
The Keratinocyte in the Picture Cutaneous Melanoma Microenvironment

{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
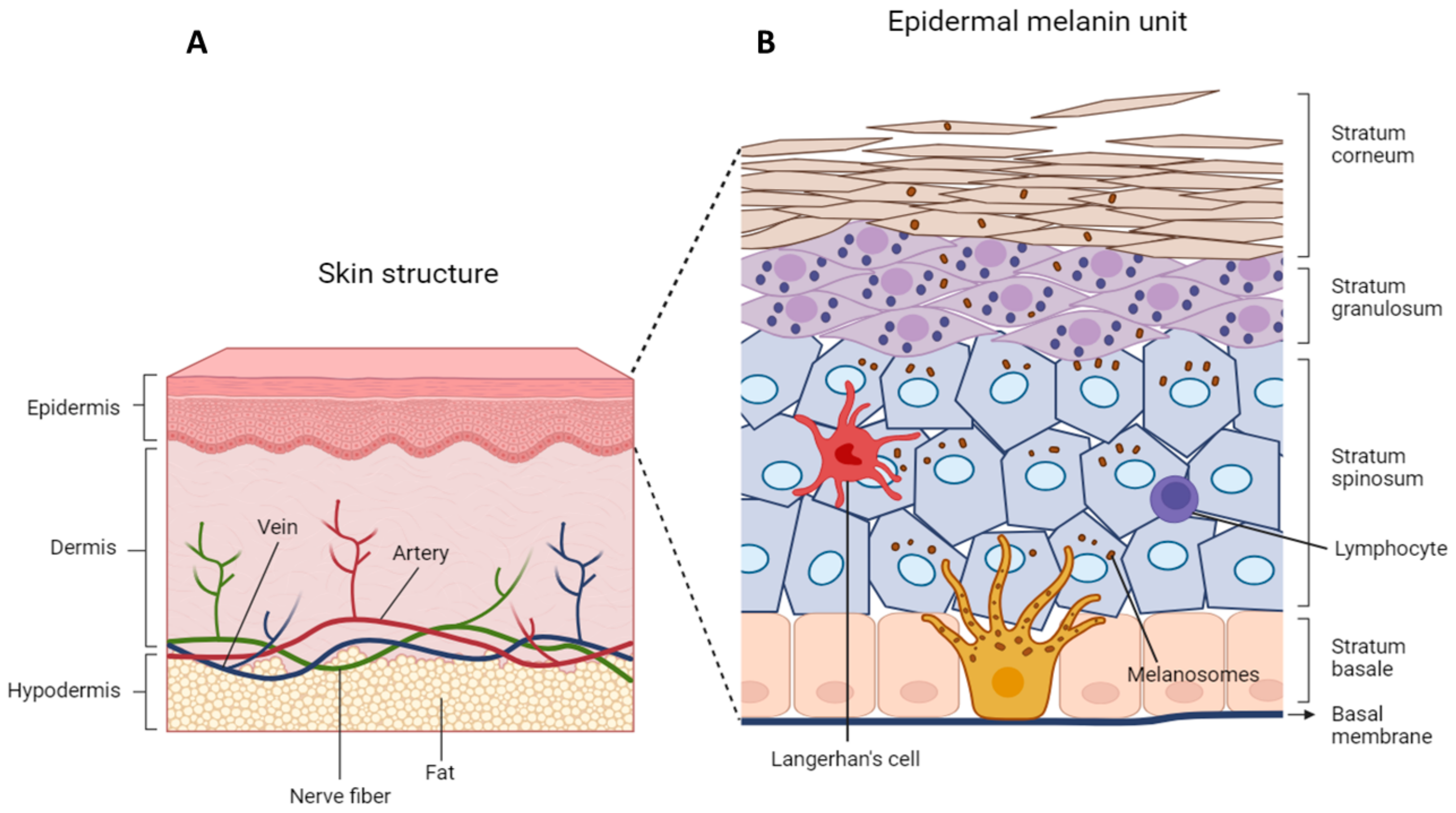
3. Skin Physiology
3.1. Skin Structure
3.2. Epidermal Organization
3.3. Epidermal–Melanin Units
3.4. Physiological Keratinocyte-Melanocyte Communication
3.4.1. Cell–Cell Contact in Melanocyte-Keratinocyte Interaction
3.4.2. Cross-Talk in the Epidermal–Melanin Unit
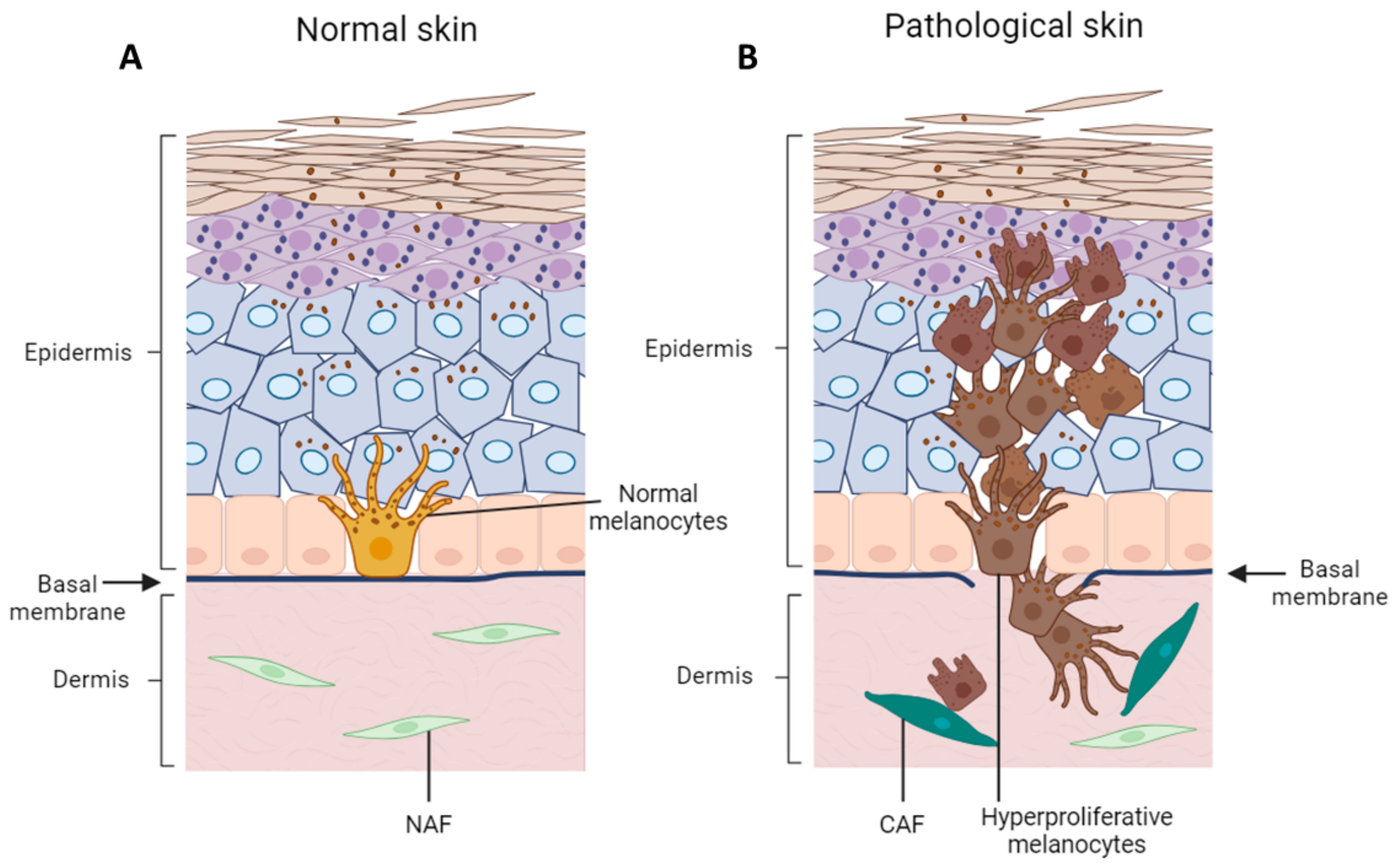
4. Remodeling of Epidermal Microenvironment during Melanoma Onset and Progression
4.1. Alteration of Melanocyte–Keratinocyte Physical Interaction during Melanoma Inception and Progression
4.2. Keratinocyte–Melanoma Cells Cross-Talk
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Long, G.V.; Swetter, S.M.; Menzies, A.M.; Gershenwald, J.E.; Scolyer, R.A. Cutaneous Melanoma. Lancet 2023, 402, 485. [Google Scholar] [CrossRef]
- Saginala, K.; Barsouk, A.; Aluru, J.S.; Rawla, P.; Barsouk, A. Epidemiology of Melanoma. Med. Sci. 2021, 9, 63. [Google Scholar] [CrossRef]
- Karimkhani, C.; Green, A.C.; Nijsten, T.; Weinstock, M.A.; Dellavalle, R.P.; Naghavi, M.; Fitzmaurice, C. The Global Burden of Melanoma: Results from the Global Burden of Disease Study 2015. Br. J. Dermatol. 2017, 177, 134. [Google Scholar] [CrossRef]
- Teixido, C.; Castillo, P.; Martinez-Vila, C.; Arance, A.; Alos, L. Molecular Markers and Targets in Melanoma. Cells 2021, 10, 2320. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Carr, S.; Smith, C.; Wernberg, J. Epidemiology and Risk Factors of Melanoma. Surg. Clin. N. Am. 2020, 100, 1–12. [Google Scholar] [CrossRef]
- Nilsen, L.T.N.; Hannevik, M.; Veierød, M.B. Ultraviolet Exposure from Indoor Tanning Devices: A Systematic Review. Br. J. Dermatol. 2016, 174, 730–740. [Google Scholar] [CrossRef] [PubMed]
- Boniol, M.; Autier, P.; Boyle, P.; Gandini, S. Cutaneous Melanoma Attributable to Sunbed use: Systematic Review and Meta-Analysis. BMJ 2012, 345, e4757. [Google Scholar] [CrossRef] [PubMed]
- Dzwierzynski, W.W. Melanoma Risk Factors and Prevention. Clin. Plast. Surg. 2021, 48, 543–550. [Google Scholar] [CrossRef]
- Bernerd, F.; Passeron, T.; Castiel, I.; Marionnet, C. The Damaging Effects of Long UVA (UVA1) Rays: A Major Challenge to Preserve Skin Health and Integrity. Int. J. Mol. Sci. 2022, 23, 8243. [Google Scholar] [CrossRef]
- Jin, S.; Padron, F.; Pfeifer, G.P. UVA Radiation, DNA Damage, and Melanoma. ACS Omega 2022, 7, 32936–32948. [Google Scholar] [CrossRef]
- Gorbunova, V.; Seluanov, A.; Mao, Z.; Hine, C. Changes in DNA Repair during Aging. Nucleic Acids Res. 2007, 35, 7466–7474. [Google Scholar] [CrossRef]
- Ren, P.; Dong, X.; Vijg, J. Age-Related Somatic Mutation Burden in Human Tissues. Front. Aging 2022, 3, 1018119. [Google Scholar] [CrossRef] [PubMed]
- Maslov, A.Y.; Vijg, J. Somatic Mutation Burden in Relation to Aging and Functional Life Span: Implications for Cellular Reprogramming and Rejuvenation. Curr. Opin. Genet. Dev. 2023, 83, 102132. [Google Scholar] [CrossRef] [PubMed]
- Wei, E.X.; Li, X.; Nan, H. Having a First-Degree Relative with Melanoma Increases Lifetime Risk of Melanoma, Squamous Cell Carcinoma, and Basal Cell Carcinoma. J. Am. Acad. Dermatol. 2019, 81, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Mohr, C.; Li, Y.; Navsaria, L.J.; Hinkston, C.L.; Margolis, D.J.; Wehner, M.R. Melanoma Risk in Skin of Color Patients with a History of a Keratinocyte Carcinoma. Br. J. Dermatol. 2023, 190, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Mercieca, L.; Aquilina, S.; Calleja, N.; Boffa, M.J. Cutaneous Melanoma More Likely to be Invasive in Fairer Skin Phototypes: A Retrospective Observational Study. Skinmed 2021, 19, 280–283. [Google Scholar] [PubMed]
- Strashilov, S.; Yordanov, A. Aetiology and Pathogenesis of Cutaneous Melanoma: Current Concepts and Advances. Int. J. Mol. Sci. 2021, 22, 6395. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.N.; Harland, M.; Bishop, D.T. The Genetics of Melanoma. Br. J. Hosp. Med. 2006, 67, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Hemminki, K.; Kharazmi, E.; Ji, J.; Sundquist, K.; Fallah, M. Multiple Primary (Even in Situ) Melanomas in a Patient Pose Significant Risk to Family Members. Eur. J. Cancer 2014, 50, 2659–2667. [Google Scholar] [CrossRef]
- Hussussian, C.J.; Struewing, J.P.; Goldstein, A.M.; Higgins, P.A.T.; Ally, D.S.; Sheahan, M.D.; Clark, W.H.; Tucker, M.A.; Dracopoli, N.C. Germline p16 Mutations in Familial Melanoma. Nat. Genet. 1994, 8, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT Promoter Mutations in Familial and Sporadic Melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Robles-Espinoza, C.D.; Harland, M.; Ramsay, A.J.; Aoude, L.G.; Quesada, V.; Ding, Z.; Pooley, K.A.; Pritchard, A.L.; Tiffen, J.C.; Petljak, M.; et al. POT1 Loss-of-Function Variants Predispose to Familial Melanoma. Nat. Genet. 2014, 46, 478–481. [Google Scholar] [CrossRef] [PubMed]
- Puntervoll, H.E.; Yang, X.R.; Vetti, H.H.; Bachmann, I.M.; Avril, M.F.; Benfodda, M.; Catricalà, C.; Dalle, S.; Duval-Modeste, A.B.; Ghiorzo, P.; et al. Melanoma Prone Families withCDK4germline Mutation: Phenotypic Profile and Associations with MC1Rvariants. J. Med. Genet. 2013, 50, 264. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, L.; Bonelli, L.; Ghiorzo, P.; Queirolo, P.; Battistuzzi, L.; Balleari, E.; Nasti, S.; Gargiulo, S.; Gliori, S.; Savoia, P.; et al. CDKN2A Mutations and MC1R Variants in Italian Patients with Single or Multiple Primary Melanoma. Pigment Cell Melanoma Res. 2008, 21, 700–709. [Google Scholar] [CrossRef]
- O’Neill, C.H.; Scoggins, C.R. Melanoma. J. Surg. Oncol. 2019, 120, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.M.; Xiao, Y.; Sampson, J.; Zhu, B.; Rotunno, M.; Bennett, H.; Wen, Y.; Jones, K.; Vogt, A.; Burdette, L.; et al. Rare Germline Variants in Known Melanoma Susceptibility Genes in Familial Melanoma. Hum. Mol. Genet. 2017, 26, 4886–4895. [Google Scholar] [CrossRef] [PubMed]
- Campos, C.; Fragoso, S.; Luís, R.; Pinto, F.; Brito, C.; Esteves, S.; Pataco, M.; Santos, S.; Machado, P.; Vicente, J.B.; et al. High-Throughput Sequencing Identifies 3 Novel Susceptibility Genes for Hereditary Melanoma. Genes 2020, 11, 403. [Google Scholar] [CrossRef]
- Castaneda-Garcia, C.; Iyer, V.; Nsengimana, J.; Trower, A.; Droop, A.; Brown, K.M.; Choi, J.; Zhang, T.; Harland, M.; Newton-Bishop, J.A.; et al. Defining Novel Causal SNPs and Linked Phenotypes at Melanoma-Associated Loci. Hum. Mol. Genet. 2022, 31, 2845–2856. [Google Scholar] [CrossRef]
- Bruno, W.; Dalmasso, B.; Barile, M.; Andreotti, V.; Elefanti, L.; Colombino, M.; Vanni, I.; Allavena, E.; Barbero, F.; Passoni, E.; et al. Predictors of Germline Status for Hereditary Melanoma: 5 Years of Multi-Gene Panel Testing within the Italian Melanoma Intergroup. ESMO Open 2022, 7, 100525. [Google Scholar] [CrossRef]
- Potjer, T.P.; Bollen, S.; Grimbergen, A.J.E.M.; van Doorn, R.; Gruis, N.A.; van Asperen, C.J.; Hes, F.J.; van der Stoep, N. Multigene Panel Sequencing of Established and Candidate Melanoma Susceptibility Genes in a Large Cohort of Dutch Non-CDKN2A/CDK4 Melanoma Families. Int. J. Cancer 2019, 144, 2453–2464. [Google Scholar] [CrossRef]
