L.; Tsuruda, P.R.; Obedencio, G.P.; Beattie, D.T. 5-HT(4) receptor agonist mediated enhancement of cognitive function in vivo and amyloid precursor protein processing in vitro: A pharmacodynamic and pharmacokinetic assessment. Neuropharmacology 2011, 61, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, G.; Sakurai, M.; Teich, A.F.; Saeed, F.; Aziz, F.; Arancio, O. 5-HT(4) receptor stimulation leads to soluble AbetaPPalpha production through MMP-9 upregulation. J Alzheimers Dis. Jad 2012, 32, 437–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castriconi, F.; Paolino, M.; Grisci, G.; Francini, C.M.; Reale, A.; Giuliani, G.; Anzini, M.; Giorgi, G.; Mennuni, L.; Sabatini, C.; et al. Development of subnanomolar-affinity serotonin 5-HT4 receptor ligands based on quinoline structures. MedChemComm 2018, 9, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Lanthier, C.; Dallemagne, P.; Lecoutey, C.; Claeysen, S.; Rochais, C. Therapeutic modulators of the serotonin 5-HT4 receptor: A patent review (2014-present). Expert Opin. Ther. Pat. 2020, 30, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Lienard, P.; Gradoz, P.; Greciet, H.; Jegham, S.; Legroux, D. Pilot scale process development of SL65.0102–10, an N-diazabicyclo[2.2.2]-octylmethyl benzamide. Org. Process Res. Dev. 2017, 21, 18–22. [Google Scholar] [CrossRef]
- Kim, M.B.; Sharpless, B.K. Cyclic sulfates containing acid-sensitive groups and chemoselective hydrolysis of sulfate esters. Tetrahedron Lett. 1989, 30, 655–658. [Google Scholar] [CrossRef]
- Perel, G.; Bliss, J.; Thomas, C.M. Carfilzomib (kyprolis): A novel proteasome inhibitor for relapsed and/or refractory multiple myeloma. Pharm. Ther. 2016, 41, 303–307. [Google Scholar]
- Niederhuber, J.; Armitage, J.; Doroshow, J.; Kastan, M.; Tepper, J. Abeloff’s Clinical Oncology, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Kim, K.B.; Crews, C.M. From epoxomicin to carfilzomib: Chemistry, biology, and medical outcomes. Nat. Prod. Rep. 2013, 30, 600–604. [Google Scholar] [CrossRef] [Green Version]
- Crystalline Peptide Epoxy Ketone Protease Inhibitors and the Synthesis of Amino Acid Keto-Epoxides. U.S. Patent US8367617B2, 5 February 2013.
- Compounds for Enzyme Inhibition. U.S. Patent US8207125B2, 26 June 2012.
- Compounds for Enzyme Inhibition. U.S. Patent US8207297B2, 26 June 2012.
- Compounds for Enzyme Inhibition. U.S. Patent US20120094930A1, 19 April 2012.
- Crystalline Peptide Epoxy Ketone Protease Inhibitors and the Synthesis of Amino Acid Keto-Epoxides. U.S. Patent US8921583B2, 30 December 2014.
- Synthesis of Amino Acid Keto-Epoxides. U.S. Patent US20050256324A1, 17 November 2005.
- Sin, N.; Kim, K.B.; Elofsson, M.; Meng, L.; Auth, H.; Kwok, B.H.B.; Crews, C.M. Total synthesis of the potent proteasome inhibitor epoxomicin: A useful tool for understanding proteasome biology. Bioorg. Med. Chem. Lett. 1999, 9, 2283–2288. [Google Scholar] [CrossRef]
- Dornan, P.K.; Anthoine, T.; Beaver, M.G.; Cheng, C.G.; Cohen, D.E.; Cui, S.; Lake, W.E.; Langille, N.F.; Lucas, S.P.; Pantel, J.; et al. Continuous process improvement in the manufacture of carfilzomib, Part 1: Process understanding and improvements in the commercial route to prepare the epoxyketone warhead. Org. Proc. Res. Dev. 2020, 24, 481–489. [Google Scholar] [CrossRef]
- Preparation Method of Intermediate Compounds of Carfilzomib and Intermediate Compounds. China Patent CN104230857A, 24 December 2014.
- Synthesis of Peptide Epoxy Ketones. U.S. Patent US20160215016A1, 2016.
- Carfilzomib Intermediate and Preparation Method Therefor, and Preparation Method for Carfilzomib. WIPO (PCT) Patent WO2015010436A1, 29 January 2015.
- Methods of Making Carfilzomib and Intermediates Thereof. U.S. Patent US20160115198A1, 28 April 2016.
- Carfilzomib Intermediate and Preparation Method Thereof, as Well as Preparation Method of Carfilzomib. China Patent CN104356197A, 18 February 2015.
