Design and Synthesis of Photoactive Iron N-Heterocyclic Carbene Complexes


Abstract
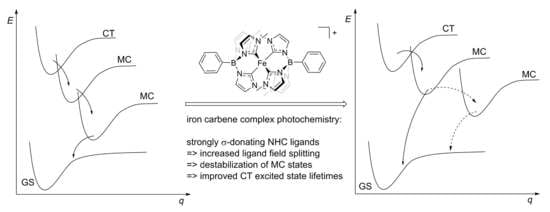
:
1. Introduction
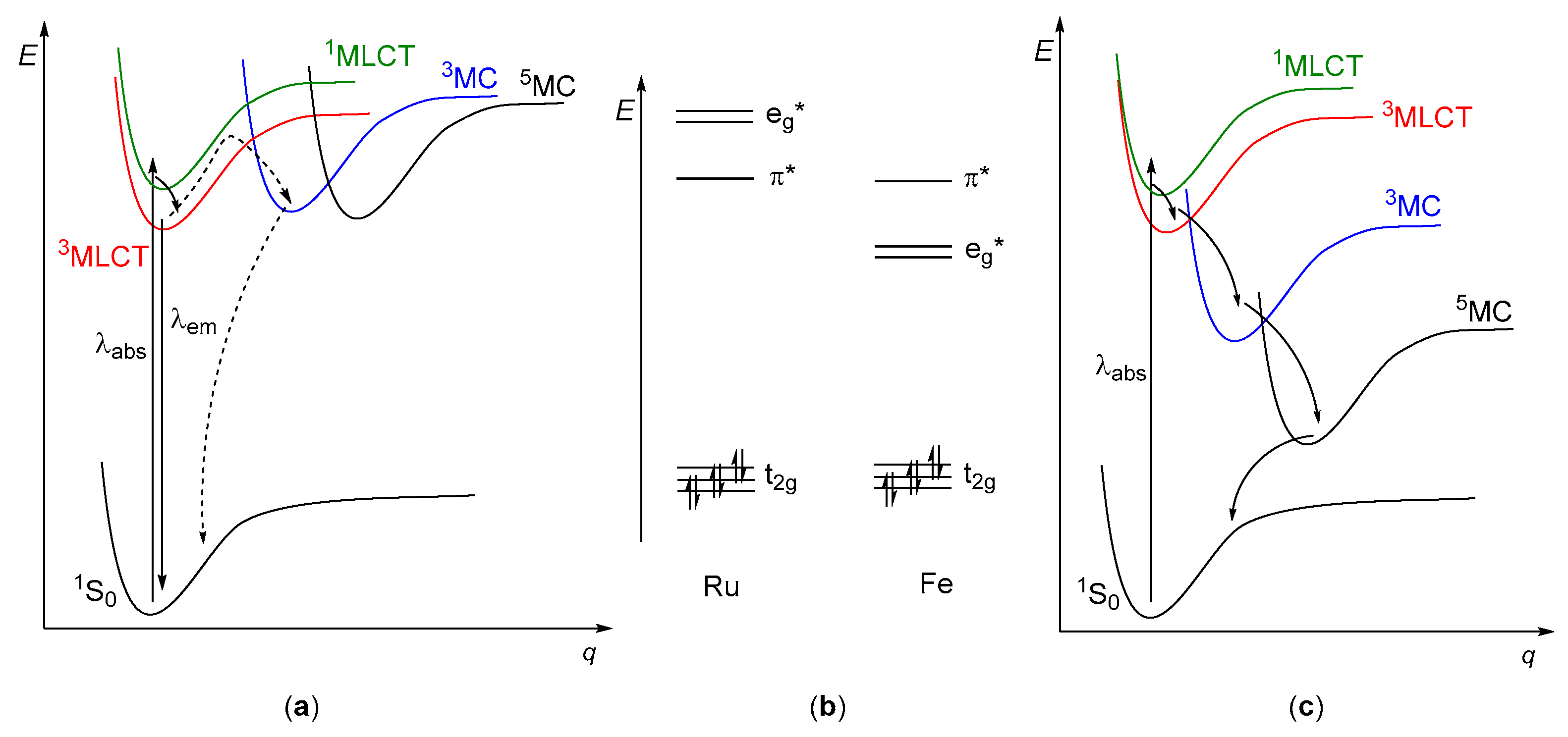
2. Fundamental Considerations
- Absorption of light over a broad range of the spectrum of the light source (visible and near-infrared in the context of solar energy conversion);
- Strong absorption of light, i.e., high molar attenuation coefficients;
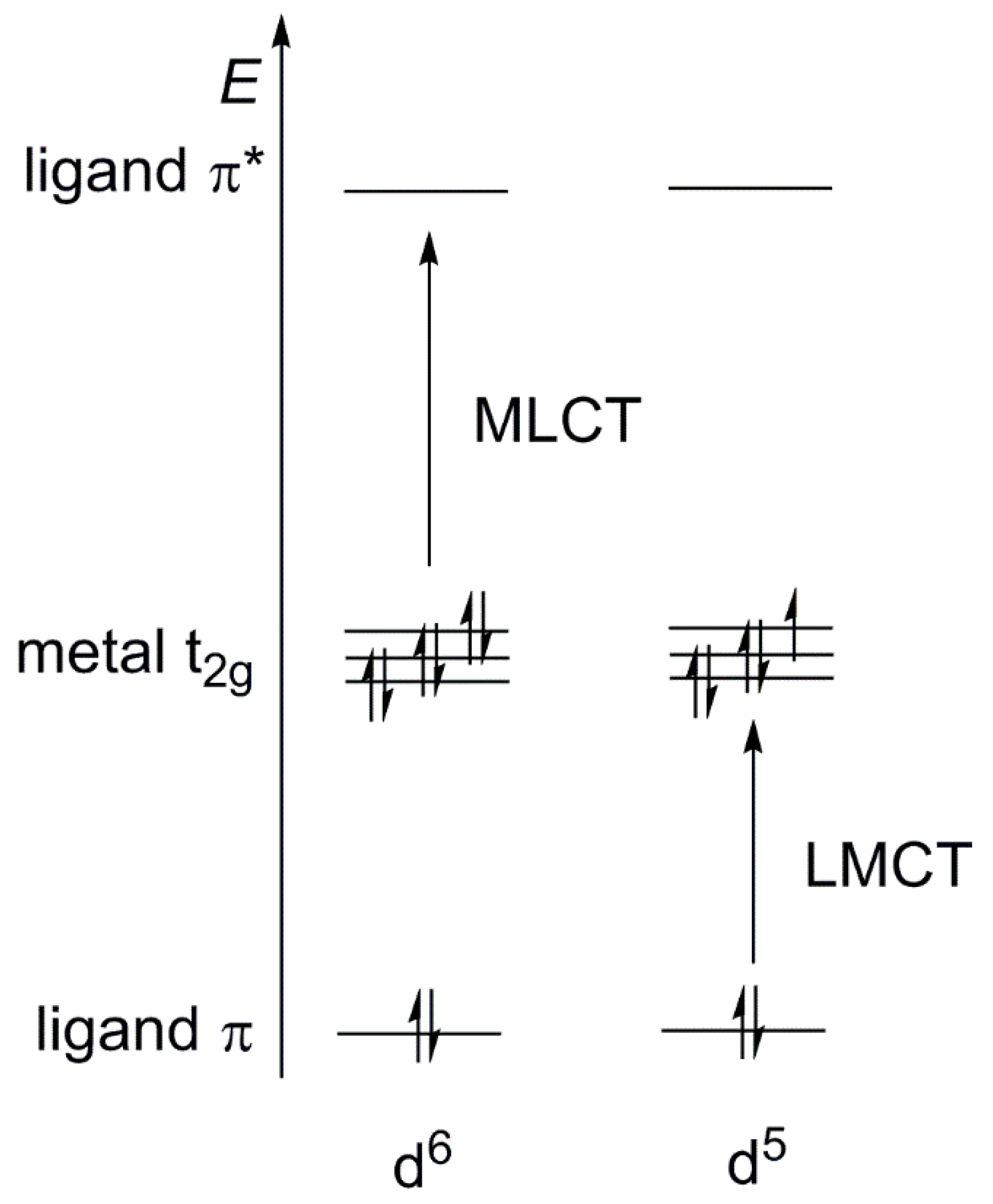
- Formation of long-lived (usually n–µs timescale) excited states of charge transfer (CT) character;
- Suitable ground and excited state redoxpotentials;
- Long-term chemical and photo-stability.