- Manganelli, M.; Guida, S.; Ferretta, A.; Pellacani, G.; Porcelli, L.; Azzariti, A.; Guida, G. Behind the Scene: Exploiting MC1R in Skin Cancer Risk and Prevention. Genes 2021, 12, 1093. [Google Scholar] [CrossRef] [PubMed]
- Tagliabue, E.; Gandini, S.; Bellocco, R.; Maisonneuve, P.; Newton-Bishop, J.; Polsky, D.; Lazovich, D.; Kanetsky, P.; Ghiorzo, P.; Gruis, N.; et al. MC1R Variants as Melanoma Risk Factors Independent of at-Risk Phenotypic Characteristics: A Pooled Analysis from the M-SKIP Project. CMAR 2018, 10, 1143. [Google Scholar] [CrossRef] [PubMed]
- Swope, V.B.; Abdel-Malek, Z.A. MC1R: Front and Center in the Bright Side of Dark Eumelanin and DNA Repair. Int. J. Mol. Sci. 2018, 19, 2667. [Google Scholar] [CrossRef] [PubMed]
- Rees, J.L. Genetics of Hair and Skin Color. Annu. Rev. Genet. 2003, 37, 67–90. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; McQuillan, M.A.; Tishkoff, S.A. Evolutionary Genetics of Skin Pigmentation in African Populations. Hum. Mol. Genet. 2021, 30, R88–R97. [Google Scholar] [CrossRef]
- Bellei, B.; Migliano, E.; Picardo, M. A Framework of Major Tumor-Promoting Signal Transduction Pathways Implicated in Melanoma-Fibroblast Dialogue. Cancers 2020, 12, 3400. [Google Scholar] [CrossRef]
- Cerdido, S.; Sánchez-Beltrán, J.; Lambertos, A.; Abrisqueta, M.; Padilla, L.; Herraiz, C.; Olivares, C.; Jiménez-Cervantes, C.; García-Borrón, J.C. A Side-by-Side Comparison of Wildtype and Variant Melanocortin 1 Receptor Signaling with Emphasis on Protection Against Oxidative Damage to DNA. Int. J. Mol. Sci. 2023, 24, 14381. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Malek, Z.A.; Ruwe, A.; Kavanagh-Starner, R.; Kadekaro, A.L.; Swope, V.; Haskell-Luevano, C.; Koikov, L.; Knittel, J.J. A-MSH Tripeptide Analogs Activate the Melanocortin 1 Receptor and Reduce UV-Induced DNA Damage in Human Melanocytes. Pigment Cell Melanoma Res. 2009, 22, 635–644. [Google Scholar] [CrossRef]
- Kadekaro, A.L.; Chen, J.; Yang, J.; Chen, S.; Jameson, J.; Swope, V.B.; Cheng, T.; Kadakia, M.; Abdel-Malek, Z. Alpha-Melanocyte–Stimulating Hormone Suppresses Oxidative Stress through a p53-Mediated Signaling Pathway in Human Melanocytes. Mol. Cancer Res. 2012, 10, 778–786. [Google Scholar] [CrossRef]
- Herraiz, C.; Journé, F.; Abdel-Malek, Z.; Ghanem, G.; Jiménez-Cervantes, C.; García-Borrón, J.C. Signaling from the Human Melanocortin 1 Receptor to ERK1 and ERK2 Mitogen-Activated Protein Kinases Involves Transactivation of cKIT. Mol. Endocrinol. 2011, 25, 138–156. [Google Scholar] [CrossRef]
- Herraiz, C.; Martínez-Vicente, I.; Maresca, V. The A-Melanocyte-Stimulating Hormone/Melanocortin-1 Receptor Interaction: A Driver of Pleiotropic Effects Beyond Pigmentation. Pigment Cell Melanoma Res. 2021, 34, 748–761. [Google Scholar] [CrossRef]
- Zhou, Y.; Cai, M. Novel Approaches to the Design of Bioavailable Melanotropins. Expert Opin. Drug Discov. 2017, 12, 1023–1030. [Google Scholar] [CrossRef]
- Caini, S.; Gandini, S.; Botta, F.; Tagliabue, E.; Raimondi, S.; Nagore, E.; Zanna, I.; Maisonneuve, P.; Newton-Bishop, J.; Polsky, D.; et al. MC1R Variants and Cutaneous Melanoma Risk According to Histological Type, Body Site, and Breslow Thickness: A Pooled Analysis from the M-SKIP Project. Melanoma Res. 2020, 30, 500–510. [Google Scholar] [CrossRef]
- Latreille, J.; Ezzedine, K.; Elfakir, A.; Ambroisine, L.; Jdid, R.; Galan, P.; Hercberg, S.; Gruber, F.; Malvy, D.; Tschachler, E.; et al. MC1R Polymorphisms and Facial Photoaging. Ann. Dermatol. Venereol. 2011, 138, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; André, M.; Adhikari, K.; Blin, M.; Bonfante, B.; Mendoza-Revilla, J.; Fuentes-Guajardo, M.; Palmal, S.; Chacón-Duque, J.C.; Hurtado, M.; et al. A Genome-Wide Association Study Identifies Novel Gene Associations with Facial Skin Wrinkling and Mole Count in Latin Americans. Br. J. Dermatol. 2021, 185, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Guida, S.; Ciardo, S.; De Pace, B.; De Carvalho, N.; Peccerillo, F.; Manfredini, M.; Farnetani, F.; Chester, J.; Kaleci, S.; Manganelli, M.; et al. The Influence of MC1R On dermal Morphological Features of Photo-Exposed Skin in Women Revealed by Reflectance Confocal Microscopy and Optical Coherence Tomography. Exp. Dermatol. 2019, 28, 1321–1327. [Google Scholar] [CrossRef] [PubMed]
- Law, M.H.; Medland, S.E.; Zhu, G.; Yazar, S.; Viñuela, A.; Wallace, L.; Shekar, S.N.; Duffy, D.L.; Bataille, V.; Glass, D.; et al. Genome-Wide Association shows that Pigmentation Genes Play a Role in Skin Aging. J. Investig. Dermatol. 2017, 137, 1887–1894. [Google Scholar] [CrossRef] [PubMed]
- Carroll, T.D.; Wong, T.; Morris, M.K.; Di Germanio, C.; Ma, Z.; Stone, M.; Ball, E.; Fritts, L.; Rustagi, A.; Simmons, G.; et al. Administration of Vaccine-Boosted COVID-19 Convalescent Plasma to SARS-CoV-2 Infected Hamsters Decreases Virus Replication in Lungs and Hastens Resolution of the Infection Despite Transiently Enhancing Disease and Lung Pathology. bioRxiv 2023. [Google Scholar] [CrossRef]
- Cirri, P.; Chiarugi, P. Cancer Associated Fibroblasts: The Dark Side of the Coin. Am. J. Cancer Res. 2011, 1, 482–497. [Google Scholar] [PubMed]
- Bellei, B.; Picardo, M. Premature Cell Senescence in Human Skin: Dual Face in Chronic Acquired Pigmentary Disorders. Ageing Res. Rev. 2020, 57, 100981. [Google Scholar] [CrossRef] [PubMed]
- Swope, V.B.; Starner, R.J.; Rauck, C.; Abdel-Malek, Z.A. Endothelin-1 and A-Melanocortin have Redundant Effects on Global Genome Repair in UV-Irradiated Human Melanocytes Despite Distinct Signaling Pathways. Pigment Cell Melanoma Res. 2020, 33, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Yin, K.; Sturm, R.A.; Smith, A.G. MC1R and NR4A Receptors in Cellular Stress and DNA Repair: Implications for UVR Protection. Exp. Dermatol. 2014, 23, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Castejón-Griñán, M.; Herraiz, C.; Olivares, C.; Jiménez-Cervantes, C.; García-Borrón, J.C. cAMP-Independent Non-Pigmentary Actions of Variant Melanocortin 1 Receptor: AKT-Mediated Activation of Protective Responses to Oxidative DNA Damage. Oncogene 2018, 37, 3631–3646. [Google Scholar] [CrossRef] [PubMed]
- Bastiaens, M.T.; Huurne, J.A.C.t.; Kielich, C.; Gruis, N.A.; Westendorp, R.G.J.; Vermeer, B.J.; Bavinck, J.N.B. Melanocortin-1 Receptor Gene Variants Determine the Risk of Nonmelanoma Skin Cancer Independently of Fair Skin and Red Hair. Am. J. Hum. Genet. 2001, 68, 884–894. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Miao, Y.; Cao, L.; Guo, L.; Cui, Y.; Yan, C.; Zeng, Z.; Xu, M.; Han, T. Activation of Melanocortin-1 Receptor Signaling in Melanoma Cells Impairs T Cell Infiltration to Dampen Antitumor Immunity. Nat. Commun. 2023, 14, 5740. [Google Scholar] [CrossRef] [PubMed]
- Guida, M.; Strippoli, S.; Ferretta, A.; Bartolomeo, N.; Porcelli, L.; Maida, I.; Azzariti, A.; Tommasi, S.; Grieco, C.; Guida, S.; et al. Detrimental Effects of Melanocortin-1 Receptor (MC1R) Variants on the Clinical Outcomes of BRAF V600 Metastatic Melanoma Patients Treated with BRAF Inhibitors. Pigment Cell Melanoma Res. 2016, 29, 679–687. [Google Scholar] [CrossRef]