- Hughes, D.L. Patent Review of Manufacturing Routes to Oncology Drugs: Carfilzomib, Osimertinib, and Venetoclax. Org Proc Res Dev. 2016, 20, 2028–2042. [Google Scholar] [CrossRef]
- Preparation Method of [(1S)-3-methyl-1-[[(2R)-2-methylepoxyethyl]carbonyl]butyl]tert-butyl Carbamate. China Patent CN104672179A, 3 June 2015.
- Daikai, K.; Kamaura, M.; Inanaga, J. Remarkable ligand effect on the enantioselectivity of the chiral lanthanum complex-catalyzed asymmetric epoxidation of enones. Tetrahedron Lett. 1998, 39, 7321–7322. [Google Scholar] [CrossRef]
- Wang, B.; Miao, C.; Wang, S.; Xia, C.; Sun, W. Manganese catalysts with C1-symmetric N4 ligand for enantioselective epoxidation of olefins. Chemistry 2012, 18, 6750–6753. [Google Scholar] [CrossRef]
- Stereoselective Synthesis of Diols and Triols by Mannich Reaction and Their Use in the Synthesis of Carfilzomib. U.S. Patent US20160194354A1, 7 July 2016.
- Yamamoto, M.; Hayashi, M.; Masaki, M.; Nohira, H. Facile synthesis of (2R,3S)-3-(4-Methoxyphenyl)glycidic esters via optical resolution of the unisolated labile free acid. Tetrahedron Asymmetry 1991, 2, 403–406. [Google Scholar] [CrossRef]
- Kanerva, L.T.; Sundholm, O. Enzymatic acylation in the resolution of methyl threo-2-Hydroxy-3-(4-met hoxyp henyl)-3-(2-X-phenylthio)propionates in organic solvents. J. Chem. Soc. Perkin Trans. 1 1993, 2407–2410. [Google Scholar] [CrossRef]
- Matsumae, H.; Furui, M.; Shibatani, T. Lipase-catalyzed asymmetric hydrolysis of 3-phenylglycidic acid ester, the key intermediate in the synthesis of diltiazem hydrochloride. J. Ferment. Bioeng. 1993, 75, 93–98. [Google Scholar] [CrossRef]
- Jacobsen, E.N.; Deng, L.; Furukawa, Y.; Martínez, L.E. Enantioselective catalytic epoxidation of cinnamate esters. Tetrahedron 1994, 50, 4323–4334. [Google Scholar] [CrossRef]
- Hashiyama, T.; Inoue, H.; Konda, M.; Takeda, M. Reaction of 3-phenylglycidic esters. Part 1. Stereoselective opening of the oxirane ring of trans-3-phenylglycidic esters with 2-nitrothiophenols and the effect of various catalysts thereon. J. Chem. Soc. Perkin Trans. 1 1984, 1725–1732. [Google Scholar] [CrossRef]
- Lynch, J.E.; Choi, W.-B.; Churchill, H.R.O.; Volante, R.P.; Reamer, R.A.; Ball, R.G. asymmetric synthesis of CDP840 by jacobsen epoxidation. An unusual syn selective reduction of an epoxide. J. Org. Chem. 1997, 62, 9223–9228. [Google Scholar] [CrossRef]
- Yamada, S.; Tsujioka, I.; Shibatani, T.; Yoshioka, R. Efficient alternative synthetic route to diltiazem via (2R, 3S)-3-(4-Methoxyphenyl)glycidamide. Chem. Pharm. Bull. 1999, 47, 146–150. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, A.; Madan, P.B.; Mohacsi, E.; O’Brien, J.P.; Todaro, L.J.; Coffen, D.L. Enantioselective synthesis of calcium channel blockers of the diltiazem group. J. Org. Chem. 1992, 57, 851–856. [Google Scholar] [CrossRef]
- Wong, O.A.; Shi, Y. Organocatalytic oxidation. Asymmetric epoxidation of olefins catalyzed by chiral ketones and iminium salts. Chem. Rev. 2008, 108, 3958–3987. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, Q.; Cornwall, R.G.; Shi, Y. Organocatalytic asymmetric epoxidation and aziridination of olefins and their synthetic applications. Chem. Rev. 2014, 114, 8199–8256. [Google Scholar] [CrossRef]
- Seki, M.; Furutani, T.; Imashiro, R.; Kuroda, T.; Yamanaka, T.; Harada, N.; Arakawa, H.; Kusama, M.; Hashiyama, T. A novel synthesis of a key intermediate for diltiazem. Tetrahedron Lett. 2001, 42, 8201–8205. [Google Scholar] [CrossRef]
- Furutani, T.; Imashiro, R.; Hatsuda, M.; Seki, M. A practical procedure for the large-scale preparation of methyl (2R,3S)-3-(4-methoxyphenyl)glycidate, a key intermediate for diltiazem. J. Org. Chem. 2002, 67, 4599–4601. [Google Scholar] [CrossRef]
- Adger, B.M.; Barkley, J.V.; Bergeron, S.; Cappi, M.W.; Flowerdew, B.E.; Jackson, M.P.; McCague, R.; Nugent, T.C.; Roberts, S.M. Improved procedure for Juliá–Colonna asymmetric epoxidation of α,β-unsaturated ketones: Total synthesis of diltiazem and Taxol TM side-chain. J. Chem. Soc. Perkin Trans. 1 1997, 23, 3501–3508. [Google Scholar] [CrossRef]
- Kleemann, A. Ullmann’s Encyclopedia of Industrial Chemistry; Wiley: Hoboken, NJ, USA, 2019. [Google Scholar]
- Shelton, R.C. Antidepressants; Springer: Berlin/Heidelberg, Germany, 2019; pp. 145–180. [Google Scholar]
- Novel Uses for Esreboxetine and Racemic Reboxetine. WIPO (PCT) Patent WO2010044016A1, 22 April 2010.