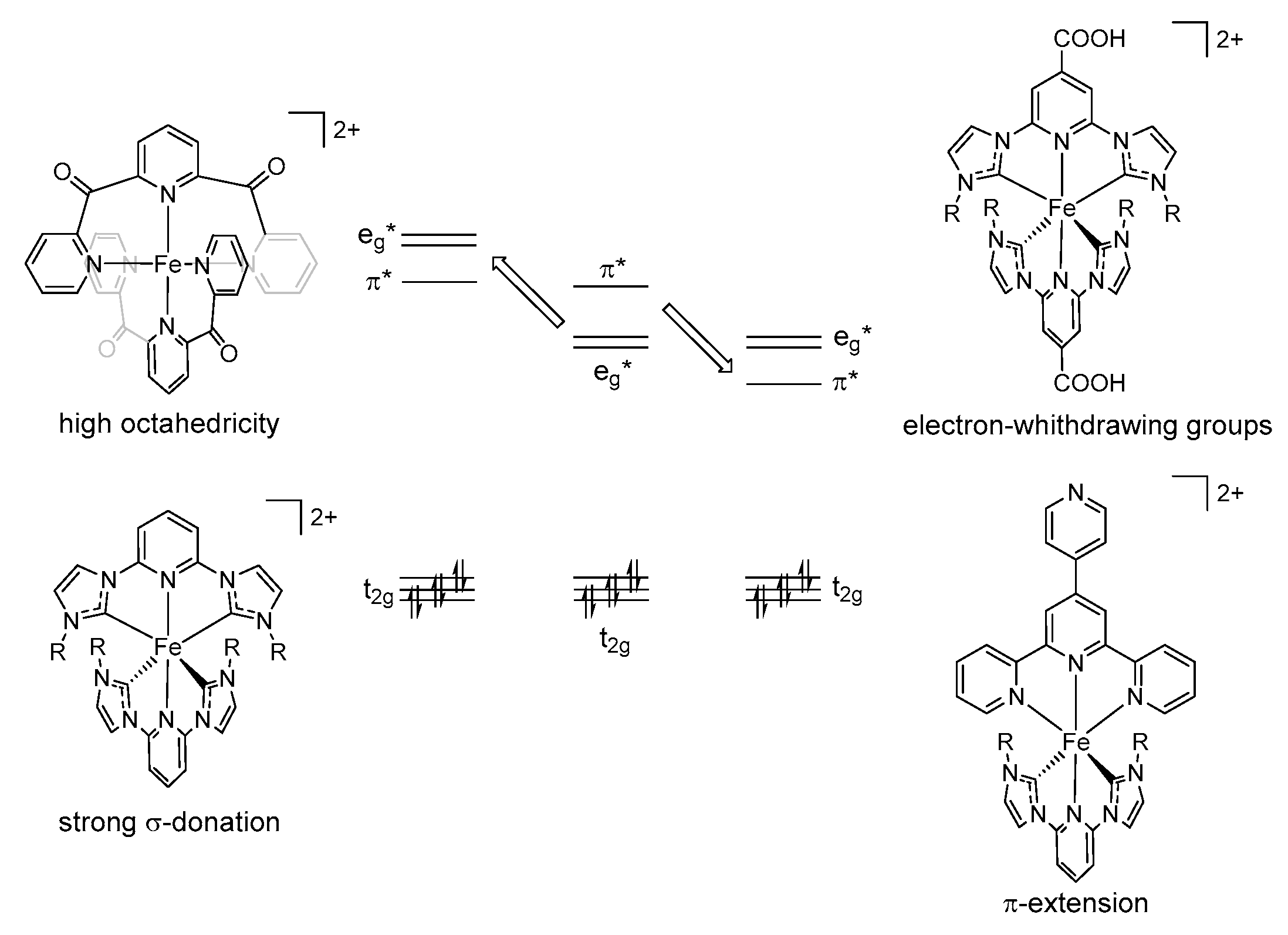
3. FeNHC Complexes with Meridional Coordinating Tridentate Ligands
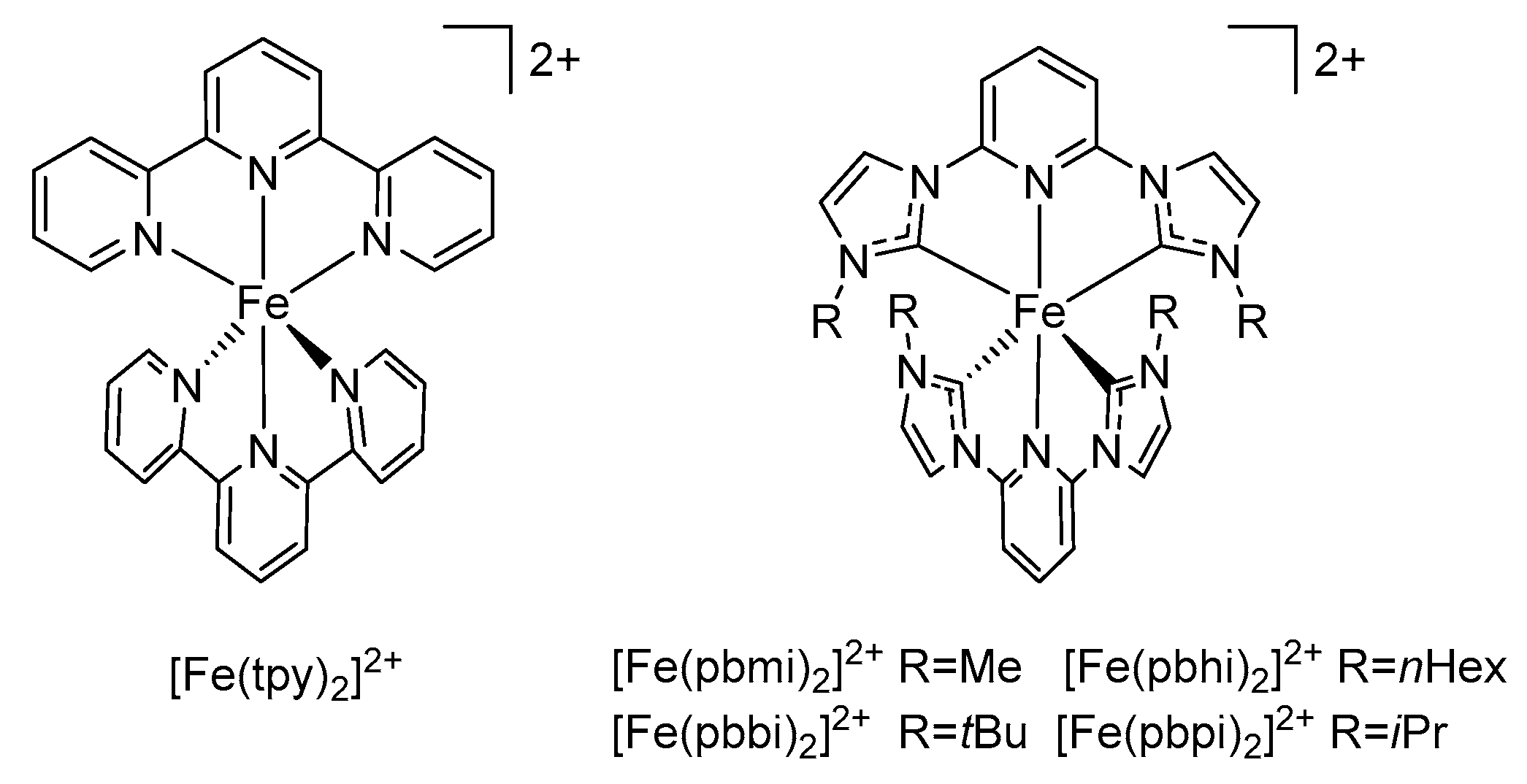
3.1. Homoleptic FeNHC Complexes with Meridional Coordinating Tridentate Ligands
3.2. Heteroleptic FeNHC Complexes with Meridional Tridentate Ligands
3.3. FeNHC Complexes with Meridional Tridentate Ligands with Anchoring Groups
3.3.1. Homoleptic FeNHC Complexes with Anchoring Groups
3.3.2. Heteroleptic FeNHC Complexes with Anchoring Groups
4. FeNHC Complexes with Bidentate Ligands
5. FeNHC Complex with Facial Coordinating Tridentate Ligand
6. Synthetic Approaches to FeNHC Complexes
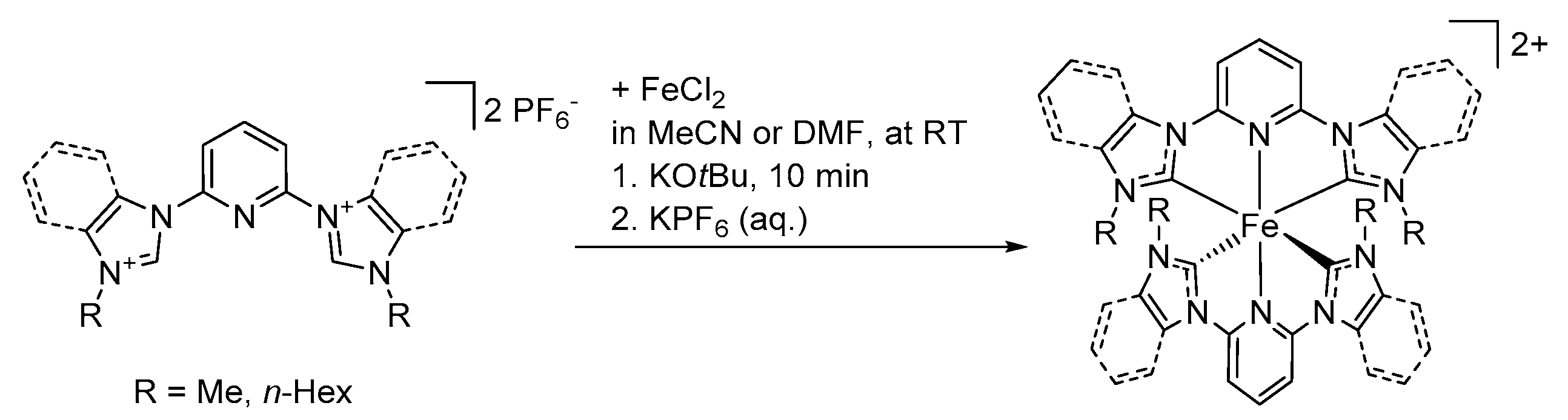
6.1. Synthesis of Homoleptic Complexes
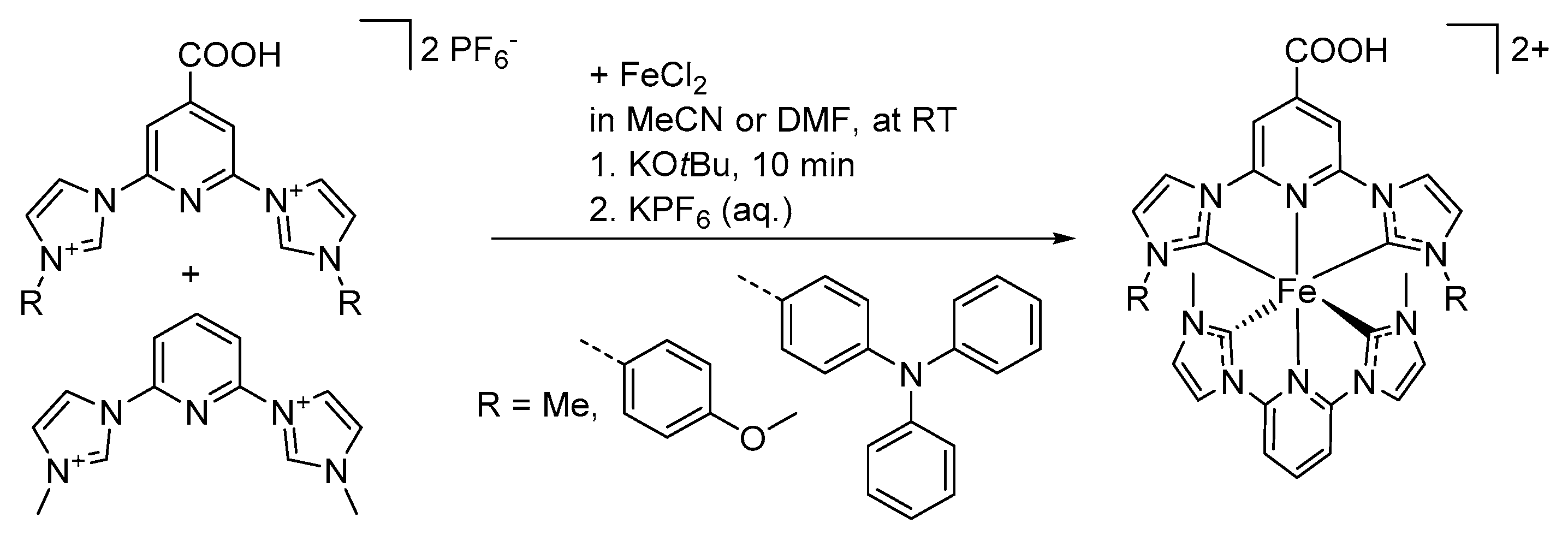
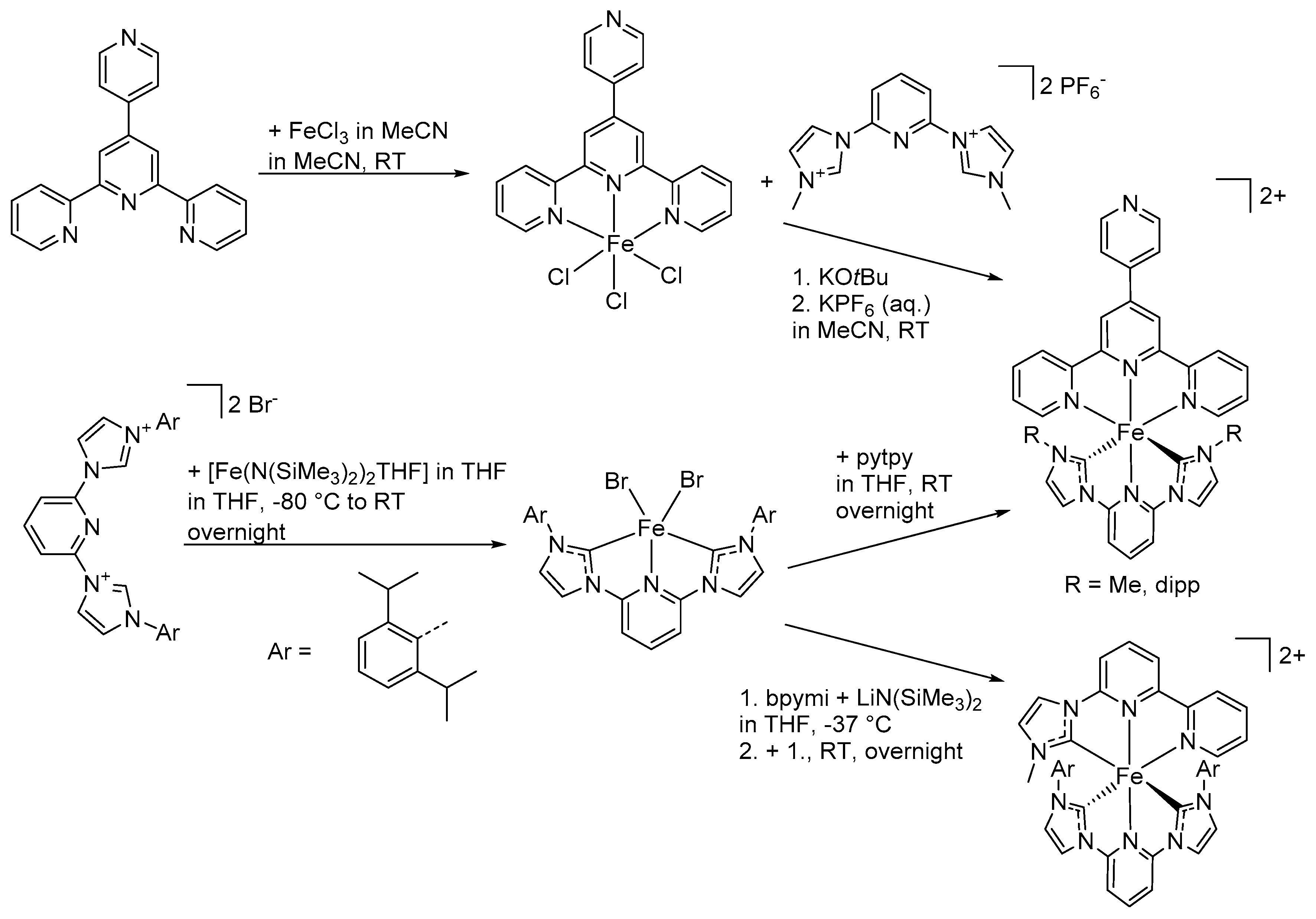
6.2. Synthesis of Heteroleptic Complexes
7. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| bpy | 2,2′-bipyridine |
| bpymi | ([2,2′-bipyridin]-6-yl)-3-methyl-imidazol-2-ylidene) |
| btz | 4,4′-bis(1,2,3-triazol-5-ylidene) |
| CoCl(dmg)2 | chloro-bis(dimethylglyoximato)-cobalt(III) |
| cpbdpai | (carboxypyridine-2,6-diyl)bis(1-N,N-diphenylanilinyl-imidazol-2-ylidene) |
| cpbai | (carboxypyridine-2,6-diyl)bis(1-p-anisyl-imidazol-2-ylidene) |
| cpbmbi | (carboxypyridine-2,6-diyl)bis(1-methyl-benzimidazol-2-ylidene) |
| cpbmi | (carboxypyridine-2,6-diyl)bis(1-methyl-imidazol-2-ylidene) |
| CT | charge transfer |
| CV | cyclic voltammetry |
| dcpp | 2,6-bis(2-carboxypyridyl)pyridine |
| ddpd | N,N′-dimethyl-N,N′-dipyridine-2-yl-pyridine-2,6-diamine |
| DFT | density functional theory |
| DMF | N,N-dimethylformamide |
| DSSC | dye-sensitized solar cell |
| EDG | electron-donating group |
| EPR | electron paramagnetic resonance |
| EWG | electron-withdrawing group |
| Fc | ferrocene |
| Fe–Co | [Fe(pbdppi)(pytpy)CoCl(dmg)]2+ |
| FeNHC | iron N-heterocyclic carbene (complex) |
| GS | ground state |
| HOMO | highest occupied molecular orbital |
| IC | internal conversion |
| ISC | intersystem crossing |
| ISC | short circuit current |
| LC | ligand centered |
| LIESST | light-induced excited state spin trapping |
| LMCT | ligand-to-metal charge transfer |
| LUMO | lowest unoccupied molecular orbital |