- Su, D.; Djureinovic, D.; Schoenfeld, D.; Marquez-Nostra, B.; Olino, K.; Jilaveanu, L.; Kluger, H. Melanocortin 1 Receptor (MC1R) Expression as a Marker of Progression in Melanoma. Res. Sq. 2023. [Google Scholar] [CrossRef]
- Sarasin, A.; Kauffmann, A. Overexpression of DNA Repair Genes is Associated with Metastasis: A New Hypothesis. Mutat. Res. 2008, 659, 49–55. [Google Scholar] [CrossRef]
- Zhang, G.; Herlyn, M. Human Nevi: No Longer Precursors of Melanomas? J. Investig. Dermatol. 2012, 132, 2133–2134. [Google Scholar] [CrossRef]
- Tsao, H.; Bevona, C.; Goggins, W.; Quinn, T. The Transformation Rate of Moles (Melanocytic Nevi) into Cutaneous Melanoma: A Population-Based Estimate. Arch. Dermatol. (1960) 2003, 139, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Shreberk-Hassidim, R.; Ostrowski, S.M.; Fisher, D.E. The Complex Interplay between Nevi and Melanoma: Risk Factors and Precursors. Int. J. Mol. Sci. 2023, 24, 3541. [Google Scholar] [CrossRef] [PubMed]
- Pollock, P.M.; Harper, U.L.; Hansen, K.S.; Yudt, L.M.; Stark, M.; Robbins, C.M.; Moses, T.Y.; Hostetter, G.; Wagner, U.; Kakareka, J.; et al. High Frequency of BRAF Mutations in Nevi. Nat. Genet. 2003, 33, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Requena, C.; Manrique, E.; Nagore, E. Update on Lentigo Maligna: Diagnostic Signs and Treatment. Actas Dermosifiliogr. 2023, 114, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Brînzea, A.; Nedelcu, R.I.; Ion, D.A.; Turcu, G.; Antohe, M.; Hodorogea, A.; Călinescu, A.; Pirici, D.; Popescu, R.; Popescu, C.M.; et al. Matrix Metalloproteinases Expression in Lentigo Maligna∕lentigo Maligna Melanoma—A Review of the Literature and Personal Experience. Rom. J. Morphol. Embryol. 2019, 60, 1091–1095. [Google Scholar] [PubMed]
- DeWane, M.E.; Kelsey, A.; Oliviero, M.; Rabinovitz, H.; Grant-Kels, J.M. Melanoma on Chronically Sun-Damaged Skin: Lentigo Maligna and Desmoplastic Melanoma. J. Am. Acad. Dermatol. 2019, 81, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Kharouf, N.; Flanagan, T.W.; Hassan, S.; Shalaby, H.; Khabaz, M.; Hassan, S.; Megahed, M.; Haikel, Y.; Santourlidis, S.; Hassan, M. Tumor Microenvironment as a Therapeutic Target in Melanoma Treatment. Cancers 2023, 15, 3147. [Google Scholar] [CrossRef]
- Zhang, G.; Ji, P.; Xia, P.; Song, H.; Guo, Z.; Hu, X.; Guo, Y.; Yuan, X.; Song, Y.; Shen, R.; et al. Identification and Targeting of Cancer-Associated Fibroblast Signature Genes for Prognosis and Therapy in Cutaneous Melanoma. Comput. Biol. Med. 2023, 167, 107597. [Google Scholar] [CrossRef]
- Anderson-Crannage, M.; Ascensión, A.M.; Ibanez-Solé, O.; Zhu, H.; Schaefer, E.; Ottomanelli, D.; Hochberg, B.; Pan, J.; Luo, W.; Tian, M.; et al. Inflammation-Mediated Fibroblast Activation and Immune Dysregulation in Collagen VII-Deficient Skin. Front. Immunol. 2023, 14, 1211505. [Google Scholar] [CrossRef]
- Wu, B.; Sodji, Q.H.; Oyelere, A.K. Inflammation, Fibrosis and Cancer: Mechanisms, Therapeutic Options and Challenges. Cancers 2022, 14, 552. [Google Scholar] [CrossRef]
- Bottazzi, B.; Riboli, E.; Mantovani, A. Aging, Inflammation and Cancer. Semin. Immunol. 2018, 40, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Chin, T.; Lee, X.E.; Ng, P.Y.; Lee, Y.; Dreesen, O. The Role of Cellular Senescence in Skin Aging and Age-Related Skin Pathologies. Front. Physiol. 2023, 14, 1297637. [Google Scholar] [CrossRef] [PubMed]
- D’Arino, A.; Caputo, S.; Eibenschutz, L.; Piemonte, P.; Buccini, P.; Frascione, P.; Bellei, B. Skin Cancer Microenvironment: What we can Learn from Skin Aging? Int. J. Mol. Sci. 2023, 24, 14043. [Google Scholar] [CrossRef]
- Chatsirisupachai, K.; Lagger, C.; de Magalhães, J.P. Age-Associated Differences in the Cancer Molecular Landscape. Trends Cancer 2022, 8, 962–971. [Google Scholar] [CrossRef]
- Nicolas, E.; Golemis, E.A.; Arora, S. POLD1: Central Mediator of DNA Replication and Repair, and Implication in Cancer and Other Pathologies. Gene 2016, 590, 128–141. [Google Scholar] [CrossRef]
- Council, M.L.; Sheinbein, D.M. Common Skin Cancers in Older Adults Approach to Diagnosis and Management. Clin. Geriatr. Med. 2024, 40, 25–36. [Google Scholar] [CrossRef]
- Papaccio, F.; Kovacs, D.; Bellei, B.; Caputo, S.; Migliano, E.; Cota, C.; Picardo, M. Profiling Cancer-Associated Fibroblasts in Melanoma. Int. J. Mol. Sci. 2021, 22, 7255. [Google Scholar] [CrossRef]
- Liu, J.; Zheng, R.; Zhang, Y.; Jia, S.; He, Y.; Liu, J. The Cross Talk between Cellular Senescence and Melanoma: From Molecular Pathogenesis to Target Therapies. Cancers 2023, 15, 2640. [Google Scholar] [CrossRef]
- Huang, J.; Heng, S.; Zhang, W.; Liu, Y.; Xia, T.; Ji, C.; Zhang, L. Dermal Extracellular Matrix Molecules in Skin Development, Homeostasis, Wound Regeneration and Diseases. Semin. Cell Dev. Biol. 2022, 128, 137–144. [Google Scholar] [CrossRef]
- Tracy, L.E.; Minasian, R.A.; Caterson, E.J. Extracellular Matrix and Dermal Fibroblast Function in the Healing Wound. Adv. Wound Care 2016, 5, 119–136. [Google Scholar] [CrossRef]
- Driskell, R.R.; Lichtenberger, B.M.; Hoste, E.; Kretzschmar, K.; Simons, B.D.; Charalambous, M.; Ferron, S.R.; Herault, Y.; Pavlovic, G.; Ferguson-Smith, A.C.; et al. Distinct Fibroblast Lineages Determine Dermal Architecture in Skin Development and Repair. Nature 2013, 504, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Tsepkolenko, A.; Tsepkolenko, V.; Dash, S.; Mishra, A.; Bader, A.; Melerzanov, A.; Giri, S. The Regenerative Potential of Skin and the Immune System. Clin. Cosmet. Investig. Dermatol. 2019, 12, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Nyström, A.; Bruckner-Tuderman, L. Matrix Molecules and Skin Biology. Semin. Cell Dev. Biol. 2019, 89, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Juarez, C.F.; Plikus, M.V. Emerging Nonmetabolic Functions of Skin Fat. Nat. Rev. Endocrinol. 2018, 14, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Nicu, C.; O’Sullivan, J.D.B.; Ramos, R.; Timperi, L.; Lai, T.; Farjo, N.; Farjo, B.; Pople, J.; Bhogal, R.; Hardman, J.A.; et al. Dermal Adipose Tissue Secretes HGF to Promote Human Hair Growth and Pigmentation. J. Investig. Dermatol. 2021, 141, 1633–1645.e13. [Google Scholar] [CrossRef] [PubMed]
- Chi, M.; Chen, J.; Ye, Y.; Tseng, H.; Lai, F.; Tay, K.H.; Jin, L.; Guo, S.T.; Jiang, C.C.; Zhang, X.D. Adipocytes Contribute to Resistance of Human Melanoma Cells to Chemotherapy and Targeted Therapy. Curr. Med. Chem. 2014, 21, 1255–1267. [Google Scholar] [CrossRef]
- Kwan, H.Y.; Fu, X.; Liu, B.; Chao, X.; Chan, C.L.; Cao, H.; Su, T.; Tse, A.K.W.; Fong, W.F.; Yu, Z. Subcutaneous Adipocytes Promote Melanoma Cell Growth by Activating the Akt Signaling Pathway: Role of Palmitic Acid. J. Biol. Chem. 2014, 289, 30525–30537. [Google Scholar] [CrossRef]
- Zhang, M.; Di Martino, J.S.; Bowman, R.L.; Campbell, N.R.; Baksh, S.C.; Simon-Vermot, T.; Kim, I.S.; Haldeman, P.; Mondal, C.; Yong-Gonzales, V.; et al. Adipocyte-Derived Lipids Mediate Melanoma Progression Via FATP Proteins. Cancer Discov. 2018, 8, 1006–1025. [Google Scholar] [CrossRef]