- Shahzad, D.; Faisal, M.; Rauf, A.; Huang, J. Synthetic story of a blockbuster drug; Reboxetine: A potent selective norepinephrine reuptake inhibitor. Org. Proc. Res. Dev. 2017, 21, 1705–1731. [Google Scholar] [CrossRef]
- Henegar, K.E.; Ball, C.T.; Horvath, C.M.; Maisto, K.D.; Mancini, S.E. Process development and scale-up for (±)-reboxetine mesylate. Org. Proc. Res. Dev. 2007, 11, 346–353. [Google Scholar] [CrossRef]
- Hayes, S.T.; Assaf, G.; Checksfield, G.; Cheung, C.; Critcher, D.; Harris, L.; Howard, R.; Mathew, S.; Regius, C.; Scotney, G.; et al. Commercial synthesis of (S,S)-reboxetine succinate: A journey to find the cheapest commercial chemistry for manufacture. Org. Proc. Res. Dev. 2011, 15, 1305–1314. [Google Scholar] [CrossRef]
- Reddy, R.S.C.; Chouthaiwale, P.V.; Suryavanshi, G.; Chavan, V.B.; Sudalai, A. Co(III)(salen)-catalyzed HKR of two stereocentered alkoxy- and azido epoxides: A concise enantioselective synthesis of (S,S)-reboxetine and (+)-epi-cytoxazone. Chem. Commun. (Camb) 2010, 46, 5012–5014. [Google Scholar] [CrossRef] [PubMed]
- Metro, T.X.; Pardo, D.G.; Cossy, J. Syntheses of (S,S)-reboxetine via a catalytic stereospecific rearrangement of beta-amino alcohols. J. Org. Chem. 2008, 73, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Brenner, E.; Baldwin, R.M.; Tamagnan, G. Asymmetric synthesis of (+)-(S,S)-reboxetine via a new (S)-2-(hydroxymethyl)morpholine preparation. Org. Lett. 2005, 7, 937–939. [Google Scholar] [CrossRef]
- Dar, A.R.; Aga, M.A.; Kumar, B.; Yousuf, S.K.; Taneja, S.C. Regioselective monochloro substitution in carbohydrates and non-sugar alcohols via Mitsunobu reaction: Applications in the synthesis of reboxetine. Org. Biomol. Chem. 2013, 11, 6195–6207. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lin, Z.W.; Zhou, Z.H.; Chen, H.B. Stereodivergent synthesis of all the four stereoisomers of antidepressant reboxetine. Org. Biomol. Chem. 2017, 15, 5395–5401. [Google Scholar] [CrossRef]
- Son, S.M.; Lee, H.K. Dynamic kinetic resolution-based asymmetric transfer hydrogenation of 2-benzoylmorpholinones and its use in concise stereoselective synthesis of all four stereoisomers of the antidepressant reboxetine. J. Org. Chem. 2013, 78, 8396–8404. [Google Scholar] [CrossRef]
- Zhou, J.; Yeung, Y.Y. Synthesis of reboxetine intermediate and carnitine acetyltransferase inhibitor via NBS-induced electrophilic multicomponent reaction. J. Org. Chem. 2014, 79, 4644–4649. [Google Scholar] [CrossRef]
- Srinivasan, K.; Siddiqui, S.; Narkhede, U.; Lahoti, R. Enantioselective synthesis of (+)-(S,S)-reboxetine. Synlett 2006, 11, 1771–1773. [Google Scholar] [CrossRef]
- Henegar, K.E.; Cebula, M. Process development for (S,S)-reboxetine succinate via a sharpless asymmetric epoxidation. Org. Proc. Res. Dev. 2007, 11, 354–358. [Google Scholar] [CrossRef]
- Assaf, G.; Checksfield, G.; Critcher, D.; Dunn, P.J.; Field, S.; Harris, L.J.; Howard, R.M.; Scotney, G.; Scott, A.; Mathew, S.; et al. The use of environmental metrics to evaluate green chemistry improvements to the synthesis of (S,S)-reboxetine succinate. Green Chem. 2012, 14, 123–129. [Google Scholar] [CrossRef]
- Aparicio, D.M.; Terán, J.L.; Gnecco, D.; Galindo, A.; Juárez, J.R.; Orea, M.L.; Mendoza, A. Application of amide-stabilized sulfur ylide reactivity to the stereodivergent synthesis of (R,S)- and (S,R)-reboxetine. Tetrahedron Asymmetry 2009, 20, 2764–2768. [Google Scholar] [CrossRef]