| MC | metal centered |
| MLCT | metal-to-ligand charge transfer |
| MO | molecular orbital |
| mpi | 3-methyl-1-(pyridin-2-yl)-imidazol-2-ylidene |
| NHC | N-heterocyclic carbene |
| NP | nanoparticle |
| NTO | natural transition orbital |
| oNTO | occupied natural transition orbital |
| pbbi | (pyridine-2,6-diyl)bis(1-tert-butyl)-imidazol-2-ylidene) |
| pbdppi | (pyridine-2,6-diyl)bis(1-(2,6-diisopropylphenyl)-imidazol-2-ylidene) |
| pbhi | (pyridine-2,6-diyl)bis(1-hexyl-imidazol-2-ylidene) |
| pbmbi | (pyridine-2,6-diyl)bis(1-methyl-benzimidazol-2-ylidene) |
| pbmi | (pyridine-2,6-diyl)bis(1-methyl-imidazol-2-ylidene) |
| pbpi | (pyridine-2,6-diyl)bis(1-isopropyl-imidazol-2-ylidene) |
| pbRi | (pyridine-2,6-diyl)bis(1-alkyl/aryl-imidazol-2-ylidene) |
| PES | potential energy surface |
| phtmib | phenyl(tris(3-methylimidazol-2-ylidene))borate |
| pmbmi | (pyrimidine-2,4-diyl)bis(1-methyl-imidazol-2-ylidene) |
| PS | photosensitizer |
| pytpy | (4′-(pyridin-4-yl)-2,2′:6′,2″-terpyridine) |
| pytpyH | 6′-(pyridin-2-yl)-[2,2′:4′,4″-terpyridin]-1″-ium |
| pytpyMe | 1″-methyl-6′-(pyridin-2-yl)-[2,2′:4′,4″-terpyridin]-1″-ium |
| pzbhbi | (pyrazine-2,6-diyl)bis(1-hexyl-benzimidazol-2-ylidene) |
| pzbhi | (pyrazine-2,6-diyl)bis(1-hexyl-imidazol-2-ylidene) |
| pzbmi | (pyrazine-2,6-diyl)bis(1-methyl-imidazol-2-ylidene) |
| QC | quantum chemical/chemistry |
| RT | room temperature |
| SCE | saturated calomel electrode |
| SEC | spectroelectrochemistry |
| TCSPC | time-correlated single photon counting |
| TD-DFT | time-dependent density functional theory |
| THF | tetrahydrofuran |
| tpiea | tris(2-(1-(pyridin-2-yl)-imidazol-2-ylidene)ethyl)amine |
| tpy | 2,2′:6′,2″-terpyridine |
| TRIS | hydro(tris(3-methylimidazol-2-ylidene))borate |
| TRWAXS | time-resolved wide-angle X-ray scattering |
| vNTO | virtual natural transition orbital |
Appendix A

| Acronym | λCT [nm] | εCT[M 1 cm−1] | τCT [ps] | Fe(III/II) vs. Fc+/Fc [V] | Fe(III/II) vs. SCE [V] | n NHC donors | Refs. |
|---|---|---|---|---|---|---|---|
| [Fe(tpy)2]2+ | 495 552 | 7500 14800 | 0.1 | 0.70 | 1.09 1 | 0 | [22] |
| [Fe(pbmi)2]2+ | 390 457 | 9100 15200 | 9 | 0.31 | 0.60 1 | 4 | [22] |