- Okumura, T.; Ohuchida, K.; Kibe, S.; Iwamoto, C.; Ando, Y.; Takesue, S.; Nakayama, H.; Abe, T.; Endo, S.; Koikawa, K.; et al. Adipose Tissue-Derived Stromal Cells are Sources of Cancer-Associated Fibroblasts and Enhance Tumor Progression by Dense Collagen Matrix. Int. J. Cancer 2019, 144, 1401–1413. [Google Scholar] [CrossRef]
- Jung, Y.Y.; Lee, Y.K.; Koo, J.S. Expression of Cancer-Associated Fibroblast-Related Proteins in Adipose Stroma of Breast Cancer. Tumour Biol. 2015, 36, 8685–8695. [Google Scholar] [CrossRef]
- Bochet, L.; Lehuédé, C.; Dauvillier, S.; Wang, Y.Y.; Dirat, B.; Laurent, V.; Dray, C.; Guiet, R.; Maridonneau-Parini, I.; Le Gonidec, S.; et al. Adipocyte-Derived Fibroblasts Promote Tumor Progression and Contribute to the Desmoplastic Reaction in Breast Cancer. Cancer Res. 2013, 73, 5657–5668. [Google Scholar] [CrossRef]
- Menon, G.K. Skin Basics; Structure and Function. In Lipids and Skin Health; Springer International Publishing: Cham, Switzerland, 2014; pp. 9–23. [Google Scholar]
- Yousef, H.; Alhajj, M.; Sharma, S. Anatomy, Skin (Integument), Epidermis. In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2023. [Google Scholar]
- Gdula, M.R.; Poterlowicz, K.; Mardaryev, A.N.; Sharov, A.A.; Peng, Y.; Fessing, M.Y.; Botchkarev, V.A. Remodeling of Three-Dimensional Organization of the Nucleus during Terminal Keratinocyte Differentiation in the Epidermis. J. Investig. Dermatol. 2013, 133, 2191–2201. [Google Scholar] [CrossRef]
- Monteleon, C.L.; Agnihotri, T.; Dahal, A.; Liu, M.; Rebecca, V.W.; Beatty, G.L.; Amaravadi, R.K.; Ridky, T.W. Lysosomes Support the Degradation, Signaling, and Mitochondrial Metabolism Necessary for Human Epidermal Differentiation. J. Investig. Dermatol. 2018, 138, 1945–1954. [Google Scholar] [CrossRef]
- Murata, T.; Honda, T.; Mostafa, A.; Kabashima, K. Stratum Corneum as Polymer Sheet: Concept and Cornification Processes. Trends Mol. Med. 2022, 28, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Kolarsick, P.A.J.; Kolarsick, M.A.; Goodwin, C. Anatomy and Physiology of the Skin. J. Dermatol. Nurses’ Assoc. 2011, 3, 203–213. [Google Scholar] [CrossRef]
- Venus, M.; Waterman, J.; McNab, I. Basic Physiology of the Skin. Surgery 2010, 28, 469–472. [Google Scholar]
- Proksch, E.; Brandner, J.M.; Jensen, J. The Skin: An Indispensable Barrier. Exp. Dermatol. 2008, 17, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Williams, I.R.; Kupper, T.S. Immunity at the Surface: Homeostatic Mechanisms of the Skin Immune System. Life Sci. 1996, 58, 1485–1507. [Google Scholar] [CrossRef] [PubMed]
- Chambers, E.S.; Vukmanovic-Stejic, M. Skin Barrier Immunity and Ageing. Immunology 2020, 160, 116–125. [Google Scholar] [CrossRef]
- Morizane, S.; Mukai, T.; Sunagawa, K.; Tachibana, K.; Kawakami, Y.; Ouchida, M. “Input/Output Cytokines” in Epidermal Keratinocytes and the Involvement in Inflammatory Skin Diseases. Front. Immunol. 2023, 14, 1239598. [Google Scholar] [CrossRef]
- Park, H.Y.; Kosmadaki, M.; Yaar, M.; Gilchrest, B.A. Cellular Mechanisms Regulating Human Melanogenesis. Cell. Mol. Life Sci. 2009, 66, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Casalou, C.; Moreiras, H.; Mayatra, J.M.; Fabre, A.; Tobin, D.J. Loss of ‘Epidermal Melanin Unit’ Integrity in Human Skin during Melanoma-Genesis. Front. Oncol. 2022, 12, 878336. [Google Scholar] [CrossRef]
- Naik, P.P.; Farrukh, S.N. Influence of Ethnicities and Skin Color Variations in Different Populations: A Review. Skin. Pharmacol. Physiol. 2022, 35, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Olson, R.L.; Gaylor, J.; Everett, M.A. Skin Color, Melanin, and Erythema. Arch. Dermatol. (1960) 1973, 108, 541–544. [Google Scholar] [CrossRef]
- Montagna, W.; Carlisle, K. The Architecture of Black and White Facial Skin. J. Am. Acad. Dermatol. 1991, 24, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Iozumi, K.; Hoganson, G.E.; Pennella, R.; Everett, M.A.; Fuller, B.B. Role of Tyrosinase as the Determinant of Pigmentation in Cultured Human Melanocytes. J. Investig. Dermatol. 1993, 100, 806–811. [Google Scholar] [CrossRef]
- Markiewicz, E.; Karaman-Jurukovska, N.; Mammone, T.; Idowu, O.C. Post-Inflammatory Hyperpigmentation in Dark Skin: Molecular Mechanism and Skincare Implications. Clin. Cosmet. Investig. Dermatol. 2022, 15, 2555–2565. [Google Scholar] [CrossRef]
- Van Den Bossche, K.; Naeyaert, J.; Lambert, J. The Quest for the Mechanism of Melanin Transfer. Traffic 2006, 7, 769–778. [Google Scholar] [CrossRef]
- Bento-Lopes, L.; Cabaço, L.C.; Charneca, J.; Neto, M.V.; Seabra, M.C.; Barral, D.C. Melanin’s Journey from Melanocytes to Keratinocytes: Uncovering the Molecular Mechanisms of Melanin Transfer and Processing. Int. J. Mol. Sci. 2023, 24, 11289. [Google Scholar] [CrossRef]
- Costin, G.; Hearing, V.J. Human Skin Pigmentation: Melanocytes Modulate Skin Color in Response to Stress. FASEB J. 2007, 21, 976–994. [Google Scholar] [CrossRef]
- Cichorek, M.; Wachulska, M.; Stasiewicz, A.; Tymińska, A. Skin Melanocytes: Biology and Development. Postep. Dermatol. Alergol. 2013, 30, 30–41. [Google Scholar] [CrossRef]
- Nordlund, J.J. The Lives of Pigment Cells. Dermatol. Clin. 1986, 4, 407–418. [Google Scholar] [CrossRef]
- Gilchrest, B.A.; Blog, F.B.; Szabo, G. Effects of Aging and Chronic Sun Exposure on Melanocytes in Human Skin. J. Investig. Dermatol. 1979, 73, 141–143. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.D.; Peles, D.; Wakamatsu, K.; Ito, S. Current Challenges in Understanding Melanogenesis: Bridging Chemistry, Biological Control, Morphology, and Function. Pigment Cell Melanoma Res. 2009, 22, 563–579. [Google Scholar] [CrossRef]
- Land, E.J.; Riley, P.A. Spontaneous Redox Reactions of Dopaquinone and the Balance between the Eumelanic and Phaeomelanic Pathways. Pigment Cell Res. 2000, 13, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Hertzman Johansson, C.; Azimi, A.; Frostvik Stolt, M.; Shojaee, S.; Wiberg, H.; Grafström, E.; Hansson, J.; Egyházi Brage, S. Association of MITF and Other Melanosome-Related Proteins with Chemoresistance in Melanoma Tumors and Cell Lines. Melanoma Res. 2013, 23, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Juntti-Berggren, L.; Lindh, U.; Berggren, P.O. Starvation is Associated with Changes in the Elemental Composition of the Pancreatic Beta-Cell. Biosci. Rep. 1991, 11, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, A.; Fisher, D.E. The Master Role of Microphthalmia-Associated Transcription Factor in Melanocyte and Melanoma Biology. Lab. Investig. 2017, 97, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Slominski, R.M.; Sarna, T.; Płonka, P.M.; Raman, C.; Brożyna, A.A.; Slominski, A.T. Melanoma, Melanin, and Melanogenesis: The Yin and Yang Relationship. Front. Oncol. 2022, 12, 842496. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.L.; Czyz, M. MITF in Melanoma: Mechanisms Behind its Expression and Activity. Cell. Mol. Life Sci. 2015, 72, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, D.; Migliano, E.; Muscardin, L.; Silipo, V.; Catricalà, C.; Picardo, M.; Bellei, B. The Role of Wnt/Β-Catenin Signaling Pathway in Melanoma Epithelial-to-Mesenchymal-Like Switching: Evidences from Patients-Derived Cell Lines. Oncotarget 2016, 7, 43295–43314. [Google Scholar] [CrossRef]