- Kirkby, N.S.; Lundberg, M.H.; Harrington, L.S.; Leadbeater, P.D.; Milne, G.L.; Potter, C.M.; Al-Yamani, M.; Adeyemi, O.; Warner, T.D.; Mitchell, J.A. Cyclooxygenase-1, not cyclooxygenase-2, is responsible for physiological production of prostacyclin in the cardiovascular system. Proc. Natl. Acad. Sci. USA 2012, 109, 17597–17602. [Google Scholar] [CrossRef] [Green Version]
- Biringer, R.G. The enzymology of the human prostanoid pathway. Mol. Biol. Rep. 2020, 47, 4569–4586. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Goleniewska, K.; Zhang, J.; Dulek, D.E.; Toki, S.; Lotz, M.T.; Newcomb, D.C.; Boswell, M.G.; Polosukhin, V.V.; Milne, G.L.; et al. Cyclooxygenase inhibition abrogates aeroallergen-induced immune tolerance by suppressing prostaglandin I2 receptor signaling. J. Allergy Clin. Immunol. 2014, 134, 698–705.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.S. Prostacyclin: A major prostaglandin in the regulation of adipose tissue development. J. Cell Physiol. 2019, 234, 3254–3262. [Google Scholar] [CrossRef]
- Li, H.Y.; McSharry, M.; Walker, D.; Johnson, A.; Kwak, J.; Bullock, B.; Neuwelt, A.; Poczobutt, J.M.; Sippel, T.R.; Keith, R.L.; et al. Targeted overexpression of prostacyclin synthase inhibits lung tumor progression by recruiting CD4+ T lymphocytes in tumors that express MHC class II. Oncoimmunology 2018, 7, e1423182. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, L.M.; Kruger, M.; Grimm, D.; Infanger, M.; Wehland, M. The prostacyclin analogue treprostinil in the treatment of pulmonary arterial hypertension. Basic Clin. Pharm. Toxicol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Hattori, K.; Tanaka, A.; Okitsu, O.; Tabuchi, S.; Taniguchi, K.; Nishio, M.; Koyama, S.; Higaki, M.; Seki, J.; Sakane, K. Discovery of diphenylcarbamate derivatives as highly potent and selective IP receptor agonists: Orally active prostacyclin mimetics. Part 3. Bioorg. Med. Chem. Lett. 2005, 15, 3091–3095. [Google Scholar] [CrossRef]
- Ohigashi, A.; Kanda, A.; Moriki, S.; Baba, Y.; Hashimoto, N.; Okada, M. Practical synthesis of PGI2 agonist: Resolution-inversion-recycle approach of its chiral intermediate. Org. Proc. Res. Dev. 2013, 17, 658–665. [Google Scholar] [CrossRef]
- Hirt, H.; Haenggi, R.; Reyes, J.; Seeger-Weibel, M.; Gallou, F. Development of a practical route for the manufacture of N-[5-(3-Imidazol-1-yl-4-methanesulfonyl-phenyl)-4-methyl-thiazol-2-yl]acetamide. Org. Proc. Res. Dev. 2008, 12, 111–115. [Google Scholar] [CrossRef]
- Sato, K.; Hyodo, M.; Aoki, M.; Zheng, X.-Q.; Noyori, R. Oxidation of sulfides to sulfoxides and sulfones with 30% hydrogen peroxide under organic solvent-and halogen-free conditions. Tetrahedron 2001, 57, 2469–2476. [Google Scholar] [CrossRef]
- Fragoso, Y.D.; Carra, A.; Macias, M.A. Cannabis and multiple sclerosis. Expert. Rev. Neurother. 2020, 20, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Inglet, S.; Winter, B.; Yost, S.E.; Entringer, S.; Lian, A.; Biksacky, M.; Pitt, R.D.; Mortensen, W. Clinical Data for the Use of Cannabis-Based Treatments: A Comprehensive Review of the Literature. Ann. Pharmacother. 2020, 54, 1109–1143. [Google Scholar] [CrossRef] [PubMed]
- Kilaru, A.; Chapman, K.D. The endocannabinoid system. Essays Biochem. 2020. [Google Scholar] [CrossRef]
- Murphy, T.; Le Foll, B. Targeting the endocannabinoid CB1 receptor to treat body weight disorders: A preclinical and clinical review of the therapeutic potential of past and present CB1 drugs. Biomolecules 2020, 10. [Google Scholar] [CrossRef]
- Stoss, P.; Merrath, P. A useful approach towards Δ9-tetrahydrocannabinol. Synlett 1991, 8, 553–554. [Google Scholar] [CrossRef]
- Synthesis of Cannabinoids. WIPO (PCT) Patent WO2002096899A1, 15 December 2002.