| [Fe(pbbi)2]2+ | 412 478 | 8400 14800 | 0.3 | 0.40 | 0.79 1 | 4 | [22] |
| [Fe(pbhi)2]2+ | 393 460 | 9000 15900 | n/a | 0.41 | 0.80 1 | 4 | [21] |
| [Fe(pbpi)2]2+ | 392 458 | 9000 15000 | 8 | 0.43 | 0.82 1 | 4 | [25,33] |
| [Fe(bpymi)2]2+ | 419 508 557 | 4100 4700 7000 | <0.1 | 0.44 | 0.83 1 | 2 | [33] |
| [Fe(pbmbi)2]2+ | 360 440 | 9000 12500 | 16 | 0.65 2 | 1.04 | 4 | [34] |
| [Fe(pzbmi)2]2+ | 389 487 | 6300 16100 | 21–25 | 0.59 2 | 0.98 | 4 | [36] |
| [Fe(pzbhi)2]2+ | 392 493 | 6200 17300 | 22 | 0.70 2 | 1.09 | 4 | [36] |
| [Fe(pzbhbi)2]2+ | 350 479 | 11000 16200 | 32 | 0.83 2 | 1.22 | 4 | [36] |
| [Fe(pmbmi)2]2+ | 406 477 | 8500 12600 | 11–12 | 0.54 2 | 0.93 | 4 | [36] |
| [Fe(pbhi)(pytpy)]2+ | 373 522 560 | 9000 9500 9000 | n/a | 0.54 | 0.93 1 | 2 | [21] |
| [Fe(pbhi)(pytpyH)]3+ | 407 525 590 | 6400 9000 11500 | n/a | 0.56 | 0.95 1 | 2 | [21] |
| [Fe(pbdppi)(tpy)]2+ | 378 502 540 | 9000 10000 9500 | <0.1 | 0.56 | 0.95 1 | 2 | [25] |
| [Fe(bpymi)(pdbppi)]2+ | 376 409 466 506 538 | 5500 6400 5200 5000 5300 | 3.6 | 0.46 | 0.85 1 | 3 | [33] |
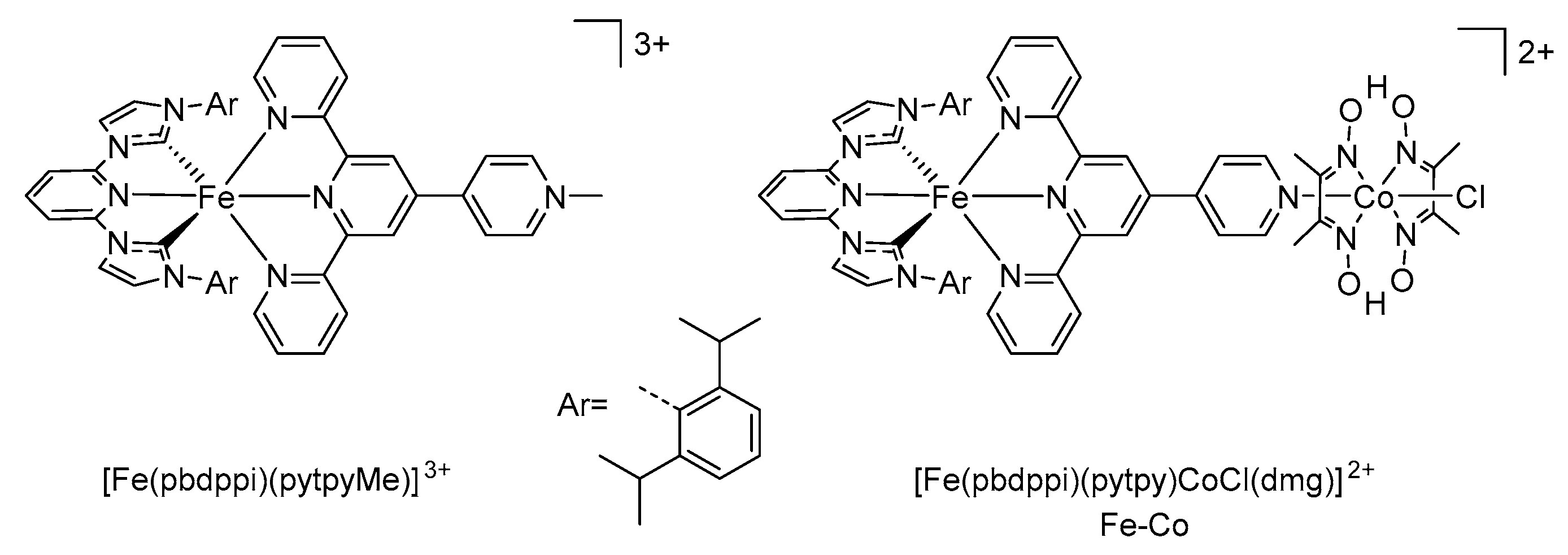
| [Fe(pbdppi)(pytpy)]2+ | 375 515 553 | 9600 10000 9800 | 1.1 | 0.60 | 0.99 1 | 2 | [37] |
| [Fe(pbdppi)(pytpyMe)]3+ | 374 411 538 586 | 6600 7000 9900 12700 | n/a | 0.66 | 1.05 1 | 2 | [37] |
| Fe–Co | 370 520 558 | 10300 9300 9400 | 1.4 | 0.63 | 1.02 1 | 2 | [37] |
| [Fe(cpbmi)2]2+ | 394 520 | 7000 16200 | 17 | 0.46 2 | 0.85 | 4 | [23,24,34] |
| [Fe(cpbmbi)2]2+ | 370 501 | 5000 12800 | 26 | 0.74 2 | 1.13 | 4 | [34] |
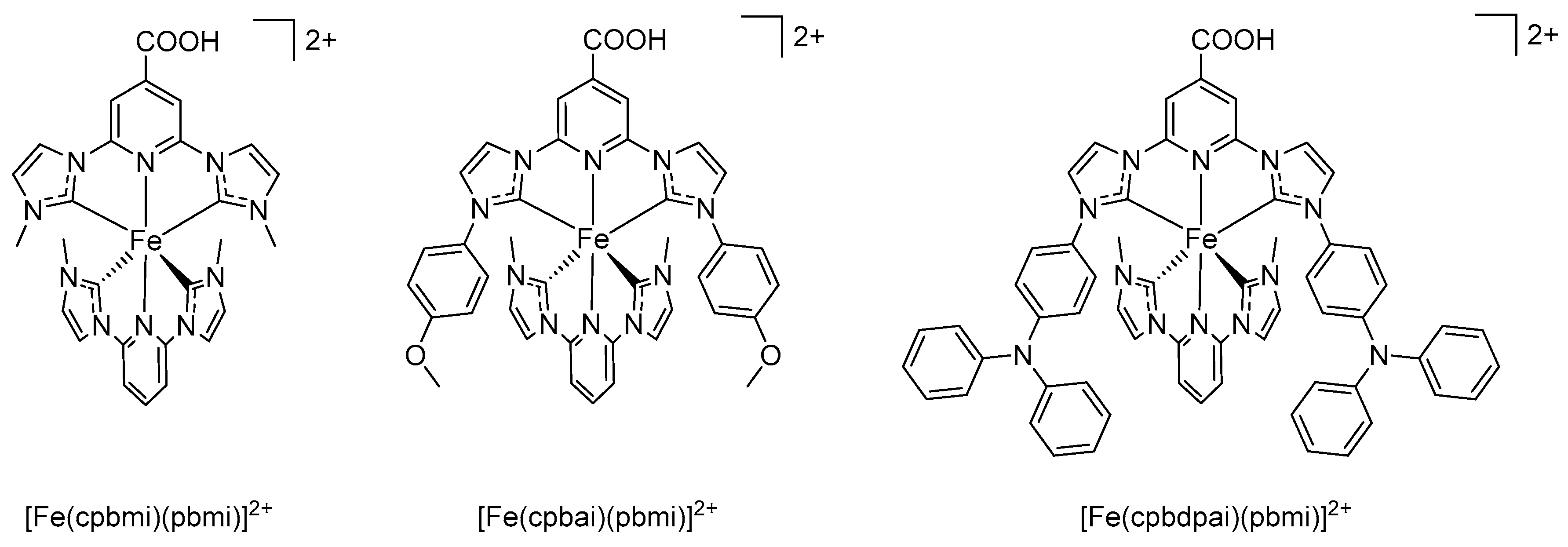
| [Fe(cpbmi)(pbmi)]2+ | 394 433 506 | 7625 7050 12650 | 14 | 0.43 2 | 0.82 | 4 | [43] |
| [Fe(cpbai)(pbmi)]2+ | 396 428 509 | 10100 8540 17700 | 10 | 0.42 2 | 0.81 | 4 | [43] |
| [Fe(cpbdpai)(pbmi)]2+ | 395 431 509 | 6115 5075 9500 | 12 | 0.41 2 | 0.82 | 4 | [43] |
| [Fe(bpy)3]2+ | 349 486 520 | 6030 6880 7980 | 0.1 | 0.68 | 1.07 1 | 0 | [49] |
| [Fe(bpy)(btz)2]2+ | 432 609 | 5080 3260 | 13 | −0.35 | 0.04 1 | 4 | [49] |
| [Fe(btz)3]2+ | 370 446 510 688 | 4600 5500 4000 2000 | 528 | −0.58 | −0.19 1 | 6 | [50,51] |
| [Fe(btz)3]3+ | 384 420 528 558 | 2400 1800 15001400 | 100 | −0.58 | −0.19 1 | 6 | [51] |
| [Fe(phtmib)2]+ | 502 | 3000 | 1960 | −1.16 | −0.77 1 | 6 | [61] |