- Hossain, S.M.; Eccles, M.R. Phenotype Switching and the Melanoma Microenvironment; Impact on Immunotherapy and Drug Resistance. Int. J. Mol. Sci. 2023, 24, 1601. [Google Scholar] [CrossRef]
- Jimbow, K.; Quevedo, W.C.; Fitzpatrick, T.B.; Szabo, G. Some Aspects of Melanin Biology: 1950–1975. J. Investig. Dermatol. 1976, 67, 72–89. [Google Scholar] [CrossRef]
- Hardman, M.J.; Liu, K.; Avilion, A.A.; Merritt, A.; Brennan, K.; Garrod, D.R.; Byrne, C. Desmosomal Cadherin Misexpression Alters Beta-Catenin Stability and Epidermal Differentiation. Mol. Cell. Biol. 2005, 25, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Charest, J.L.; Jennings, J.M.; King, W.P.; Kowalczyk, A.P.; García, A.J. Cadherin-Mediated Cell-Cell Contact Regulates Keratinocyte Differentiation. J. Investig. Dermatol. 2009, 129, 564–572. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, C.; Kiel, C. Cell Adhesion Molecules in Normal Skin and Melanoma. Biomolecules 2021, 11, 1213. [Google Scholar] [CrossRef]
- Tang, A.; Eller, M.S.; Hara, M.; Yaar, M.; Hirohashi, S.; Gilchrest, B.A. E-Cadherin is the Major Mediator of Human Melanocyte Adhesion to Keratinocytes In Vitro. J. Cell Sci. 1994, 107 Pt 4, 983–992. [Google Scholar] [CrossRef]
- Hung, C.; Chiang, H.; Lo, H.; Jian, J.; Wu, W. E-Cadherin and its Downstream Catenins are Proteolytically Cleaved in Human HaCaT Keratinocytes Exposed to UVB. Exp. Dermatol. 2006, 15, 315–321. [Google Scholar] [CrossRef]
- Gambichler, T.; Rotterdam, S.; Tigges, C.; Altmeyer, P.; Bechara, F.G. Impact of Ultraviolet Radiation on the Expression of Marker Proteins of Gap and Adhesion Junctions in Human Epidermis. Photodermatol. Photoimmunol. Photomed. 2008, 24, 318–321. [Google Scholar] [CrossRef]
- Jamal, S.; Schneider, R.J. UV-Induction of Keratinocyte Endothelin-1 Downregulates E-Cadherin in Melanocytes and Melanoma Cells. J. Clin. Investig. 2002, 110, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Jiang, S.; Miao, F.; Lei, T. sPmel17 Secreted by Ultraviolet B-Exposed Melanocytes Alters the Intercellular Adhesion of Keratinocytes. Oxid. Med. Cell. Longev. 2022, 2022, 1856830. [Google Scholar] [CrossRef]
- Shain, A.H.; Bastian, B.C. From Melanocytes to Melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef]
- Huber, O.; Bierkamp, C.; Kemler, R. Cadherins and Catenins in Development. Curr. Opin. Cell Biol. 1996, 8, 685–691. [Google Scholar] [CrossRef]
- Ramani, V.; Teshima, T.; Tamura, K.; Chung, J.; Kobayashi, M.; Cruz, P.D.; Ariizumi, K. Melanoma-Derived Soluble DC-HIL/GPNMB Promotes Metastasis by Excluding T-Lymphocytes from the Pre-Metastatic Niches. J. Investig. Dermatol. 2018, 138, 2443–2451. [Google Scholar] [CrossRef]
- Tomihari, M.; Hwang, S.; Chung, J.; Cruz, P.D.; Ariizumi, K. Gpnmb is a Melanosome-Associated Glycoprotein that Contributes to Melanocyte/Keratinocyte Adhesion in a RGD-Dependent Fashion. Exp. Dermatol. 2009, 18, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Biswas, K.B.; Takahashi, A.; Mizutani, Y.; Takayama, S.; Ishitsuka, A.; Yang, L.; Yang, F.; Iddamalgoda, A.; Katayama, I.; Inoue, S. GPNMB is Expressed in Human Epidermal Keratinocytes but Disappears in the Vitiligo Lesional Skin. Sci. Rep. 2020, 10, 4930. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Kuroda, Y.; Yang, L.; Lai, S.; Mizutani, Y.; Iddamalgoda, A.; Guo, J.; Yamamoto, A.; Murase, D.; Takahashi, Y.; et al. GPNMB Extracellular Fragment Protects Melanocytes from Oxidative Stress by Inhibiting AKT Phosphorylation Independent of CD44. Int. J. Mol. Sci. 2021, 22, 10843. [Google Scholar] [CrossRef]
- Arnette, C.R.; Roth-Carter, Q.R.; Koetsier, J.L.; Broussard, J.A.; Burks, H.E.; Cheng, K.; Amadi, C.; Gerami, P.; Johnson, J.L.; Green, K.J. Keratinocyte Cadherin Desmoglein 1 Controls Melanocyte Behavior through Paracrine Signaling. Pigment Cell Melanoma Res. 2020, 33, 305–317. [Google Scholar] [CrossRef]
- Hsu, M.; Andl, T.; Li, G.; Meinkoth, J.L.; Herlyn, M. Cadherin Repertoire Determines Partner-Specific Gap Junctional Communication during Melanoma Progression. J. Cell Sci. 2000, 113 Pt 9, 1535–1542. [Google Scholar] [CrossRef]
- Haass, N.K.; Wladykowski, E.; Kief, S.; Moll, I.; Brandner, J.M. Differential Induction of Connexins 26 and 30 in Skin Tumors and their Adjacent Epidermis. J. Histochem. Cytochem. 2006, 54, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.Z.; Jiang, J.X. Gap Junction and Hemichannel-Independent Actions of Connexins on Cell and Tissue Functions—An Update. FEBS Lett. 2014, 588, 1186–1192. [Google Scholar] [CrossRef]
- Bellei, B.; Mastrofrancesco, A.; Briganti, S.; Aspite, N.; Ale-Agha, N.; Sies, H.; Picardo, M. Ultraviolet A Induced Modulation of Gap Junctional Intercellular Communication by P38 MAPK Activation in Human Keratinocytes. Exp. Dermatol. 2008, 17, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Dedrick, R.L.; Walicke, P.; Garovoy, M. Anti-Adhesion Antibodies Efalizumab, a Humanized Anti-CD11a Monoclonal Antibody. Transpl. Immunol. 2002, 9, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, P.R.; Ho, T.; Abdel-Malek, Z.A. Participation of Keratinocyte- and Fibroblast-Derived Factors in Melanocyte Homeostasis, the Response to UV, and Pigmentary Disorders. Pigment Cell Melanoma Res. 2021, 34, 762–776. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Fukunaga-Kalabis, M.; Herlyn, M. Crosstalk in Skin: Melanocytes, Keratinocytes, Stem Cells, and Melanoma. J. Cell Commun. Signal. 2016, 10, 191–196. [Google Scholar] [CrossRef]
- Hirobe, T. Keratinocytes Regulate the Function of Melanocytes. Dermatol. Sin. 2014, 32, 200–204. [Google Scholar] [CrossRef]
- Haass, N.K.; Smalley, K.S.M.; Li, L.; Herlyn, M. Adhesion, Migration and Communication in Melanocytes and Melanoma. Pigment Cell Res. 2005, 18, 150–159. [Google Scholar] [CrossRef]
- Imokawa, G. Autocrine and Paracrine Regulation of Melanocytes in Human Skin and in Pigmentary Disorders. Pigment Cell Res. 2004, 17, 96–110. [Google Scholar] [CrossRef]
- Schiller, M.; Brzoska, T.; Böhm, M.; Metze, D.; Scholzen, T.E.; Rougier, A.; Luger, T.A. Solar-Simulated Ultraviolet Radiation-Induced Upregulation of the Melanocortin-1 Receptor, Proopiomelanocortin, and Alpha-Melanocyte-Stimulating Hormone in Human Epidermis In Vivo. J. Investig. Dermatol. 2004, 122, 468–476. [Google Scholar]
- Swope, V.B.; Medrano, E.E.; Smalara, D.; Abdel-Malek, Z.A. Long-Term Proliferation of Human Melanocytes is Supported by the Physiologic Mitogens Alpha-Melanotropin, Endothelin-1, and Basic Fibroblast Growth Factor. Exp. Cell Res. 1995, 217, 453–459. [Google Scholar] [CrossRef]
- Murase, D.; Hachiya, A.; Amano, Y.; Ohuchi, A.; Kitahara, T.; Takema, Y. The Essential Role of p53 in Hyperpigmentation of the Skin Via Regulation of Paracrine Melanogenic Cytokine Receptor Signaling. J. Biol. Chem. 2009, 284, 4343–4353. [Google Scholar] [CrossRef]
- Lübbe, J.; Reichel, M.; Burg, G.; Kleihues, P. Absence of p53 Gene Mutations in Cutaneous Melanoma. J. Investig. Dermatol. 1994, 102, 819–821. [Google Scholar] [CrossRef]