- Cannabinoid Active Pharmaceutical Ingredient for Improved Dosage Forms. WIPO (PCT) Patent WO2006133941A2, 21 December 2006.
- Cabaj, J.E.; Lukesh, J.M.; Pariza, R.J.; Zizelman, P.M. Large-scale preparation of (+)-p-Menth-2-ene-1,8-diol, a key intermediate in the synthesis of Δ-9-tetrahydrocannabinol. Org. Process Res. Dev. 2009, 13, 358–361. [Google Scholar] [CrossRef]
- Limbani, B.; Bera, S.; Mondal, D. Synthetic advancement of neuraminidase inhibitor “Tamiflu”. ChemistrySelect 2020, 5, 6083–6122. [Google Scholar] [CrossRef]
- Andraos, J. Global green chemistry metrics analysis algorithm and spreadsheets: Evaluation of the material efficiency performances of synthesis plans for oseltamivir phosphate (Tamiflu) as a test case. Org. Proc. Res. Dev. 2009, 13, 161–185. [Google Scholar] [CrossRef]
- Shie, J.J.; Fang, J.M.; Wong, C.H. A concise and flexible synthesis of the potent anti-influenza agents Tamiflu and tamiphosphor. Angew. Chem. 2008, 47, 5788–5791. [Google Scholar] [CrossRef]
- Bhowmik, S.; Batra, S. Morita-baylis-hillman approach toward formal total synthesis of Tamiflu and total synthesis of gabaculine. Eur. J. Org. Chem. 2013, 71, 7145–7151. [Google Scholar] [CrossRef]
- Hayashi, Y.; Ogasawara, S. Time economical total synthesis of (-)-oseltamivir. Org. Lett. 2016, 18, 3426–3429. [Google Scholar] [CrossRef] [PubMed]
- Karpf, M.; Trussardi, R. Efficient access to oseltamivir phosphate (Tamiflu) via the O-trimesylate of shikimic acid ethyl ester. Angew. Chem. 2009, 48, 5760–5762. [Google Scholar] [CrossRef] [PubMed]
- Kongkathip, B.; Akkarasamiyo, S.; Kongkathip, N. A new and efficient asymmetric synthesis of oseltamivir phosphate (Tamiflu) from d-glucose. Tetrahedron 2015, 71, 2393–2399. [Google Scholar] [CrossRef]
- Li, H.; Shen, S.-J.; Zhu, C.-L.; Xu, H. Enantioselective synthesis of oseltamivir phosphate (Tamiflu) via the iron-catalyzed stereoselective olefin diazidation. J. Am. Chem. Soc. 2018, 140, 10619–10626. [Google Scholar] [CrossRef] [PubMed]
- Nie, L.D.; Shi, X.X.; Ko, K.H.; Lu, W.D. A short and practical synthesis of oseltamivir phosphate (Tamiflu) from (-)-shikimic acid. J. Org. Chem. 2009, 74, 3970–3973. [Google Scholar] [CrossRef]
- Trost, B.M.; Zhang, T. Development of a concise synthesis of (-)-oseltamivir (Tamiflu). Chemistry 2011, 17, 3630–3643. [Google Scholar] [CrossRef] [Green Version]
- Rawat, V.; Dey, S.; Sudalai, A. Synthesis of the anti-influenza agent (-)-oseltamivir free base and (-)-methyl 3-epi-shikimate. Org. Biomol. Chem. 2012, 10, 3988–3990. [Google Scholar] [CrossRef]
- Federspiel, M.; Fischer, R.; Hennig, M.; Mair, H.-J.; Oberhauser, T.; Rimmler, G.; Albiez, T.; Bruhin, J.; Estermann, H.; Gandert, C.; et al. Industrial synthesis of the key precursor in the synthesis of the anti-influenza drug oseltamivir phosphate (Ro 64–0796/002, GS-4104–02): Ethyl (3R,4S,5S)-4,5-epoxy-3-(1-ethyl-propoxy)-cyclohex-1-ene-1-carboxylate. Org. Proc. Res. Dev. 1999, 3, 266–274. [Google Scholar] [CrossRef]