| [Fe(mpi) 3]2+ | 360 430 | 4500 12000 | 15–20 3 | 0.26 2 | 0.65 | 3 | [55,56] |
| [Fetpiea]2+ | 369 438 | 5200 8000 | 15–20 3 | 0.22 2 | 0.61 | 3 | [55,56] |
References
- Stephenson, C.R.J.; Yoon, T.P.; MacMillan, D.W.C. (Eds.) Visible Light Photocatalysis in Organic Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2018. [Google Scholar]
- Yuan, Y.-J.; Yu, Z.-T.; Chen, D.-Q.; Zou, Z.-G. Metal-complex chromophores for solar hydrogen generation. Chem. Soc. Rev. 2017, 46, 603–631. [Google Scholar] [CrossRef]
- Yamamoto, M.; Tanaka, K. Artificial molecular photosynthetic systems: Towards efficient photoelectrochemical water oxidation. ChemPlusChem 2016, 81, 1028–1044. [Google Scholar] [CrossRef]
- Junge, H.; Rockstroh, N.; Fischer, S.; Brückner, A.; Ludwig, R.; Lochbrunner, S.; Kühn, O.; Beller, M. Light to hydrogen: Photocatalytic hydrogen generation from water with molecularly-defined iron complexes. Inorganics 2017, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Armaroli, N.; Balzani, V. Solar electricity and solar fuels: Status and perspectives in the context of the energy transition. Chem. A Eur. J. 2016, 22, 32–57. [Google Scholar] [CrossRef] [PubMed]
- Balzani, V.; Campagna, S. (Eds.) Photochemistry and Photophysics of Coordination Compounds I; Springer: Berlin/Heidelberg, Germany, 2007. [Google Scholar] [CrossRef]
- Campagna, S.; Puntoriero, F.; Nastasi, F.; Bergamini, G.; Balzani, V. Photochemistry and Photophysics of Coordination Compounds II; Springer: Berlin/Heidelberg, Germany, 2007. [Google Scholar] [CrossRef]
- Wenger, O.S. Photoactive complexes with earth-abundant metals. J. Am. Chem. Soc. 2018, 140, 13522–13533. [Google Scholar] [CrossRef] [Green Version]
- Hockin, B.M.; Li, C.; Robertson, N.; Zysman-Colman, E. Photoredox catalysts based on earth-abundant metal complexes. Catal. Sci. Technol. 2019, 9, 889–915. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Browne, W.R. Photochemistry of iron complexes. Coord. Chem. Rev. 2018, 374, 15–35. [Google Scholar] [CrossRef]
- Wikipedia—Abundance of Elements in Earth’s Crust (n.d.). Available online: https://en.wikipedia.org/wiki/Abundance_of_elements_in_Earth%27s_crust (accessed on 11 October 2019).