- Choi, S.; Bin, B.; Kim, W.; Lee, E.; Lee, T.R.; Cho, E. Exposure of Human Melanocytes to UVB Twice and Subsequent Incubation Leads to Cellular Senescence and Senescence-Associated Pigmentation through the Prolonged p53 Expression. J. Dermatol. Sci. 2018, 90, 303–312. [Google Scholar] [CrossRef]
- Wu, C.; Lan, C.E.; Chiou, M.; Yu, H. Basic Fibroblast Growth Factor Promotes Melanocyte Migration Via Increased Expression of p125(FAK) on Melanocytes. Acta Derm. Venereol. 2006, 86, 498–502. [Google Scholar] [CrossRef]
- Shi, H.; Lin, B.; Huang, Y.; Wu, J.; Zhang, H.; Lin, C.; Wang, Z.; Zhu, J.; Zhao, Y.; Fu, X.; et al. Basic Fibroblast Growth Factor Promotes Melanocyte Migration Via Activating PI3K/Akt-Rac1-FAK-JNK and ERK Signaling Pathways. IUBMB Life 2016, 68, 735–747. [Google Scholar] [CrossRef]
- Halaban, R.; Rubin, J.S.; Funasaka, Y.; Cobb, M.; Boulton, T.; Faletto, D.; Rosen, E.; Chan, A.; Yoko, K.; White, W. Met and Hepatocyte Growth Factor/Scatter Factor Signal Transduction in Normal Melanocytes and Melanoma Cells. Oncogene 1992, 7, 2195–2206. [Google Scholar]
- Weidner, K.M.; Di Cesare, S.; Sachs, M.; Brinkmann, V.; Behrens, J.; Birchmeier, W. Interaction between Gab1 and the C-Met Receptor Tyrosine Kinase is Responsible for Epithelial Morphogenesis. Nature 1996, 384, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Ponzetto, C.; Bardelli, A.; Zhen, Z.; Maina, F.; dalla Zonca, P.; Giordano, S.; Graziani, A.; Panayotou, G.; Comoglio, P.M. A Multifunctional Docking Site Mediates Signaling and Transformation by the Hepatocyte Growth Factor/Scatter Factor Receptor Family. Cell 1994, 77, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Mildner, M.; Mlitz, V.; Gruber, F.; Wojta, J.; Tschachler, E. Hepatocyte Growth Factor Establishes Autocrine and Paracrine Feedback Loops for the Protection of Skin Cells After UV Irradiation. J. Investig. Dermatol. 2007, 127, 2637–2644. [Google Scholar] [CrossRef] [PubMed]
- Grichnik, J.M.; Burch, J.A.; Burchette, J.; Shea, C.R. The SCF/KIT Pathway Plays a Critical Role in the Control of Normal Human Melanocyte Homeostasis. J. Investig. Dermatol. 1998, 111, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Scott, G.; Ewing, J.; Ryan, D.; Abboud, C. Stem Cell Factor Regulates Human Melanocyte-Matrix Interactions. Pigment Cell Res. 1994, 7, 44–51. [Google Scholar] [CrossRef]
- Imokawa, G.; Yada, Y.; Miyagishi, M. Endothelins Secreted from Human Keratinocytes are Intrinsic Mitogens for Human Melanocytes. J. Biol. Chem. 1992, 267, 24675–24680. [Google Scholar] [CrossRef]
- Imokawa, G.; Yada, Y.; Kimura, M. Signalling Mechanisms of Endothelin-Induced Mitogenesis and Melanogenesis in Human Melanocytes. Biochem. J. 1996, 314 Pt 1, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Ujfaludi, Z.; Tuzesi, A.; Majoros, H.; Rothler, B.; Pankotai, T.; Boros, I.M. Coordinated Activation of a Cluster of MMP Genes in Response to UVB Radiation. Sci. Rep. 2018, 8, 2660. [Google Scholar] [CrossRef]
- Yoshihisa, Y.; Norisugi, O.; Matsunaga, K.; Nishihira, J.; Shimizu, T. Involvement of MIF in Basement Membrane Damage in Chronically UVB-Exposed Skin in Mice. PLoS ONE 2014, 9, e89569. [Google Scholar] [CrossRef] [PubMed]
- Purwar, R.; Kraus, M.; Werfel, T.; Wittmann, M. Modulation of Keratinocyte-Derived MMP-9 by IL-13: A Possible Role for the Pathogenesis of Epidermal Inflammation. J. Investig. Dermatol. 2008, 128, 59–66. [Google Scholar] [CrossRef]
- Wang, X.; Bi, Z.; Chu, W.; Wan, Y. IL-1 Receptor Antagonist Attenuates MAP Kinase/AP-1 Activation and MMP1 Expression in UVA-Irradiated Human Fibroblasts Induced by Culture Medium from UVB-Irradiated Human Skin Keratinocytes. Int. J. Mol. Med. 2005, 16, 1117–1124. [Google Scholar] [CrossRef]
- Lee, M.J.; Oh, J.; Park, C.; Kim, K.H.; Lee, D.H.; Chung, J.H. Galanin Contributes to Ultraviolet Irradiation-Induced Inflammation in Human Skin. Exp. Dermatol. 2017, 26, 744–747. [Google Scholar] [CrossRef]
- Harsha, A.; Stojadinovic, O.; Brem, H.; Sehara-Fujisawa, A.; Wewer, U.; Loomis, C.A.; Blobel, C.P.; Tomic-Canic, M. ADAM12: A Potential Target for the Treatment of Chronic Wounds. J. Mol. Med. 2008, 86, 961–969. [Google Scholar] [CrossRef]
- Oh, S.T.; Schramme, A.; Stark, A.; Tilgen, W.; Gutwein, P.; Reichrath, J. Overexpression of ADAM 10 and ADAM 12 in Lesional Psoriatic Skin. Br. J. Dermatol. 2008, 158, 1371–1373. [Google Scholar] [CrossRef]
- Abbes, A.; Zayani, Y.; Zidi, W.; Hammami, M.B.; Mebazaa, A.; El Euch, D.; Ben Ammar, A.; Sanhaji, H.; El May, M.V.; Mokni, M.; et al. Matrix Metalloproteinase-7 could be a Predictor for Acute Inflammation in Psoriatic Patients. Cytokine 2020, 134, 155195. [Google Scholar] [CrossRef]
- Suomela, S.; Kariniemi, A.L.; Snellman, E.; Saarialho-Kere, U. Metalloelastase (MMP-12) and 92-kDa Gelatinase (MMP-9) as Well as their Inhibitors, TIMP-1 and -3, are Expressed in Psoriatic Lesions. Exp. Dermatol. 2001, 10, 175–183. [Google Scholar] [CrossRef]
- Boukhedouni, N.; Martins, C.; Darrigade, A.; Drullion, C.; Rambert, J.; Barrault, C.; Garnier, J.; Jacquemin, C.; Thiolat, D.; Lucchese, F.; et al. Type-1 Cytokines Regulate MMP-9 Production and E-Cadherin Disruption to Promote Melanocyte Loss in Vitiligo. JCI Insight 2020, 5, e133772. [Google Scholar]
- Su, M.; Miao, F.; Jiang, S.; Shi, Y.; Luo, L.; He, X.; Wan, J.; Xu, S.; Lei, T. Role of the p53-TRPM1/miR-211-MMP9 Axis in UVB-induced Human Melanocyte Migration and its Potential in Repigmentation. Int. J. Mol. Med. 2020, 45, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Parsad, D.; Kanwar, A.J.; Kaul, D. Altered Levels of Ets-1 Transcription Factor and Matrix Metalloproteinases in Melanocytes from Patients with Vitiligo. Br. J. Dermatol. 2011, 165, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Valyi-Nagy, I.T.; Hirka, G.; Jensen, P.J.; Shih, I.M.; Juhasz, I.; Herlyn, M. Undifferentiated Keratinocytes Control Growth, Morphology, and Antigen Expression of Normal Melanocytes through Cell-Cell Contact. Lab. Investig. 1993, 69, 152–159. [Google Scholar]
- Le Varlet, B.; Chaudagne, C.; Saunois, A.; Barré, P.; Sauvage, C.; Berthouloux, B.; Meybeck, A.; Dumas, M.; Bonté, F. Age-Related Functional and Structural Changes in Human Dermo-Epidermal Junction Components. J. Investig. Dermatol. Symp. Proc. 1998, 3, 172–179. [Google Scholar] [CrossRef]
- Craven, N.M.; Watson, R.E.; Jones, C.J.; Shuttleworth, C.A.; Kielty, C.M.; Griffiths, C.E. Clinical Features of Photodamaged Human Skin are Associated with a Reduction in Collagen VII. Br. J. Dermatol. 1997, 137, 344–350. [Google Scholar] [CrossRef]
- Bosset, S.; Bonnet-Duquennoy, M.; Barré, P.; Chalon, A.; Lazou, K.; Kurfurst, R.; Bonté, F.; Schnébert, S.; Disant, F.; Le Varlet, B.; et al. Decreased Expression of Keratinocyte Beta1 Integrins in Chronically Sun-Exposed Skin In Vivo. Br. J. Dermatol. 2003, 148, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Duan, E. Fighting Against Skin Aging: The Way from Bench to Bedside. Cell Transplant. 2018, 27, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Miskolczi, Z.; Smith, M.P.; Rowling, E.J.; Ferguson, J.; Barriuso, J.; Wellbrock, C. Collagen Abundance Controls Melanoma Phenotypes through Lineage-Specific Microenvironment Sensing. Oncogene 2018, 37, 3166–3182. [Google Scholar] [CrossRef]