- Rohloff, J.C.; Kent, K.M.; Postich, M.J.; Becker, M.W.; Chapman, H.H.; Kelly, D.E.; Lew, W.; Louie, M.S.; McGee, L.R.; Prisbe, E.J.; et al. Practical total synthesis of the anti-influenza drug GS-4104. J. Org. Chem. 1998, 63, 4545–4550. [Google Scholar] [CrossRef]
- Chandran, S.S.; Yi, J.; Draths, K.M.; von Daeniken, R.; Weber, W.; Frost, J.W. Phosphoenolpyruvate availability and the biosynthesis of shikimic acid. Biotechnol. Prog. 2008, 19, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Carr, R.; Ciccone, F.; Gabel, R.; Guinn, M.; Johnston, D.; Mastriona, J.; Vandermeer, T.; Groaning, M. Streamlined process for the esterification and ketalization of shikimic acid en route to the key precursor for oseltamivir phosphate (TamifluTM). Green Chem. 2008, 10, 743–745. [Google Scholar] [CrossRef]
- Abrecht, S.; Federspiel, M.C.; Estermann, H.; Fischer, R.; Karpf, M.; Mair, H.-J.; Oberhauser, T.; Rimmler, G.; Trussadi, R.; Zutter, U. The synthetic-technical development of oseltamivir phosphate TamifluTM: A race against time. Chimia 2007, 61, 93–99. [Google Scholar] [CrossRef]
- Karpf, M.; Trussardi, R. New, azide-free transformation of epoxides into 1,2-diamino compounds: Synthesis of the anti-influenza neuraminidase inhibitor oseltamivir phosphate (Tamiflu). J. Org. Chem. 2001, 66, 2044–2051. [Google Scholar] [CrossRef] [PubMed]
- Harrington, P.J.; Brown, J.D.; Foderaro, T.; Hughes, R.C. Research and development of a second-generation process for oseltamivir phosphate, prodrug for a neuraminidase inhibitor. Org. Proc. Res. Dev. 2004, 8, 86–91. [Google Scholar] [CrossRef]
- Lu, D.Y.; Wu, H.Y.; Yarla, N.S.; Xu, B.; Ding, J.; Lu, T.R. HAART in HIV/AIDS treatments: Future trends. Infect. Disord. Drug Targets. 2018, 18, 15–22. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Osswald, H.L.; Prato, G. Recent progress in the development of HIV-1 protease inhibitors for the treatment of HIV/AIDS. J. Med. Chem. 2016, 59, 5172–5208. [Google Scholar] [CrossRef] [Green Version]
- Lv, Z.; Chu, Y.; Wang, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV Aids (Auckl) 2015, 7, 95–104. [Google Scholar] [CrossRef] [Green Version]
- Voshavar, C. Protease inhibitors for the treatment of HIV/AIDS: Recent advances and future challenges. Curr. Top. Med. Chem. 2019, 19, 1571–1598. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Bilcer, G.; Schiltz, G. Syntheses of FDA approved HIV protease inhibitors. Synthesis (Stuttg) 2001, 2001, 2203–2229. [Google Scholar] [CrossRef]
- Reider, P.J. Advances in AIDS chemotherapy: The asymmetric synthesis of CRIXIVAN®. Chimia 1997, 51, 306–308. [Google Scholar]
