
Emerging Lipid Targets in Glioblastoma
 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
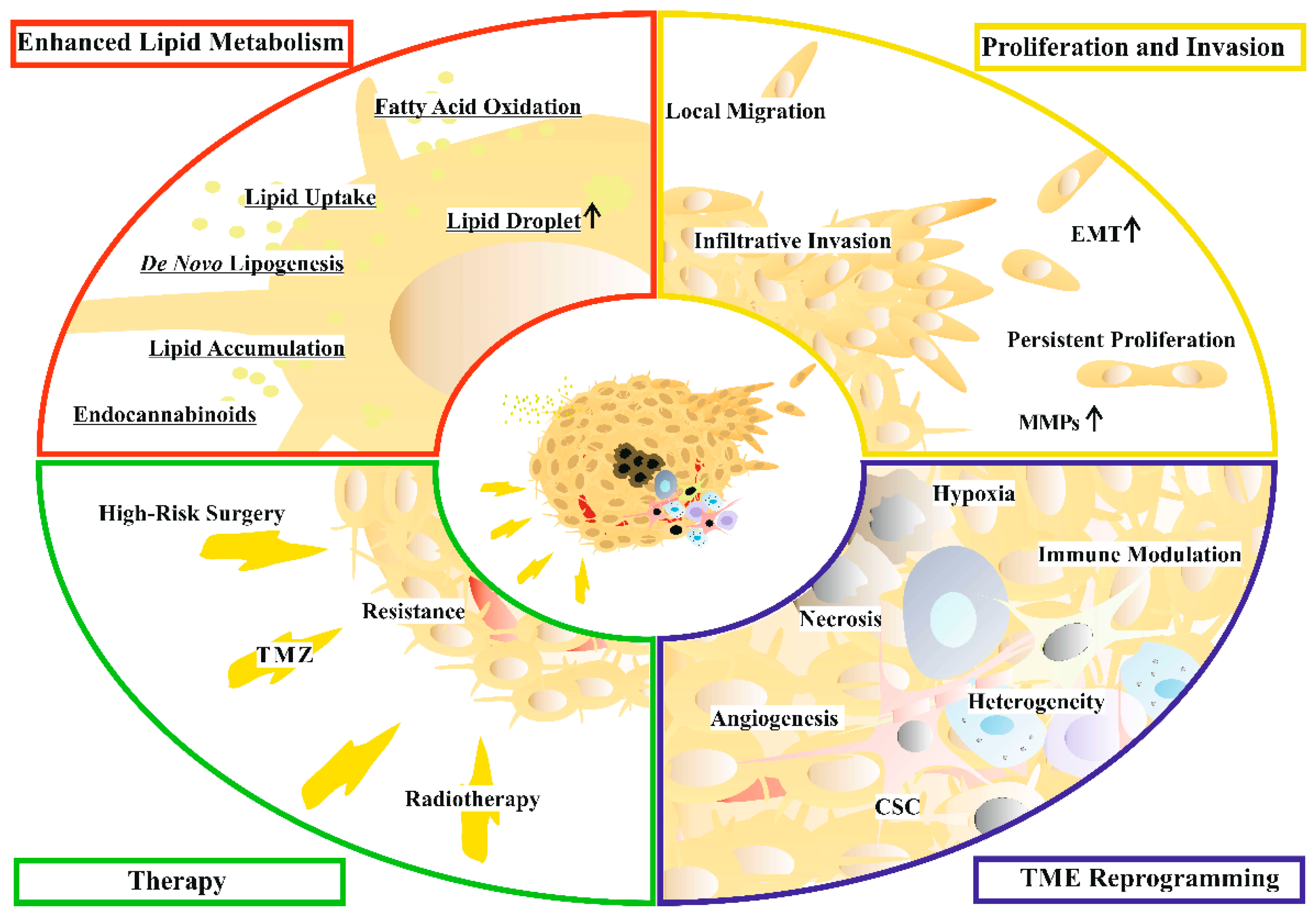
2. Glioblastoma
3. Lipids of the Healthy Brain
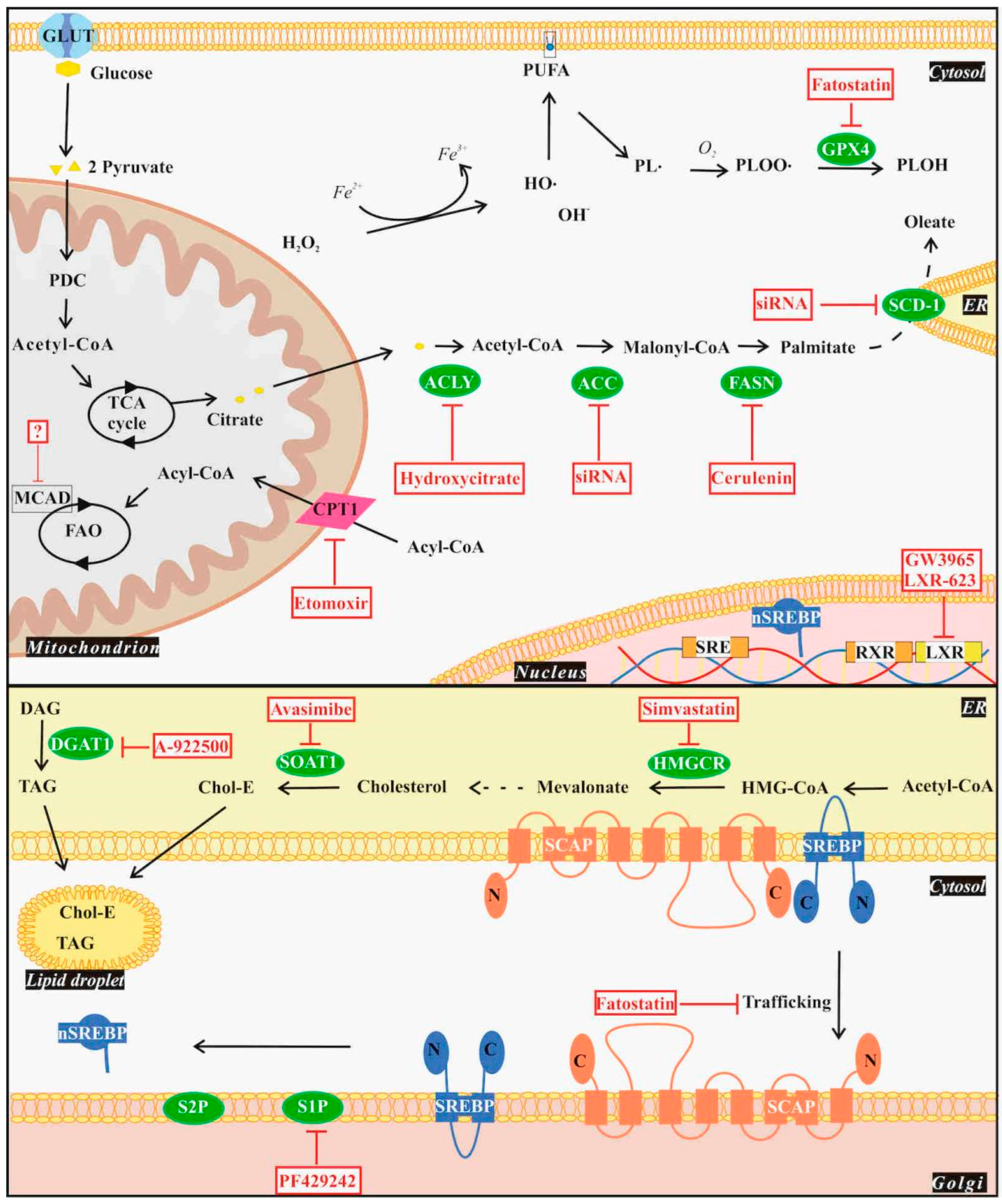
4. Lipid Metabolism in Glioblastoma Cells
4.1. De Novo Lipogenesis
4.2. Lipid Uptake and Storage
4.3. Fatty Acid Oxidation
4.4. Ferroptosis
5. Lipids with Special Impact on Glioblastoma Progression
5.1. Prostaglandins
5.2. Lysophosphatidic Acid
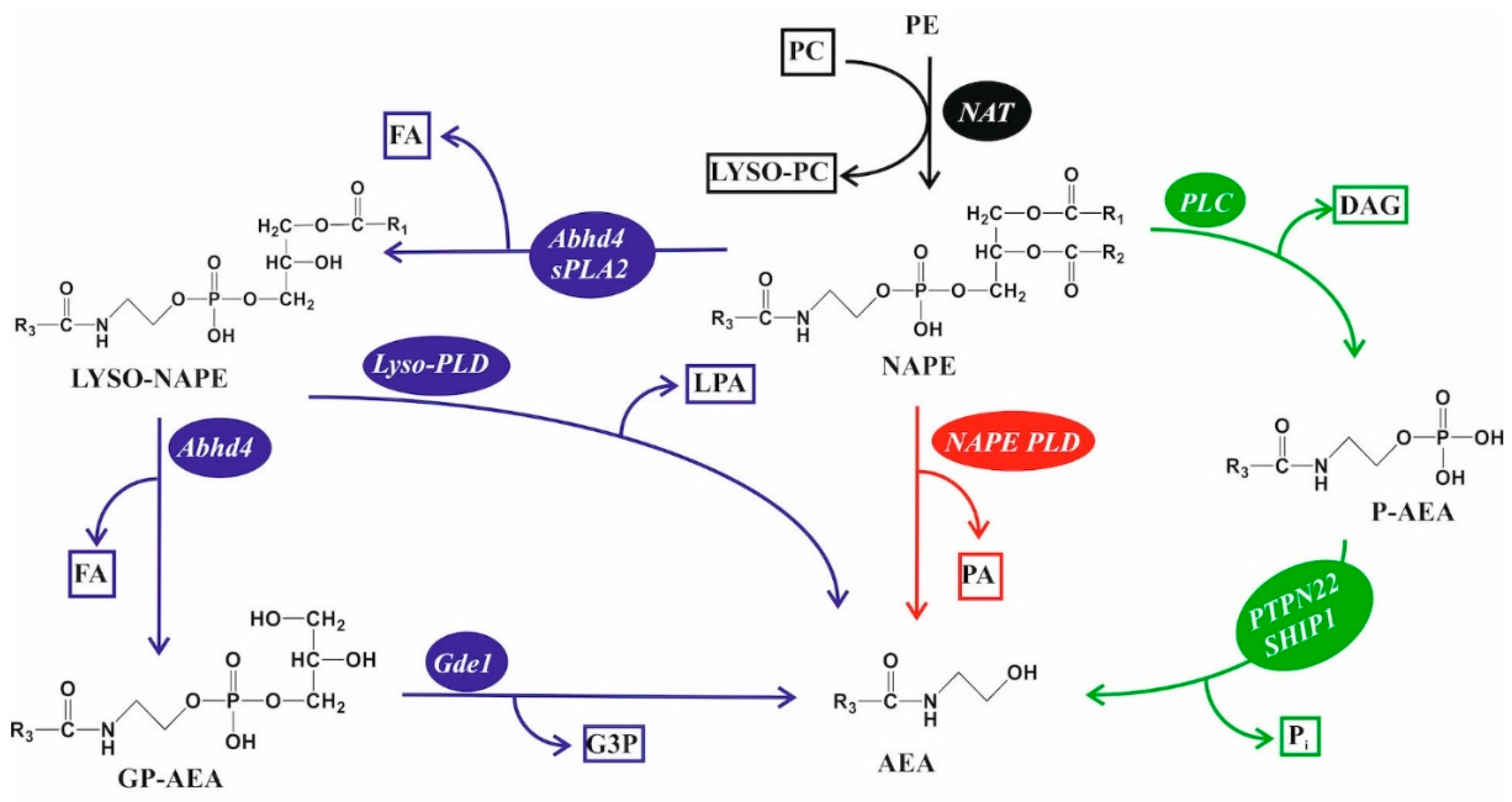
5.3. Endocannabinoid Lipids
6. Clinical Tests of Lipid-Targeting Drugs for Glioblastoma Treatment

{kind=link}
{kind=link}
{kind=link}
| NCT No. | Drug | Target | Combined with | Phase | Outcome | Reference |
|---|---|---|---|---|---|---|
| NCT03032484 | TVB-2640 (Denifanstat) | FASN | Bevacizumab | II | Improved PFS6 | [70] |
| NCT05118776 | ASC40 | FASN | Bevacizumab | III | Ongoing | - |
| NCT02029573 | Atorvastatin | HMGCR | Radiotherapy +TMZ | II | No benefit | [149] |
| NCT00047294 | Celecoxib | COX-2 | Thalidomide +TMZ | II | No benefit | [150] |
| Case study (15 patients) | CBD | Cannabinoid pathway | Radiotherapy +TMZ | - | Improved OS | [130] |
| NCT03529448 | TN-TC11G (THC+CBD) | Radiotherapy +TMZ | I | Ongoing | - | |
| NCT01812616 | Nabiximols | TMZ | Ib | Improved OS | [131] |
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gould, J. Breaking down the epidemiology of brain cancer. Nature 2018, 561, S40. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Lapointe, S.; Perry, A.; Butowski, N.A. Primary brain tumours in adults. Lancet 2018, 392, 432–446. [Google Scholar] [CrossRef]
- Boire, A.; Brastianos, P.K.; Garzia, L.; Valiente, M. Brain metastasis. Nat. Rev. Cancer 2020, 20, 4–11. [Google Scholar] [CrossRef]
- Chatterjee, S.; Nizamani, F.A.; Nürnberger, A.; Speck, O. Classification of brain tumours in MR images using deep spatiospatial models. Sci. Rep. 2022, 12, 1505. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Ostrom, Q.T.; Kruchko, C.; Patil, N.; Tihan, T.; Cioffi, G.; Fuchs, H.E.; Waite, K.A.; Jemal, A.; Siegel, R.L. Brain and other central nervous system tumor statistics, 2021. CA Cancer J. Clin. 2021, 71, 381–406. [Google Scholar] [CrossRef]
- Müller Bark, J.; Kulasinghe, A.; Chua, B.; Day, B.W.; Punyadeera, C. Circulating biomarkers in patients with glioblastoma. Br. J. Cancer 2020, 122, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.; Pfister, S.M.; Reifenberger, G. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Schaff, L.R.; Mellinghoff, I.K. Glioblastoma and other primary brain malignancies in adults: A review. JAMA 2023, 329, 574–587. [Google Scholar] [CrossRef]
- Grochans, S.; Cybulska, A.M.; Simińska, D.; Korbecki, J.; Kojder, K.; Chlubek, D.; Baranowska-Bosiacka, I. Epidemiology of Glioblastoma Multiforme–Literature Review. Cancers 2022, 14, 2412. [Google Scholar] [CrossRef]