- Kirkpatrick, S.J.; Wang, R.K.; Duncan, D.D.; Kulesz-Martin, M.; Lee, K. Imaging the Mechanical Stiffness of Skin Lesions by in Vivo Acousto-Optical Elastography. Opt. Express 2006, 14, 9770–9779. [Google Scholar] [CrossRef]
- Naylor, E.C.; Watson, R.E.B.; Sherratt, M.J. Molecular Aspects of Skin Ageing. Maturitas 2011, 69, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Taloni, A.; Alemi, A.A.; Ciusani, E.; Sethna, J.P.; Zapperi, S.; La Porta, C.A.M. Mechanical Properties of Growing Melanocytic Nevi and the Progression to Melanoma. PLoS ONE 2014, 9, e94229. [Google Scholar] [CrossRef] [PubMed]
- Napoli, S.; Scuderi, C.; Gattuso, G.; Bella, V.D.; Candido, S.; Basile, M.S.; Libra, M.; Falzone, L. Functional Roles of Matrix Metalloproteinases and their Inhibitors in Melanoma. Cells 2020, 9, 1151. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Beck, I.M.; Gadesmann, J.; Karschuk, N.; Paschen, A.; Proksch, E.; Djonov, V.; Reiss, K.; Sedlacek, R. MMP19 is Upregulated during Melanoma Progression and Increases Invasion of Melanoma Cells. Mod. Pathol. 2010, 23, 511–521. [Google Scholar] [CrossRef]
- Hofmann, U.B.; Westphal, J.R.; Zendman, A.J.; Becker, J.C.; Ruiter, D.J.; van Muijen, G.N. Expression and Activation of Matrix Metalloproteinase-2 (MMP-2) and its Co-Localization with Membrane-Type 1 Matrix Metalloproteinase (MT1-MMP) Correlate with Melanoma Progression. J. Pathol. 2000, 191, 245–256. [Google Scholar] [CrossRef]
- Salemi, R.; Falzone, L.; Madonna, G.; Polesel, J.; Cinà, D.; Mallardo, D.; Ascierto, P.A.; Libra, M.; Candido, S. MMP-9 as a Candidate Marker of Response to BRAF Inhibitors in Melanoma Patients with BRAFV600E Mutation Detected in Circulating-Free DNA. Front. Pharmacol. 2018, 9, 856. [Google Scholar] [CrossRef]
- Guarneri, C.; Bevelacqua, V.; Polesel, J.; Falzone, L.; Cannavò, P.S.; Spandidos, D.A.; Malaponte, G.; Libra, M. NF-κB Inhibition is Associated with OPN/MMP-9 Downregulation in Cutaneous Melanoma. Oncol. Rep. 2017, 37, 737–746. [Google Scholar] [CrossRef]
- Frank, A.; David, V.; Aurelie, T.; Florent, G.; William, H.; Philippe, B. Regulation of MMPs during Melanoma Progression: From Genetic to Epigenetic. Anticancer Agents Med. Chem. 2012, 12, 773–782. [Google Scholar] [CrossRef]
- Moustakas, A. TGF-Beta Targets PAX3 to Control Melanocyte Differentiation. Dev. Cell 2008, 15, 797–799. [Google Scholar] [CrossRef]
- Brenner, M.; Degitz, K.; Besch, R.; Berking, C. Differential Expression of Melanoma-Associated Growth Factors in Keratinocytes and Fibroblasts by Ultraviolet A and Ultraviolet B Radiation. Br. J. Dermatol. 2005, 153, 733–739. [Google Scholar] [CrossRef]
- Lee, S.B.; Schramme, A.; Doberstein, K.; Dummer, R.; Abdel-Bakky, M.S.; Keller, S.; Altevogt, P.; Oh, S.T.; Reichrath, J.; Oxmann, D.; et al. ADAM10 is Upregulated in Melanoma Metastasis Compared with Primary Melanoma. J. Investig. Dermatol. 2010, 130, 763–773. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Hearing, V.J. The Roles of ADAMs Family Proteinases in Skin Diseases. Enzym. Res. 2011, 2011, 482498. [Google Scholar] [CrossRef] [PubMed]
- Mazurkiewicz, J.; Simiczyjew, A.; Dratkiewicz, E.; Kot, M.; Pietraszek-Gremplewicz, K.; Wilk, D.; Ziętek, M.; Matkowski, R.; Nowak, D. Melanoma Stimulates the Proteolytic Activity of HaCaT Keratinocytes. Cell Commun. Signal. 2022, 20, 146. [Google Scholar] [CrossRef]
- Loh, C.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef]
- Karim, R.; Tse, G.; Putti, T.; Scolyer, R.; Lee, S. The Significance of the Wnt Pathway in the Pathology of Human Cancers. Pathology 2004, 36, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Dorsky, R.I.; Raible, D.W.; Moon, R.T. Direct Regulation of Nacre, a Zebrafish MITF Homolog Required for Pigment Cell Formation, by the Wnt Pathway. Genes Dev. 2000, 14, 158–162. [Google Scholar] [CrossRef]
- Bellei, B.; Pitisci, A.; Catricalà, C.; Larue, L.; Picardo, M. Wnt/Β-Catenin Signaling is Stimulated by A-Melanocyte-Stimulating Hormone in Melanoma and Melanocyte Cells: Implication in Cell Differentiation. Pigment Cell Melanoma Res. 2011, 24, 309–325. [Google Scholar] [CrossRef]
- Gallagher, S.J.; Rambow, F.; Kumasaka, M.; Champeval, D.; Bellacosa, A.; Delmas, V.; Larue, L. Beta-Catenin Inhibits Melanocyte Migration but Induces Melanoma Metastasis. Oncogene 2013, 32, 2230–2238. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Yasumoto, K.; Takeda, K.; Takahashi, K.; Yamamoto, H.; Shibahara, S. Microphthalmia-Associated Transcription Factor in the Wnt Signaling Pathway. Pigment Cell Res. 2003, 16, 261–265. [Google Scholar] [CrossRef]
- Untiveros, G.; Dezi, L.; Gillette, M.; Sidor, J.; Strizzi, L. Normal Skin Cells Increase Aggressiveness of Cutaneous Melanoma by Promoting Epithelial-to-Mesenchymal Transition Via Nodal and Wnt Activity. Int. J. Mol. Sci. 2021, 22, 11719. [Google Scholar] [CrossRef]
- Chien, A.J.; Moore, E.C.; Lonsdorf, A.S.; Kulikauskas, R.M.; Rothberg, B.G.; Berger, A.J.; Major, M.B.; Hwang, S.T.; Rimm, D.L.; Moon, R.T. Activated Wnt/Beta-Catenin Signaling in Melanoma is Associated with Decreased Proliferation in Patient Tumors and a Murine Melanoma Model. Proc. Natl. Acad. Sci. USA 2009, 106, 1193–1198. [Google Scholar] [CrossRef]
- Widlund, H.R.; Horstmann, M.A.; Price, E.R.; Cui, J.; Lessnick, S.L.; Wu, M.; He, X.; Fisher, D.E. Beta-Catenin-Induced Melanoma Growth Requires the Downstream Target Microphthalmia-Associated Transcription Factor. J. Cell Biol. 2002, 158, 1079–1087. [Google Scholar] [CrossRef]
- Arozarena, I.; Bischof, H.; Gilby, D.; Belloni, B.; Dummer, R.; Wellbrock, C. In Melanoma, Beta-Catenin is a Suppressor of Invasion. Oncogene 2011, 30, 4531–4543. [Google Scholar] [CrossRef]
- Bachmann, I.M.; Straume, O.; Puntervoll, H.E.; Kalvenes, M.B.; Akslen, L.A. Importance of P-Cadherin, Beta-Catenin, and Wnt5a/Frizzled for Progression of Melanocytic Tumors and Prognosis in Cutaneous Melanoma. Clin. Cancer Res. 2005, 11, 8606–8614. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, X.; Pang, D.; Wang, Y.; Bai, J.; Tian, F.; Han, D.; Shi, S.; Hu, L. Nomogram Incorporating the WNT/Β-Catenin Signaling Pathway for Predicting the Survival of Cutaneous Melanoma. Int. J. Gen. Med. 2021, 14, 2751–2761. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Koetsier, J.L.; Sirico, A.; Agidi, A.T.; Antonini, D.; Missero, C.; Green, K.J. The Desmosomal Protein Desmoglein 1 Aids Recovery of Epidermal Differentiation After Acute UV Light Exposure. J. Investig. Dermatol. 2014, 134, 2154–2162. [Google Scholar] [CrossRef] [PubMed]
- El Kharbili, M.; Cario, M.; Béchetoille, N.; Pain, C.; Boucheix, C.; Degoul, F.; Masse, I.; Berthier-Vergnes, O. Tspan8 Drives Melanoma Dermal Invasion by Promoting ProMMP-9 Activation and Basement Membrane Proteolysis in a Keratinocyte-Dependent Manner. Cancers 2020, 12, 1297. [Google Scholar] [CrossRef]
- Berthier-Vergnes, O.; Barbollat-Boutrand, L.; Pommier, R.M.; de la Fouchardière, A.; Combemale, P.; Grimont, M.; Lopez-Ramirez, N.; Caramel, J.; Dalle, S.; Perrot, J.; et al. Tetraspanin8 Expression Predicts an Increased Metastatic Risk and is Associated with Cancer-Related Death in Human Cutaneous Melanoma. Mol. Cancer 2021, 20, 127. [Google Scholar] [CrossRef]