- Gallou, I.; Senanayake, C.H. Cis-1-Amino-2-indanol in drug design and applications to asymmetric processes. Chem. Rev. 2006, 106, 2843–2874. [Google Scholar] [CrossRef] [PubMed]
- Tani, K.B.M.C. A practical synthesis and biological evaluation of 9-halogenated PGF analogues. Bioorg. Med. Chem. 2002, 10, 1883–1894. [Google Scholar] [CrossRef]
- Kimura, T.; Yamamoto, N.; Suzuki, Y.; Kawano, K.; Norimine, Y.; Ito, K.; Nagato, S.; Iimura, Y.; Yonaga, M. Practical synthesis of chiral emopamil left hand as a bioactive motif. J. Org. Chem. 2002, 67, 6228–6231. [Google Scholar] [CrossRef] [PubMed]
- Voight, E.A.; Yin, H.; Downing, S.V.; Calad, S.A.; Matsuhashi, H.; Giordano, I.; Hennessy, A.J.; Goodman, R.M.; Wood, J.L. Target-directed synthesis of antibacterial drug candidate GSK966587. Org. Lett. 2010, 12, 3422–3425. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.; Davies, M.R.; Finney, F.J.L.; Geen, G.R.; Kincey, P.M.; Mann, I. S The effect of catalyst loading and donor ligands in the Mn(III) salen catalysed chiral epoxidation of chromenes: Synthesis of BRL 55834. Tetrahedron Lett. 1996, 37, 3895–3898. [Google Scholar] [CrossRef]
- Magano, J.; Acciacca, A.; Beylin, V.; Spence, J.; Dunn, P.; Hughes, M. Synthesis of the potassium channel opener (3S,4R)-3,4-Dihydro-4-(2,3-dihydro-2-methyl-3-oxo-pyridazin-6-Yl)oxy-3-hydroxy-6-(3-hydroxyphenyl)sulphonyl-2,2,3-trimethyl-2H-benzo[b]pyran. Synth. Commun. 2007, 37, 3569–3578. [Google Scholar] [CrossRef]
- Day, D.P.; Sellars, P.B. Recent advances in iminium-salt-catalysed asymmetric epoxidation. Eur. J. Org. Chem. 2017, 6, 1034–1044. [Google Scholar] [CrossRef] [Green Version]
- Weiss, K.M.; Tsogoeva, S.B. Enantioselective epoxidation of electron-deficient olefins: An organocatalytic approach. Chem. Rec. 2011, 11, 18–39. [Google Scholar] [CrossRef]
- Ager, D.J.; Anderson, K.; Oblinger, E.; Shi, Y.; VanderRoest, J. An epoxidation approach to a chiral lactone: Application of the shi epoxidation. Org. Proc. Res. Dev. 2007, 11, 44–51. [Google Scholar] [CrossRef]
- Borbely, A.; Thoreau, F.; Figueras, E.; Kadri, M.; Coll, J.L.; Boturyn, D.; Sewald, N. Synthesis and biological characterization of monomeric and tetrameric RGD-cryptophycin conjugates. Chemistry 2020, 26, 2602–2605. [Google Scholar] [CrossRef] [PubMed]
- Figueras, E.; Borbely, A.; Ismail, M.; Frese, M.; Sewald, N. Novel unit B cryptophycin analogues as payloads for targeted therapy. Beilstein J. Org. Chem. 2018, 14, 1281–1286. [Google Scholar] [CrossRef]
- Hoard, D.W.; Moher, E.D.; Martinelli, M.J.; Norman, B.H. Synthesis of cryptophycin 52 using the Shi epoxidation. Org. Lett. 2002, 4, 1813–1815. [Google Scholar] [CrossRef]