- Poon, M.T.; Sudlow, C.L.; Figueroa, J.D.; Brennan, P.M. Longer-term (≥2 years) survival in patients with glioblastoma in population-based studies pre-and post-2005: A systematic review and meta-analysis. Sci. Rep. 2020, 10, 11622. [Google Scholar] [CrossRef] [PubMed]
- Tykocki, T.; Eltayeb, M. Ten-year survival in glioblastoma. A systematic review. J. Clin. Neurosci. 2018, 54, 7–13. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Cloughesy, T.; Perry, J.R.; Wick, W. Standards of care for treatment of recurrent glioblastoma—Are we there yet? Neuro-Oncology 2013, 15, 4–27. [Google Scholar] [CrossRef]
- Birzu, C.; French, P.; Caccese, M.; Cerretti, G.; Idbaih, A.; Zagonel, V.; Lombardi, G. Recurrent glioblastoma: From molecular landscape to new treatment perspectives. Cancers 2020, 13, 47. [Google Scholar] [CrossRef]
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, QLD, Australia, 2017. [Google Scholar]
- Cervio, A.; Piedimonte, F.; Salaberry, J.; Alcorta, S.C.; Salvat, J.; Diez, B.; Sevlever, G. Bone metastases from secondary glioblastoma multiforme: A case report. J. Neuro-Oncol. 2001, 52, 141–148. [Google Scholar] [CrossRef]
- Sidaway, P. Glioblastoma subtypes revisited. Nat. Rev. Clin. Oncol. 2017, 14, 587. [Google Scholar] [CrossRef]
- Zhang, P.; Xia, Q.; Liu, L.; Li, S.; Dong, L. Current opinion on molecular characterization for GBM classification in guiding clinical diagnosis, prognosis, and therapy. Front. Mol. Biosci. 2020, 7, 562798. [Google Scholar] [CrossRef] [PubMed]
- Kanderi, T.; Gupta, V. Glioblastoma multiforme. In StatPearls [Internet]; StatPearls Publishing: St. Petersburg, FL, USA, 2021. [Google Scholar]
- Alzial, G.; Renoult, O.; Paris, F.; Gratas, C.; Clavreul, A.; Pecqueur, C. Wild-type isocitrate dehydrogenase under the spotlight in glioblastoma. Oncogene 2022, 41, 613–621. [Google Scholar] [CrossRef]
- Armento, A.; Ehlers, J.; Schötterl, S.; Naumann, U. Molecular Mechanisms of Glioma Cell Motility; Exon Publications: Brisbane, QLD, Australia, 2017; pp. 73–93. [Google Scholar]
- Watkins, S.; Sontheimer, H. Hydrodynamic cellular volume changes enable glioma cell invasion. J. Neurosci. 2011, 31, 17250–17259. [Google Scholar] [CrossRef]
- Nakada, M.; Okada, Y.; Yamashita, J. The role of matrix metalloproteinases in glioma invasion. Front. Biosci.-Landmark 2003, 8, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Pu, W.; Qiu, J.; Riggins, G.J.; Parat, M.-O. Matrix protease production, epithelial-to-mesenchymal transition marker expression and invasion of glioblastoma cells in response to osmotic or hydrostatic pressure. Sci. Rep. 2020, 10, 2634. [Google Scholar] [CrossRef] [PubMed]
- Iwadate, Y. Epithelial-mesenchymal transition in glioblastoma progression. Oncol. Lett. 2016, 11, 1615–1620. [Google Scholar] [CrossRef]