- Navarrete, M.; Salazar-Onfray, F.; Tittarelli, A. Flow Cytometry Evaluation of Gap Junction-Mediated Intercellular Communication between Cytotoxic T Cells and Target Tumor Cells. Methods Mol. Biol. 2021, 2346, 225–236. [Google Scholar]
- Tittarelli, A.; Mendoza-Naranjo, A.; Farías, M.; Guerrero, I.; Ihara, F.; Wennerberg, E.; Riquelme, S.; Gleisner, A.; Kalergis, A.; Lundqvist, A.; et al. Gap Junction Intercellular Communications Regulate NK Cell Activation and Modulate NK Cytotoxic Capacity. J. Immunol. 2014, 192, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Naranjo, A.; Cormie, P.; Serrano, A.E.; Wang, C.M.; Thrasivoulou, C.; Sutcliffe, J.E.S.; Gilmartin, D.J.; Tsui, J.; Serena, T.E.; Phillips, A.R.J.; et al. Overexpression of the Gap Junction Protein Cx43 as found in Diabetic Foot Ulcers can Retard Fibroblast Migration. Cell Biol. Int. 2012, 36, 661–667. [Google Scholar] [CrossRef]
- Trosko, J.E.; Ruch, R.J. Cell-Cell Communication in Carcinogenesis. Front. Biosci. 1998, 3, 208. [Google Scholar] [CrossRef]
- Tittarelli, A.; Guerrero, I.; Tempio, F.; Gleisner, M.A.; Avalos, I.; Sabanegh, S.; Ortíz, C.; Michea, L.; López, M.N.; Mendoza-Naranjo, A.; et al. Overexpression of Connexin 43 Reduces Melanoma Proliferative and Metastatic Capacity. Br. J. Cancer 2015, 113, 259–267. [Google Scholar] [CrossRef]
- Scatolini, M.; Patel, A.; Grosso, E.; Mello-Grand, M.; Ostano, P.; Coppo, R.; Vitiello, M.; Venesio, T.; Zaccagna, A.; Pisacane, A.; et al. GJB5 Association with BRAF Mutation and Survival in Cutaneous Malignant Melanoma. Br. J. Dermatol. 2022, 186, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Läubli, H.; Borsig, L. Selectins Promote Tumor Metastasis. Semin. Cancer Biol. 2010, 20, 169–177. [Google Scholar] [CrossRef]
- Shih, I.M.; Elder, D.E.; Hsu, M.Y.; Herlyn, M. Regulation of Mel-CAM/MUC18 Expression on Melanocytes of Different Stages of Tumor Progression by Normal Keratinocytes. Am. J. Pathol. 1994, 145, 837–845. [Google Scholar]
- Brose, M.S.; Volpe, P.; Feldman, M.; Kumar, M.; Rishi, I.; Gerrero, R.; Einhorn, E.; Herlyn, M.; Minna, J.; Nicholson, A.; et al. BRAF and RAS Mutations in Human Lung Cancer and Melanoma. Cancer Res. 2002, 62, 6997–7000. [Google Scholar]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF Gene in Human Cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Stefanato, C.M.; Yaar, M.; Bhawan, J.; Phillips, T.J.; Kosmadaki, M.G.; Botchkarev, V.; Gilchrest, B.A. Modulations of Nerve Growth Factor and Bcl-2 in Ultraviolet-Irradiated Human Epidermis. J. Cutan. Pathol. 2003, 30, 351–357. [Google Scholar] [CrossRef]
- Li, G.; Schaider, H.; Satyamoorthy, K.; Hanakawa, Y.; Hashimoto, K.; Herlyn, M. Downregulation of E-Cadherin and Desmoglein 1 by Autocrine Hepatocyte Growth Factor during Melanoma Development. Oncogene 2001, 20, 8125–8135. [Google Scholar] [CrossRef] [PubMed]
- Noujarède, J.; Carrié, L.; Garcia, V.; Grimont, M.; Eberhardt, A.; Mucher, E.; Genais, M.; Schreuder, A.; Carpentier, S.; Ségui, B.; et al. Sphingolipid Paracrine Signaling Impairs Keratinocyte Adhesion to Promote Melanoma Invasion. Cell Rep. 2023, 42, 113586. [Google Scholar] [CrossRef]
- Albinet, V.; Bats, M.-L.; Huwiler, A.; Rochaix, P.; Chevreau, C.; Ségui, B.; Levade, T.; Andrieu-Abadie, N. Dual Role of Sphingosine Kinase-1 in Promoting the Differentiation of Dermal Fibroblasts and the Dissemination of Melanoma Cells. Oncogene 2014, 33, 3364–3373. [Google Scholar] [CrossRef] [PubMed]
- Mancianti, M.L.; Herlyn, M.; Weil, D.; Jambrosic, J.; Rodeck, U.; Becker, D.; Diamond, L.; Clark, W.H.; Koprowski, H. Growth and Phenotypic Characteristics of Human Nevus Cells in Culture. J. Investig. Dermatol. 1988, 90, 134–141. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.W.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.A.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-Associated Senescence-Like Cell Cycle Arrest of Human Naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef]
- Sadangi, S.; Milosavljevic, K.; Castro-Perez, E.; Lares, M.; Singh, M.; Altameemi, S.; Beebe, D.J.; Ayuso, J.M.; Setaluri, V. Role of the Skin Microenvironment in Melanomagenesis: Epidermal Keratinocytes and Dermal Fibroblasts Promote BRAF Oncogene-Induced Senescence Escape in Melanocytes. Cancers 2022, 14, 1233. [Google Scholar] [CrossRef] [PubMed]
- Tagore, M.; Hergenreder, E.; Perlee, S.C.; Cruz, N.M.; Menocal, L.; Suresh, S.; Chan, E.; Baron, M.; Melendez, S.; Dave, A.; et al. GABA Regulates Electrical Activity and Tumor Initiation in Melanoma. Cancer Discov. 2023, 13, 2270–2291. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Messer, A.R.; Amitai-Lange, A.; Melamed, Z.; Ohana, R.; Bell, R.E.; Kapitansky, O.; Lerman, G.; Greenberger, S.; Khaled, M.; et al. Interactions of Melanoma Cells with Distal Keratinocytes Trigger Metastasis Via Notch Signaling Inhibition of MITF. Mol. Cell 2015, 59, 664–676. [Google Scholar] [CrossRef]
- Hou, J.; Karin, M.; Sun, B. Targeting Cancer-Promoting Inflammation—Have Anti-Inflammatory Therapies Come of Age? Nat. Rev. Clin. Oncol. 2021, 18, 261–279. [Google Scholar] [CrossRef]
- Sheng, Y.; Liu, J.; Zhang, M.; Zheng, S. Unveiling the Link between Inflammasomes and Skin Cutaneous Melanoma: Insights into Expression Patterns and Immunotherapy Response Prediction. Math. Biosci. Eng. 2023, 20, 19912–19928. [Google Scholar] [CrossRef] [PubMed]
- Kučera, J.; Strnadová, K.; Dvořánková, B.; Lacina, L.; Krajsová, I.; Štork, J.; Kovářová, H.; Skalníková, H.K.; Vodička, P.; Motlík, J.; et al. Serum Proteomic Analysis of Melanoma Patients with Immunohistochemical Profiling of Primary Melanomas and Cultured Cells: Pilot Study. Oncol. Rep. 2019, 42, 1793–1804. [Google Scholar] [CrossRef] [PubMed]
- Smatlik, N.; Drexler, S.K.; Burian, M.; Röcken, M.; Yazdi, A.S. ASC Speck Formation After Inflammasome Activation in Primary Human Keratinocytes. Oxid. Med. Cell. Longev. 2021, 2021, 7914829. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Awoyemi, A.A.; Fahy, K.E.; Thapa, P.; Borchers, C.; Wu, B.Y.; McGlone, C.L.; Schmeusser, B.; Sattouf, Z.; Rohan, C.A.; et al. Keratinocyte-Derived Microvesicle Particles Mediate Ultraviolet B Radiation-Induced Systemic Immunosuppression. J. Clin. Investig. 2021, 131, e144963. [Google Scholar] [CrossRef]
- Katiyar, S.K. UV-Induced Immune Suppression and Photocarcinogenesis: Chemoprevention by Dietary Botanical Agents. Cancer Lett. 2007, 255, 1–11. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marrapodi, R.; Bellei, B. The Keratinocyte in the Picture Cutaneous Melanoma Microenvironment. Cancers 2024, 16, 913. https://doi.org/10.3390/cancers16050913
Marrapodi R, Bellei B. The Keratinocyte in the Picture Cutaneous Melanoma Microenvironment. Cancers. 2024; 16(5):913. https://doi.org/10.3390/cancers16050913
Chicago/Turabian StyleMarrapodi, Ramona, and Barbara Bellei. 2024. "The Keratinocyte in the Picture Cutaneous Melanoma Microenvironment" Cancers 16, no. 5: 913. https://doi.org/10.3390/cancers16050913