- Lindh, L.; Chábera, P.; Rosemann, N.W.; Uhlig, J.; Wärnmark, K.; Yartsev, A.; Sundström, V.; Persson, P. Photophysics and Photochemistry of Iron Carbene Complexes for Solar Energy Conversion and Photocatalysis. Catalysts. in press.
- Wenger, O.S. Is Iron the new ruthenium? Chem. A Eur. J. 2019, 25, 6043–6052. [Google Scholar] [CrossRef] [Green Version]
- McCusker, J.K. Electronic structure in the transition metal block and its implications for light harvesting. Science 2019, 363, 484–488. [Google Scholar] [CrossRef]
- Fink, D.W.; Ohnesorge, W.E. Temperature effects on charge-transfer luminescence intensity of some transition metal ion chelates. J. Am. Chem. Soc. 1969, 91, 4995–4998. [Google Scholar] [CrossRef]
- Decurtins, S.; Gutlich, P.; Hasselbach, K.M.; Hauser, A.; Spiering, H. Light-induced excited-spin-state trapping in iron(II) spin-crossover systems. Optical spectroscopic and magnetic susceptibility study. Inorg. Chem. 1985, 24, 2174–2178. [Google Scholar] [CrossRef]
- Kahn, O. Spin-transition polymers: From molecular materials toward memory devices. Science 1998, 279, 44–48. [Google Scholar] [CrossRef]
- Gütlich, P.; Gaspar, A.B.; Garcia, Y. Spin state switching in iron coordination compounds. Beilstein J. Org. Chem. 2013, 9, 342–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamula, L.L.; Brown, A.M.; Guo, D.; McCusker, J.K. Synthesis and characterization of a high-symmetry ferrous polypyridyl complex: Approaching the 5 T2/3 T1 crossing point for Fe II. Inorg. Chem. 2014, 53, 15–17. [Google Scholar] [CrossRef] [PubMed]
- Duchanois, T.; Etienne, T.; Beley, M.; Assfeld, X.; Perpète, E.A.; Monari, A.; Gros, P.C. Heteroleptic pyridyl-carbene Iron complexes with tuneable electronic properties. Eur. J. Inorg. Chem. 2014, 2014, 3747–3753. [Google Scholar] [CrossRef]
- Liu, Y.; Harlang, T.; Canton, S.E.; Chábera, P.; Suárez-Alcántara, K.; Fleckhaus, A.; Vithanage, D.A.; Göransson, E.; Corani, A.; Lomoth, R.; et al. Towards longer-lived metal-to-ligand charge transfer states of iron(ii) complexes: An N-heterocyclic carbene approach. Chem. Commun. 2013, 49, 6412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duchanois, T.; Etienne, T.; Cebrián, C.; Liu, L.; Monari, A.; Beley, M.; Assfeld, X.; Haacke, S.; Gros, P.C. An Iron-based photosensitizer with extended excited-state lifetime: Photophysical and photovoltaic properties. Eur. J. Inorg. Chem. 2015, 2015, 2469–2477. [Google Scholar] [CrossRef]
- Harlang, T.C.B.; Liu, Y.; Gordivska, O.; Fredin, L.A.; Ponseca, C.S.; Huang, P.; Chábera, P.; Kjaer, K.S.; Mateos, H.; Uhlig, J.; et al. Iron sensitizer converts light to electrons with 92% yield. Nat. Chem. 2015, 7, 883–889. [Google Scholar] [CrossRef] [Green Version]
- Zimmer, P.; Müller, P.; Burkhardt, L.; Schepper, R.; Neuba, A.; Steube, J.; Dietrich, F.; Flörke, U.; Mangold, S.; Gerhards, M.; et al. N-Heterocyclic carbene complexes of Iron as photosensitizers for light-induced water reduction. Eur. J. Inorg. Chem. 2017, 2017, 1504–1509. [Google Scholar] [CrossRef]
- Clayden, J.; Greeves, N.; Warren, S. Organic Chemistry, 2nd ed.; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Fatur, S.M.; Shepard, S.G.; Higgins, R.F.; Shores, M.P.; Damrauer, N.H. A synthetically tunable system to control MLCT excited-state lifetimes and spin states in Iron(II) polypyridines. J. Am. Chem. Soc. 2017, 139, 4493–4505. [Google Scholar] [CrossRef]
- Winkler, J.R.; Creutz, C.; Sutin, N. Solvent tuning of the excited-state properties of (2,2′-bipyridine)tetracyanoferrate(II): Direct observation of a metal-to-ligand charge-transfer excited state of Iron(II). J. Am. Chem. Soc. 1987, 109, 3470–3471. [Google Scholar] [CrossRef]
- Mengel, A.K.C.; Förster, C.; Breivogel, A.; Mack, K.; Ochsmann, J.R.; Laquai, F.; Ksenofontov, V.; Heinze, K. A heteroleptic push-pull substituted Iron(II) bis(tridentate) complex with low-energy charge-transfer states. Chem. A Eur. J. 2015, 21, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Persson, P.; Sundström, V.; Wärnmark, K. Fe N-Heterocyclic carbene complexes as promising photosensitizers. Acc. Chem. Res. 2016, 49, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Leshchev, D.; Harlang, T.C.B.; Fredin, L.A.; Khakhulin, D.; Liu, Y.; Biasin, E.; Laursen, M.G.; Newby, G.E.; Haldrup, K.; Nielsen, M.M.; et al. Tracking the picosecond deactivation dynamics of a photoexcited iron carbene complex by time-resolved X-ray scattering. Chem. Sci. 2018, 9, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Fredin, L.A.; Pápai, M.; Rozsályi, E.; Vankó, G.; Wärnmark, K.; Sundström, V.; Persson, P. Exceptional excited-state lifetime of an Iron(II)—N-Heterocyclic carbene complex explained. J. Phys. Chem. Lett. 2014, 5, 2066–2071. [Google Scholar] [CrossRef]
- Zimmer, P.; Burkhardt, L.; Friedrich, A.; Steube, J.; Neuba, A.; Schepper, R.; Müller, P.; Flörke, U.; Huber, M.; Lochbrunner, S.; et al. The connection between NHC ligand count and photophysical properties in Fe(II) photosensitizers: An experimental study. Inorg. Chem. 2018, 57, 360–373. [Google Scholar] [CrossRef]