- Kim, S.; Park, S.-J.; Chowdhury, T.; Hong, J.-I.; Ahn, J.; Jeong, T.Y.; Yu, H.J.; Shin, Y.-K.; Ku, J.-L.; Park, J.B. Subcellular progression of mesenchymal transition identified by two discrete synchronous cell lines derived from the same glioblastoma. Cell. Mol. Life Sci. 2022, 79, 181. [Google Scholar] [CrossRef]
- Hou, L.C.; Veeravagu, A.; Hsu, A.R.; Victor, C. Recurrent glioblastoma multiforme: A review of natural history and management options. Neurosurg. Focus 2006, 20, E3. [Google Scholar] [CrossRef]
- Fernandes, C.; Costa, A.; Osório, L.; Lago, R.C.; Linhares, P.; Carvalho, B.; Caeiro, C. Current Standards of Care in Glioblastoma Therapy; Exon Publications: Brisbane, QLD, Australia, 2017; pp. 197–241. [Google Scholar]
- Holland, E.C. Glioblastoma multiforme: The terminator. Proc. Natl. Acad. Sci. USA 2000, 97, 6242–6244. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of temozolomide resistance in glioblastoma-a comprehensive review. Cancer Drug Resist. 2021, 4, 17–43. [Google Scholar] [CrossRef]
- Tanaka, S.; Louis, D.N.; Curry, W.T.; Batchelor, T.T.; Dietrich, J. Diagnostic and therapeutic avenues for glioblastoma: No longer a dead end? Nat. Rev. Clin. Oncol. 2013, 10, 14–26. [Google Scholar] [CrossRef]
- Cohen, M.H.; Shen, Y.L.; Keegan, P.; Pazdur, R. FDA drug approval summary: Bevacizumab (Avastin®) as treatment of recurrent glioblastoma multiforme. Oncol. 2009, 14, 1131–1138. [Google Scholar] [CrossRef]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin®) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef]
- O’Brien, J.S.; Sampson, E.L. Lipid composition of the normal human brain: Gray matter, white matter, and myelin. J. Lipid Res. 1965, 6, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Premarathna, A.; Jayasooriya, A.; Sinclair, A. The role of lipids in the brain. In Advances in Dietary Lipids and Human Health; Li, D., Ed.; Academic Press: Cambridge, MA, USA, 2022; pp. 27–50. [Google Scholar]
- Su, X.Q.; Wang, J.; Sinclair, A.J. Plasmalogens and Alzheimer’s disease: A review. Lipids Health Dis. 2019, 18, 100. [Google Scholar] [CrossRef]
- Naudí, A.; Cabré, R.; Jové, M.; Ayala, V.; Gonzalo, H.; Portero-Otín, M.; Ferrer, I.; Pamplona, R. Lipidomics of human brain aging and Alzheimer’s disease pathology. Int. Rev. Neurobiol. 2015, 122, 133–189. [Google Scholar]
- Arenas, F.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Intracellular cholesterol trafficking and impact in neurodegeneration. Front. Mol. Neurosci. 2017, 10, 382. [Google Scholar] [CrossRef]
- Vitali, C.; Wellington, C.L.; Calabresi, L. HDL and cholesterol handling in the brain. Cardiovasc. Res. 2014, 103, 405–413. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell 2015, 6, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Pitas, R.E.; Boyles, J.K.; Lee, S.H.; Foss, D.; Mahley, R.W. Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim. Biophys. Acta (BBA)-Lipids Lipid Metab. 1987, 917, 148–161. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Horrocks, L.A.; Farooqui, T. Glycerophospholipids in brain: Their metabolism, incorporation into membranes, functions, and involvement in neurological disorders. Chem. Phys. Lipids 2000, 106, 1–29. [Google Scholar] [CrossRef]