- Altmann, K.-H.; Bold, G.; Caravatti, G.; Denni, D.; Flörsheimer, A.; Schmidt, A.; Rihs, G.; Wartmann, M. The total synthesis and biological assessment of trans-epothilone, A. Helv. Chim. Acta. 2002, 85, 4086–4110. [Google Scholar] [CrossRef]
- Method of Synthesis of 17β-hydroxy-11β-[4-(dimethylamino)-phenyl]-17α-(prop-1-ynyl)-estra-4,9-diene-one. Russia Patent RU2165938C1, 27 April 2001.
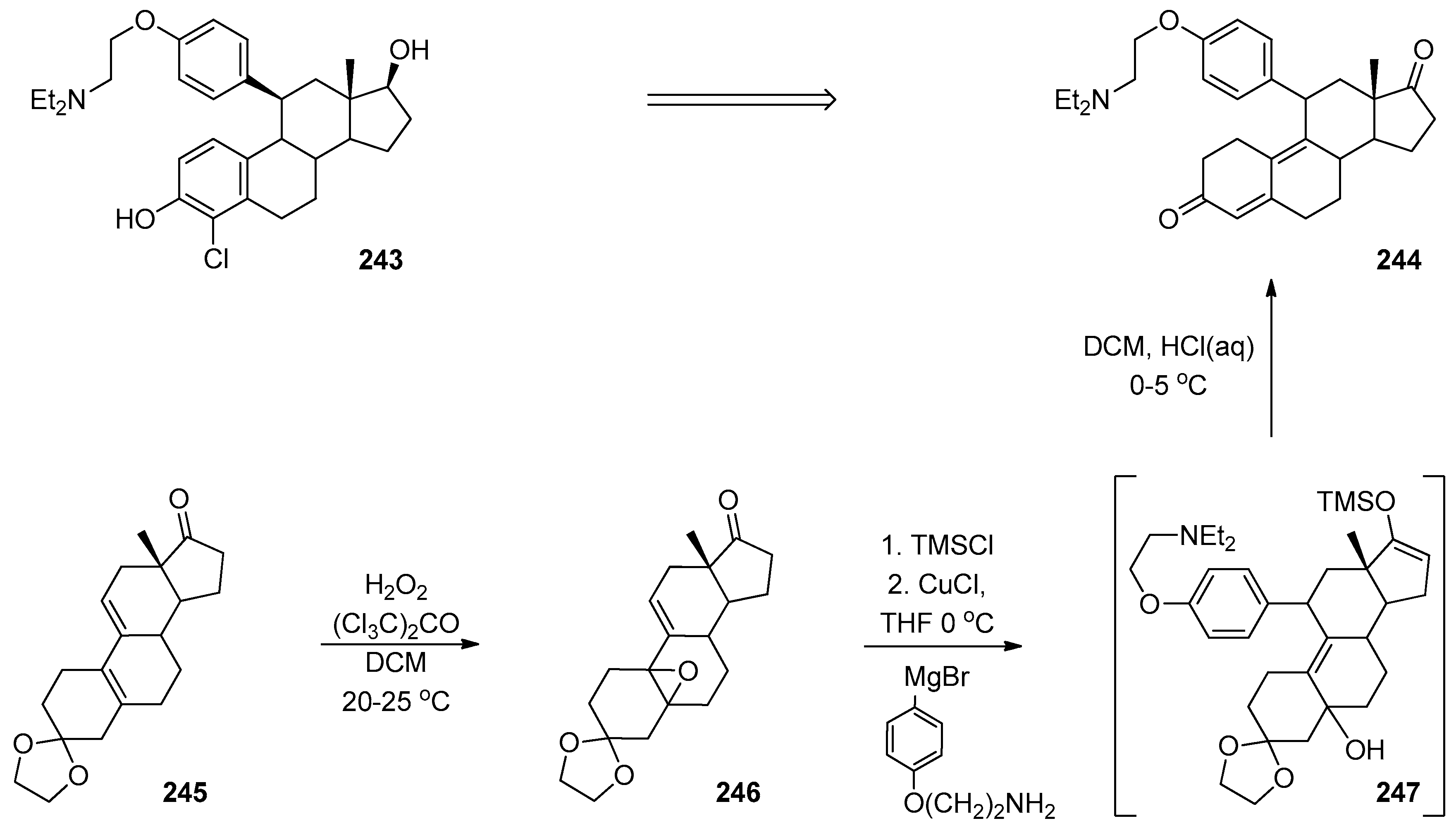
- Prat, D.; Benedetti, F.; Franc Girard, G.; Nait Bouda, L.; Larkin, J.; Wehrey, C.; Lenay, J. Industrial synthesis of 4-chloro,11β-arylestradiol: How to circumvent a poor diastereoselectivity. Org. Proc. Res. Dev. 2004, 8, 219–228. [Google Scholar] [CrossRef]
- Page, B.P.C.; Rassias, G.A.; Bethell, D.; Schilling, M.B. A new system for catalytic asymmetric epoxidation using iminium salt catalysts. J. Org. Chem. 1998, 63, 2774–2777. [Google Scholar] [CrossRef]
- Page, B.P.C.; Rassias, G.A.; Barros, D.; Ardakani, A.; Buckley, B.; Bethell, D.; Smith, T.A.; Slawin, A.M. Functionalized iminium salt systems for catalytic asymmetric epoxidation. J. Org. Chem. 2001, 66, 6926–6931. [Google Scholar] [CrossRef]
- Page, B.P.C.; Bartlett, C.J.; Chan, Y.; Day, D.; Parker, P.; Buckley, B.R.; Rassias, G.A.; Slawin, A.M.; Allin, S.M.; Lacour, J.; et al. Asymmetric epoxidation using iminium salt organocatalysts featuring dynamically controlled atropoisomerism. J. Org. Chem. 2012, 77, 6128–6138. [Google Scholar] [CrossRef]
- Page, B.P.C.; Pearce, C.A.; Chan, Y.; Parker, P.; Buckley, B.R.; Rassias, G.A.; Elsegood, M.R. Atropo- and diastereoselective construction of tetracyclic biphenylazepinium salts derived from aminoalcohols: Use as catalysts in enantioselective asymmetric epoxidation. J. Org. Chem. 2015, 80, 8036–8045. [Google Scholar] [CrossRef] [Green Version]
- Page, B.P.C.; Buckley, B.R.; Heaney, H.; Blacker, A.J. Asymmetric epoxidation of cis-alkenes mediated by iminium salts: Highly enantioselective synthesis of levcromakalim. Org. Lett. 2005, 7, 375–377. [Google Scholar] [CrossRef]


































































© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moschona, F.; Savvopoulou, I.; Tsitopoulou, M.; Tataraki, D.; Rassias, G. Epoxide Syntheses and Ring-Opening Reactions in Drug Development. Catalysts 2020, 10, 1117. https://doi.org/10.3390/catal10101117
Moschona F, Savvopoulou I, Tsitopoulou M, Tataraki D, Rassias G. Epoxide Syntheses and Ring-Opening Reactions in Drug Development. Catalysts. 2020; 10(10):1117. https://doi.org/10.3390/catal10101117
Chicago/Turabian StyleMoschona, Fotini, Ioanna Savvopoulou, Maria Tsitopoulou, Despoina Tataraki, and Gerasimos Rassias. 2020. "Epoxide Syntheses and Ring-Opening Reactions in Drug Development" Catalysts 10, no. 10: 1117. https://doi.org/10.3390/catal10101117