- Liu, L.; Duchanois, T.; Etienne, T.; Monari, A.; Beley, M.; Assfeld, X.; Haacke, S.; Gros, P.C. A new record excited state 3MLCT lifetime for metalorganic iron(II) complexes. Phys. Chem. Chem. Phys. 2016, 18, 12550–12556. [Google Scholar] [CrossRef]
- Gaggioli, C.A.; Bistoni, G.; Ciancaleoni, G.; Tarantelli, F.; Belpassi, L.; Belanzoni, P. Modulating the bonding properties of N-Heterocyclic Carbenes (NHCs): A systematic charge-displacement analysis. Chem. A Eur. J. 2017, 23, 7558–7569. [Google Scholar] [CrossRef]
- Darari, M.; Domenichini, E.; Francés-Monerris, A.; Cebrián, C.; Magra, K.; Beley, M.; Pastore, M.; Monari, A.; Assfeld, X.; Haacke, S.; et al. Iron(II) complexes with diazinyl-NHC ligands: Impact of π-deficiency of the azine core on photophysical properties. Dalton Trans. 2019. [Google Scholar] [CrossRef]
- Zimmer, P.; Burkhardt, L.; Schepper, R.; Gosztola, D.; Zheng, K.; Neuba, A.; Flörke, U.; Wölper, C.; Schoch, R.; Gawelda, W.; et al. Towards noble-metal free dyads: Ground and excited state tuning by a cobalt dimethylglyoxime catalyst connected to an iron N-heterocyclic carbene photosensitizer. Eur. J. Inorg. Chem. 2018. [Google Scholar] [CrossRef] [Green Version]
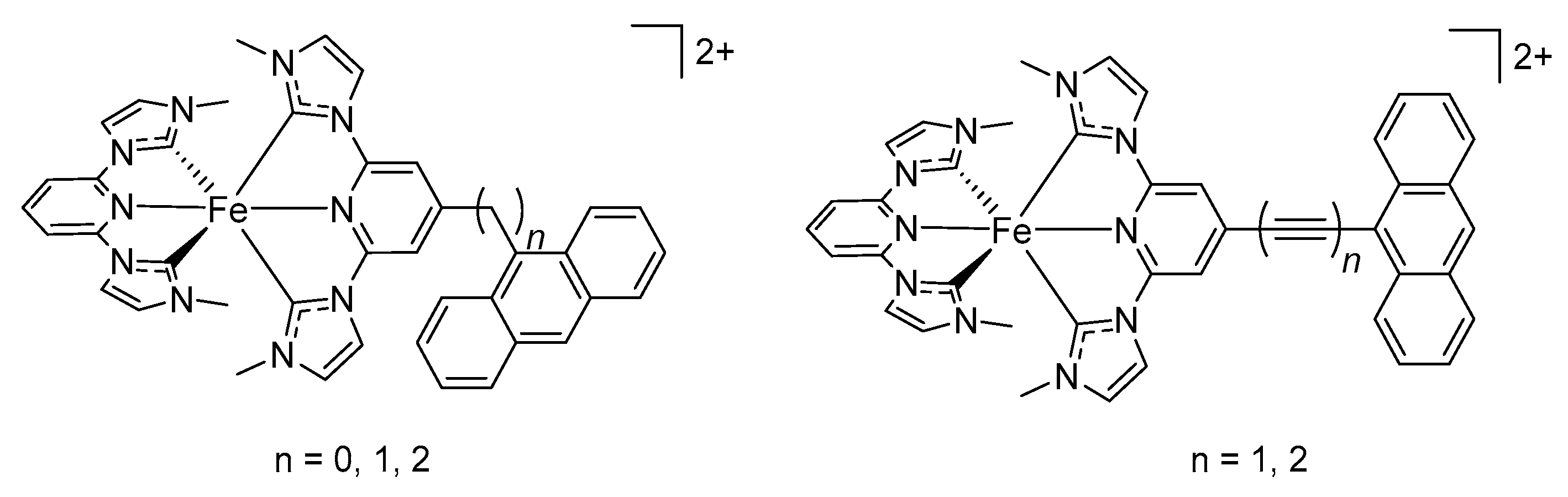
- Francés-Monerris, A.; Gros, P.C.; Pastore, M.; Assfeld, X.; Monari, A. Photophysical properties of bichromophoric Fe(II) complexes bearing an aromatic electron acceptor. Chem. Acc. 2019, 138, 86. [Google Scholar] [CrossRef]
- Ferrere, S.; Gregg, B.A. Photosensitization of TiO2 by [FeII(2,2′-bipyridine-4,4′-dicarboxylic acid)2(CN)2]: Band selective electron injection from ultra-short-lived excited states. J. Am. Chem. Soc. 1998, 120, 843–844. [Google Scholar] [CrossRef]
- Ferrere, S. New photosensitizers based upon [Fe(L)2(CN)2] and [Fe(L)3] (L = Substituted 2,2′-Bipyridine): Yields for the photosensitization of TiO2 and effects on the band selectivity. Chem. Mater. 2000, 12, 1083–1089. [Google Scholar] [CrossRef]
- Abrahamsson, M. Solar energy conversion using iron polypyridyl type photosensitizers—A viable route for the future? Photochemistry 2017, 44, 285–295. [Google Scholar] [CrossRef]
- Fredin, L.A.; Wärnmark, K.; Sundström, V.; Persson, P. Molecular and interfacial calculations of Iron(II) light harvesters. ChemSusChem 2016, 9, 667–675. [Google Scholar] [CrossRef]
- Pastore, M.; Duchanois, T.; Liu, L.; Monari, A.; Assfeld, X.; Haacke, S.; Gros, P.C. Interfacial charge separation and photovoltaic efficiency in Fe(ii)-carbene sensitized solar cells. Phys. Chem. Chem. Phys. 2016, 18, 28069–28081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpacheva, M.; Housecroft, C.E.; Constable, E.C. Electrolyte tuning in dye-sensitized solar cells with N-heterocyclic carbene (NHC) Iron(II) sensitizers. Beilstein J. Nanotechnol. 2018, 9, 3069–3078. [Google Scholar] [CrossRef] [Green Version]
- Karpacheva, M.; Wyss, V.; Housecroft, C.E.; Constable, E.C. There Is a Future for N-Heterocyclic Carbene Iron(II) Dyes in Dye-Sensitized Solar Cells: Improving Performance through Changes in the Electrolyte. Materials 2019, 12, 4181. [Google Scholar] [CrossRef] [Green Version]
- Marchini, E.; Darari, M.; Lazzarin, L.; Boaretto, R.; Argazzi, R.; Bignozzi, C.A.; Gros, P.C.; Caramori, S. Recombination and regeneration dynamics in FeNHC(ii)-sensitized solar cells. Chem. Commun. 2020, 56, 2–5. [Google Scholar] [CrossRef]
- Duchanois, T.; Liu, L.; Pastore, M.; Monari, A.; Cebrián, C.; Trolez, Y.; Darari, M.; Magra, K.; Francés-Monerris, A.; Domenichini, E.; et al. NHC-based Iron sensitizers for DSSCs. Inorganics 2018, 6, 63. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wärnmark, K. Fe complexes as photosensitizers for dye-sensitized solar cells. In Emerging Photovoltaic Technologies: Photophysics and Devices, 1st ed.; Ponseca, C.S., Ed.; Jenny Standford Publishing: Singapore, 2020. [Google Scholar]
- Liu, Y.; Kjaer, K.S.; Fredin, L.A.; Chábera, P.; Harlang, T.; Canton, S.E.; Lidin, S.; Zhang, J.; Lomoth, R.; Bergquist, K.-E.; et al. A heteroleptic ferrous complex with mesoionic bis(1,2,3-triazol-5-ylidene) ligands: Taming the MLCT excited state of Iron(II). Chem. A Eur. J. 2015, 21, 3628–3639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chábera, P.; Kjaer, K.S.; Prakash, O.; Honarfar, A.; Liu, Y.; Fredin, L.A.; Harlang, T.C.B.; Lidin, S.; Uhlig, J.; Sundström, V.; et al. Fe II hexa N-Heterocyclic carbene complex with a 528 ps metal-to-ligand charge-transfer excited-state lifetime. J. Phys. Chem. Lett. 2018, 9, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Chábera, P.; Liu, Y.; Prakash, O.; Thyrhaug, E.; Nahhas, A.; Honarfar, A.; Essén, S.; Fredin, L.A.; Harlang, T.C.B.; Kjær, K.S.; et al. A low-spin Fe(iii) complex with 100-ps ligand-to-metal charge transfer photoluminescence. Nature 2017, 543, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Tatsuno, H.; Kjaer, K.S.; Kunnus, K.; Harlang, T.C.B.; Timm, C.; Guo, M.; Chàbera, P.; Fredin, L.A.; Hartsock, R.W.; Reinhard, M.E.; et al. Hot Branching Dynamics in a Light-Harvesting Iron Carbene Complex Revealed by Ultrafast X-Ray Emission Spectroscopy. Angew. Chemie Int. Ed. 2020, 59, 364–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ericson, F.; Honarfar, A.; Prakash, O.; Tatsuno, H.; Fredin, L.A.; Handrup, K.; Chabera, P.; Gordivska, O.; Kjær, K.S.; Liu, Y.; et al. Electronic structure and excited state properties of iron carbene photosensitizers—A combined X-ray absorption and quantum chemical investigation. Chem. Phys. Lett. 2017, 683, 559–566. [Google Scholar] [CrossRef]