- Sun, G.Y.; Xu, J.; Jensen, M.D.; Simonyi, A. Phospholipase A2 in the central nervous system: Implications for neurodegenerative diseases. J. Lipid Res. 2004, 45, 205–213. [Google Scholar] [CrossRef]
- Hayaishi, O. Molecular mechanisms of sleep–wake regulation: A role of prostaglandin D2. Philos. Trans. R. Soc. London Ser. B Biol. Sci. 2000, 355, 275–280. [Google Scholar] [CrossRef]
- Poitelon, Y.; Kopec, A.M.; Belin, S. Myelin fat facts: An overview of lipids and fatty acid metabolism. Cells 2020, 9, 812. [Google Scholar] [CrossRef] [PubMed]
- Alessenko, A.V.; Albi, E. Exploring sphingolipid implications in neurodegeneration. Front. Neurol. 2020, 11, 437. [Google Scholar] [CrossRef]
- Chen, C. Homeostatic regulation of brain functions by endocannabinoid signaling. Neural Regen. Res. 2015, 10, 691–692. [Google Scholar] [CrossRef]
- Alexandre, J.; Carmo, H.; Carvalho, F.; Silva, J.P. Synthetic cannabinoids and their impact on neurodevelopmental processes. Addict. Biol. 2020, 25, e12824. [Google Scholar] [CrossRef]
- Lu, H.-C.; Mackie, K. Review of the endocannabinoid system. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2021, 6, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Scherma, M.; Masia, P.; Satta, V.; Fratta, W.; Fadda, P.; Tanda, G. Brain activity of anandamide: A rewarding bliss? Acta Pharmacol. Sin. 2019, 40, 309–323. [Google Scholar] [CrossRef]
- Cristino, L.; Bisogno, T.; Di Marzo, V. Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat. Rev. Neurol. 2020, 16, 9–29. [Google Scholar] [CrossRef]
- Gopal, K.; Grossi, E.; Paoletti, P.; Usardi, M. Lipid composition of human intracranial tumors: A biochemical study. Acta Neurochir. 1963, 11, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Garcia Corrales, A.V.; Haidar, M.; Bogie, J.F.; Hendriks, J.J. Fatty acid synthesis in glial cells of the cns. Int. J. Mol. Sci. 2021, 22, 8159. [Google Scholar] [CrossRef] [PubMed]
- Dietschy, J.M. Central nervous system: Cholesterol turnover, brain development and neurodegeneration. Biol. Chem. 2009, 390, 287–293. [Google Scholar] [CrossRef]
- Lewis, C.; Brault, C.; Peck, B.; Bensaad, K.; Griffiths, B.; Mitter, R.; Chakravarty, P.; East, P.; Dankworth, B.; Alibhai, D. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid-and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene 2015, 34, 5128–5140. [Google Scholar] [CrossRef] [PubMed]
- Shakya, S.; Gromovsky, A.D.; Hale, J.S.; Knudsen, A.M.; Prager, B.; Wallace, L.C.; Penalva, L.O.; Brown, H.A.; Kristensen, B.W.; Rich, J.N. Altered lipid metabolism marks glioblastoma stem and non-stem cells in separate tumor niches. Acta Neuropathol. Commun. 2021, 9, 101. [Google Scholar] [CrossRef]
- Snaebjornsson, M.T.; Janaki-Raman, S.; Schulze, A. Greasing the wheels of the cancer machine: The role of lipid metabolism in cancer. Cell Metab. 2020, 31, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Miska, J.; Chandel, N.S. Targeting fatty acid metabolism in glioblastoma. J. Clin. Investig. 2023, 133, e163448. [Google Scholar] [CrossRef] [PubMed]
- Wallace, M.; Metallo, C.M. Tracing insights into de novo lipogenesis in liver and adipose tissues. Proc. Semin. Cell Dev. Biol. 2020, 108, 65–71. [Google Scholar] [CrossRef]
- Romero-Garcia, S.; Lopez-Gonzalez, J.S.; B’ ez-Viveros, J.L.; Aguilar-Cazares, D.; Prado-Garcia, H. Tumor cell metabolism: An integral view. Cancer Biol. Ther. 2011, 12, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Mashima, T.; Seimiya, H.; Tsuruo, T. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br. J. Cancer 2009, 100, 1369–1372. [Google Scholar] [CrossRef]
- Sun, T.; Hayakawa, K.; Bateman, K.S.; Fraser, M.E. Identification of the citrate-binding site of human ATP-citrate lyase using X-ray crystallography. J. Biol. Chem. 2010, 285, 27418–27428. [Google Scholar] [CrossRef]
- Tong, L. Acetyl-coenzyme A carboxylase: Crucial metabolic enzyme and attractive target for drug discovery. Cell. Mol. Life Sci. CMLS 2005, 62, 1784–1803. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Beckner, M.E.; Fellows-Mayle, W.; Zhang, Z.; Agostino, N.R.; Kant, J.A.; Day, B.W.; Pollack, I.F. Identification of ATP citrate lyase as a positive regulator of glycolytic function in glioblastomas. Int. J. Cancer 2010, 126, 2282–2295. [Google Scholar] [CrossRef]
- Jones, J.E.; Esler, W.P.; Patel, R.; Lanba, A.; Vera, N.B.; Pfefferkorn, J.A.; Vernochet, C. Inhibition of acetyl-CoA carboxylase 1 (ACC1) and 2 (ACC2) reduces proliferation and de novo lipogenesis of EGFRvIII human glioblastoma cells. PLoS ONE 2017, 12, e0169566. [Google Scholar] [CrossRef]
- Zhao, W.; Kridel, S.; Thorburn, A.; Kooshki, M.; Little, J.; Hebbar, S.A.; Robbins, M. Fatty acid synthase: A novel target for antiglioma therapy. Br. J. Cancer 2006, 95, 869–878. [Google Scholar] [CrossRef]
- Yasumoto, Y.; Miyazaki, H.; Vaidyan, L.K.; Kagawa, Y.; Ebrahimi, M.; Yamamoto, Y.; Ogata, M.; Katsuyama, Y.; Sadahiro, H.; Suzuki, M. Inhibition of fatty acid synthase decreases expression of stemness markers in glioma stem cells. PLoS ONE 2016, 11, e0147717. [Google Scholar] [CrossRef] [PubMed]
- Kelly, W.; Diaz Duque, A.E.; Michalek, J.; Konkel, B.; Caflisch, L.; Chen, Y.; Pathuri, S.C.; Madhusudanannair-Kunnuparampil, V.; Floyd, J.; Brenner, A. Phase II Investigation of TVB-2640 (Denifanstat) with Bevacizumab in Patients with First Relapse High-Grade Astrocytoma. Clin. Cancer Res. 2023, 29, 2419–2425. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.; Espenshade, P.J. Sterol regulatory element-binding protein (SREBP) cleavage regulates Golgi-to-endoplasmic reticulum recycling of SREBP cleavage-activating protein (SCAP). J. Biol. Chem. 2014, 289, 7547–7557. [Google Scholar] [CrossRef] [PubMed]