- Chábera, P.; Fredin, L.A.; Kjær, K.S.; Rosemann, N.W.; Lindh, L.; Prakash, O.; Liu, Y.; Wärnmark, K.; Uhlig, J.; Sundström, V.; et al. Band-selective dynamics in charge-transfer excited iron carbene complexes. Faraday Discuss. 2019, 216, 191–210. [Google Scholar] [CrossRef] [Green Version]
- Francés-Monerris, A.; Magra, K.; Darari, M.; Cebrián, C.; Beley, M.; Domenichini, E.; Haacke, S.; Pastore, M.; Assfeld, X.; Gros, P.C.; et al. Synthesis and computational study of a pyridylcarbene Fe(II) complex: Unexpected effects of fac/mer isomerism in metal-to-ligand triplet potential energy surfaces. Inorg. Chem. 2018, 57, 10431–10441. [Google Scholar] [CrossRef] [PubMed]
- Magra, K.; Domenichini, E.; Francés-Monerris, A.; Cebrián, C.; Beley, M.; Darari, M.; Pastore, M.; Monari, A.; Assfeld, X.; Haacke, S.; et al. Impact of the fac/mer Isomerism on the excited-state dynamics of pyridyl-carbene Fe(II) complexes. Inorg. Chem. 2019, 58, 5069–5081. [Google Scholar] [CrossRef] [Green Version]
- Forshaw, A.P.; Bontchev, R.P.; Smith, J.M. Oxidation of the tris(carbene)borate complex PhB(MeIm)3MnI(CO)3 to MnIV[PhB(MeIm)3]2(OTf)2. Inorg. Chem. 2007, 46, 3792–3794. [Google Scholar] [CrossRef]
- Kernbach, U.; Ramm, M.; Luger, P.; Fehlhammer, W.P. A chelating triscarbene ligand and its hexacarbene Iron complex. Angew. Chem. Int. Ed. Engl. 1996, 35, 310–312. [Google Scholar] [CrossRef]
- Muñoz, S.B.; Foster, W.K.; Lin, H.J.; Margarit, C.G.; Dickie, D.A.; Smith, J.M. Tris(carbene)borate ligands featuring imidazole-2-ylidene, benzimidazol-2-ylidene, and 1,3,4-triazol-2-ylidene donors. Evaluation of donor properties in four-coordinate {NiNO}10 complexes. Inorg. Chem. 2012, 51, 12660–12668. [Google Scholar] [CrossRef] [Green Version]
- Forshaw, A.P.; Smith, J.M.; Ozarowski, A.; Krzystek, J.; Smirnov, D.; Zvyagin, S.A.; Harris, T.D.; Karunadasa, H.I.; Zadrozny, J.M.; Schnegg, A.; et al. Low-spin hexacoordinate Mn(III): Synthesis and spectroscopic investigation of homoleptic tris(pyrazolyl)borate and tris(carbene)borate complexes. Inorg. Chem. 2013, 52, 144–159. [Google Scholar] [CrossRef] [PubMed]
- Kjær, K.S.; Kaul, N.; Prakash, O.; Chábera, P.; Rosemann, N.W.; Honarfar, A.; Gordivska, O.; Fredin, L.A.; Bergquist, K.; Häggström, L.; et al. Luminescence and reactivity of a charge-transfer excited iron complex with nanosecond lifetime. Science 2019, 363, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhang, W.-Z.; Liu, C.-H.; Qu, J.-P.; Lu, X.-B. CO2 adducts of N-Heterocyclic carbenes: Thermal stability and catalytic activity toward the coupling of CO2 with epoxides. J. Org. Chem. 2008, 73, 8039–8044. [Google Scholar] [CrossRef] [PubMed]
- Chábera, P.; Lindh, L.; Rosemann, N.W.; Prakash, O.; Uhlig, J.; Yartsev, A.; Wärnmark, K.; Sundström, V.; Persson, P. Photofunctionality of Iron (III) N-Heterocyclic Carbenes and Related d5 Transition Metal Complexes. Coord. Chem. Rev. 2020, in press. [Google Scholar]
- Dixon, I.M.; Khan, S.; Alary, F.; Boggio-Pasqua, M.; Heully, J.L. Probing the photophysical capability of mono and bis(cyclometallated) Fe(ii) polypyridine complexes using inexpensive ground state DFT. Dalt. Trans. 2014, 43, 15898–15905. [Google Scholar] [CrossRef]
- Dixon, I.M.; Alary, F.; Boggio-Pasqua, M.; Heully, J.L. Reversing the relative 3MLCT-3MC order in Fe(II) complexes using cyclometallating ligands: A computational study aiming at luminescent Fe(II) complexes. Dalt. Trans. 2015, 44, 13498–13503. [Google Scholar] [CrossRef]
- Mukherjee, S.; Bowman, D.N.; Jakubikova, E. Cyclometalated Fe(II) complexes as sensitizers in dye-sensitized solar cells. Inorg. Chem. 2015, 54, 560–569. [Google Scholar] [CrossRef]
- Ashley, D.C.; Mukherjee, S.; Jakubikova, E. Designing air-stable cyclometalated Fe(ii) complexes: Stabilization via electrostatic effects. Dalt. Trans. 2019, 48, 374–378. [Google Scholar] [CrossRef]
- Steube, J.; Burkhardt, L.; Päpcke, A.; Moll, J.; Zimmer, P.; Schoch, R.; Wölper, C.; Heinze, K.; Lochbrunner, S.; Bauer, M. Excited state kinetics of an air-stable cyclometalated iron(II) complex. Chem. A Eur. J. 2019. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.D.; Lozada, I.B.; Kolodziej, C.; Burda, C.; Newman, K.M.E.; van Lierop, J.; Davis, R.L.; Herbert, D.E. Iron(ii) coordination complexes with panchromatic absorption and nanosecond charge-transfer excited state lifetimes. Nat. Chem. 2019, 11, 1144–1150. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaufhold, S.; Wärnmark, K. Design and Synthesis of Photoactive Iron N-Heterocyclic Carbene Complexes. Catalysts 2020, 10, 132. https://doi.org/10.3390/catal10010132
Kaufhold S, Wärnmark K. Design and Synthesis of Photoactive Iron N-Heterocyclic Carbene Complexes. Catalysts. 2020; 10(1):132. https://doi.org/10.3390/catal10010132
Chicago/Turabian StyleKaufhold, Simon, and Kenneth Wärnmark. 2020. "Design and Synthesis of Photoactive Iron N-Heterocyclic Carbene Complexes" Catalysts 10, no. 1: 132. https://doi.org/10.3390/catal10010132