- Osborne, T.F. Sterol regulatory element-binding proteins (SREBPs): Key regulators of nutritional homeostasis and insulin action. J. Biol. Chem. 2000, 275, 32379–32382. [Google Scholar] [CrossRef]
- Geng, F.; Cheng, X.; Wu, X.; Yoo, J.Y.; Cheng, C.; Guo, J.Y.; Mo, X.; Ru, P.; Hurwitz, B.; Kim, S.-H. Inhibition of SOAT1 Suppresses Glioblastoma Growth via Blocking SREBP-1–Mediated LipogenesisTargeting SOAT1 to Treat Glioblastoma. Clin. Cancer Res. 2016, 22, 5337–5348. [Google Scholar] [CrossRef]
- Caruana, B.T.; Skoric, A.; Brown, A.J.; Lutze-Mann, L.H. Site-1 protease, a novel metabolic target for glioblastoma. Biochem. Biophys. Res. Commun. 2017, 490, 760–766. [Google Scholar] [CrossRef]
- Medes, G.; Thomas, A.; Weinhouse, S. Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res. 1953, 13, 27–29. [Google Scholar]
- Chen, Y.; Zhang, J.; Cui, W.; Silverstein, R.L. CD36, a signaling receptor and fatty acid transporter that regulates immune cell metabolism and fate. J. Exp. Med. 2022, 219, e20211314. [Google Scholar] [CrossRef] [PubMed]
- Hale, J.S.; Otvos, B.; Sinyuk, M.; Alvarado, A.G.; Hitomi, M.; Stoltz, K.; Wu, Q.; Flavahan, W.; Levison, B.; Johansen, M.L. Cancer stem cell-specific scavenger receptor CD36 drives glioblastoma progression. Stem Cells 2014, 32, 1746–1758. [Google Scholar] [CrossRef] [PubMed]
- Furuhashi, M.; Hotamisligil, G.S. Fatty acid-binding proteins: Role in metabolic diseases and potential as drug targets. Nat. Rev. Drug Discov. 2008, 7, 489–503. [Google Scholar] [CrossRef]
- Li, H.; Lv, B.; Bi, Y. FABP4 accelerates glioblastoma cell growth and metastasis through Wnt10b signalling. Eur. Rev. Med. Pharmacol. Sci 2018, 22, 7807–7818. [Google Scholar]
- Wang, J.; Zhao, S.; Sun, J.; Wang, X.; Guan, M.; Yin, J.; Tang, B. Oncogenic role and potential regulatory mechanism of fatty acid binding protein 5 based on a pan-cancer analysis. Sci. Rep. 2023, 13, 4060. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Diehn, M.; Watson, N.; Bollen, A.W.; Aldape, K.D.; Nicholas, M.K.; Lamborn, K.R.; Berger, M.S.; Botstein, D.; Brown, P.O. Gene expression profiling reveals molecularly and clinically distinct subtypes of glioblastoma multiforme. Proc. Natl. Acad. Sci. USA 2005, 102, 5814–5819. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.-S.; Mak, C.; Githaka, J.M.; Glubrecht, D.; Jeon, P.; Godbout, R. Role of brain fatty acid binding protein in glioblastoma microtube formation. Cancer Res. 2023, 83, 1736. [Google Scholar] [CrossRef]
- Guo, X.; Zhou, S.; Yang, Z.; Li, Z.-A.; Hu, W.; Dai, L.; Liang, W.; Wang, X. Cholesterol metabolism and its implication in glioblastoma therapy. J. Cancer 2022, 13, 1745–1757. [Google Scholar] [CrossRef]
- Chang, T.; Chang, C.C.; Cheng, D. Acyl-coenzyme A: Cholesterol acyltransferase. Annu. Rev. Biochem. 1997, 66, 613–638. [Google Scholar] [CrossRef]
- Wu, H.; Jiang, H.; Lu, D.; Xiong, Y.; Qu, C.; Zhao, D.; Mahmood, A.; Chopp, M. Effect of simvastatin on glioma cell proliferation, migration and apoptosis. Neurosurgery 2009, 65, 1087–1097. [Google Scholar] [CrossRef]
- Ralhan, I.; Chang, C.-L.; Lippincott-Schwartz, J.; Ioannou, M.S. Lipid droplets in the nervous system. J. Cell Biol. 2021, 220, e202102136. [Google Scholar] [CrossRef] [PubMed]
- Farmer, B.C.; Walsh, A.E.; Kluemper, J.C.; Johnson, L.A. Lipid droplets in neurodegenerative disorders. Front. Neurosci. 2020, 14, 742. [Google Scholar] [CrossRef] [PubMed]
- Farese, R.V.; Walther, T.C. Lipid droplets finally get a little RESPECT. Cell 2009, 139, 855–860. [Google Scholar] [CrossRef]
- Harris, C.A.; Haas, J.T.; Streeper, R.S.; Stone, S.J.; Kumari, M.; Yang, K.; Han, X.; Brownell, N.; Gross, R.W.; Zechner, R. DGAT enzymes are required for triacylglycerol synthesis and lipid droplets in adipocytes [S]. J. Lipid Res. 2011, 52, 657–667. [Google Scholar] [CrossRef]
- Paton, C.M.; Ntambi, J.M. Biochemical and physiological function of stearoyl-CoA desaturase. Am. J. Physiol.-Endocrinol. Metab. 2009, 297, E28–E37. [Google Scholar] [CrossRef] [PubMed]
- Taïb, B.; Aboussalah, A.M.; Moniruzzaman, M.; Chen, S.; Haughey, N.J.; Kim, S.F.; Ahima, R.S. Lipid accumulation and oxidation in glioblastoma multiforme. Sci. Rep. 2019, 9, 19593. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, A.R.; Hill, R.; Pilkington, G.J.; Madureira, P.A. The role of hypoxia in glioblastoma invasion. Cells 2017, 6, 45. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, H.K. Current understanding of hypoxia in glioblastoma multiforme and its response to immunotherapy. Cancers 2022, 14, 1176. [Google Scholar] [CrossRef]
- Opstad, K.S.; Bell, B.A.; Griffiths, J.R.; Howe, F.A. An investigation of human brain tumour lipids by high-resolution magic angle spinning 1H MRS and histological analysis. NMR Biomed. Int. J. Devoted Dev. Appl. Magn. Reson. In Vivo 2008, 21, 677–685. [Google Scholar] [CrossRef]
- Laurenti, G.; Benedetti, E.; D’angelo, B.; Cristiano, L.; Cinque, B.; Raysi, S.; Alecci, M.; Cerù, M.; Cifone, M.; Galzio, R. Hypoxia induces peroxisome proliferator-activated receptor α (PPARα) and lipid metabolism peroxisomal enzymes in human glioblastoma cells. J. Cell. Biochem. 2011, 112, 3891–3901. [Google Scholar] [CrossRef]
- Offer, S.; Menard, J.A.; Pérez, J.E.; de Oliveira, K.G.; Indira Chandran, V.; Johansson, M.C.; Bång-Rudenstam, A.; Siesjö, P.; Ebbesson, A.; Hedenfalk, I. Extracellular lipid loading augments hypoxic paracrine signaling and promotes glioma angiogenesis and macrophage infiltration. J. Exp. Clin. Cancer Res. 2019, 38, 241. [Google Scholar] [CrossRef]
- Cheng, X.; Geng, F.; Pan, M.; Wu, X.; Zhong, Y.; Wang, C.; Tian, Z.; Cheng, C.; Zhang, R.; Puduvalli, V. Targeting DGAT1 ameliorates glioblastoma by increasing fat catabolism and oxidative stress. Cell Metab. 2020, 32, 229–242.e8. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Yan, Y.; Xu, Z.; Zeng, S.; Qian, L.; Huo, L.; Li, X.; Sun, L.; Gong, Z. SCD1 confers temozolomide resistance to human glioma cells via the Akt/GSK3β/β-catenin signaling axis. Front. Pharmacol. 2018, 8, 960. [Google Scholar] [CrossRef] [PubMed]
- Morais, C.M.; Cardoso, A.M.; Araújo, A.R.D.; Reis, A.; Domingues, P.; Domingues, M.R.M.; de Lima, M.C.P.; Jurado, A.S. Stearoyl CoA Desaturase-1 Silencing in Glioblastoma Cells: Phospholipid Remodeling and Cytotoxicity Enhanced upon Autophagy Inhibition. Int. J. Mol. Sci. 2022, 23, 13014. [Google Scholar] [CrossRef] [PubMed]
- Rudling, M.J.; Angelin, B.; Peterson, C.O.; Collins, V.P. Low density lipoprotein receptor activity in human intracranial tumors and its relation to the cholesterol requirement. Cancer Res. 1990, 50, 483–487. [Google Scholar] [PubMed]
- Villa, G.R.; Hulce, J.J.; Zanca, C.; Bi, J.; Ikegami, S.; Cahill, G.L.; Gu, Y.; Lum, K.M.; Masui, K.; Yang, H. An LXR-cholesterol axis creates a metabolic co-dependency for brain cancers. Cancer Cell 2016, 30, 683–693. [Google Scholar] [CrossRef]
- Guo, D.; Reinitz, F.; Youssef, M.; Hong, C.; Nathanson, D.; Akhavan, D.; Kuga, D.; Amzajerdi, A.N.; Soto, H.; Zhu, S. An LXR Agonist Promotes Glioblastoma Cell Death through Inhibition of an EGFR/AKT/SREBP-1/LDLR–Dependent PathwayEGFR Signaling Regulates Cholesterol Metabolism. Cancer Discov. 2011, 1, 442–456. [Google Scholar] [CrossRef]
- Cirillo, A.; Di Salle, A.; Petillo, O.; Melone, M.A.; Grimaldi, G.; Bellotti, A.; Torelli, G.; de’Santi, M.S.; Cantatore, G.; Marinelli, A. High grade glioblastoma is associated with aberrant expression of ZFP57, a protein involved in gene imprinting, and of CPT1A and CPT1C that regulate fatty acid metabolism. Cancer Biol. Ther. 2014, 15, 735–741. [Google Scholar] [CrossRef]
- Puca, F.; Yu, F.; Bartolacci, C.; Pettazzoni, P.; Carugo, A.; Huang-Hobbs, E.; Liu, J.; Zanca, C.; Carbone, F.; Del Poggetto, E. Medium-chain acyl CoA dehydrogenase protects mitochondria from lipid peroxidation in glioblastoma. Cancer Discov. 2021, 11, 2904. [Google Scholar] [CrossRef]
- Lin, H.; Patel, S.; Affleck, V.S.; Wilson, I.; Turnbull, D.M.; Joshi, A.R.; Maxwell, R.; Stoll, E.A. Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro-Oncology 2017, 19, 43–54. [Google Scholar] [CrossRef]
- Pike, L.S.; Smift, A.L.; Croteau, N.J.; Ferrick, D.A.; Wu, M. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim. Biophys. Acta (BBA)-Bioenerg. 2011, 1807, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, S.; He, G.; Chen, T.; Li, X.; Liang, Y.; Wu, W.; Weng, L.; Feng, J.; Gao, Z.; Yang, K. Emerging role of ferroptosis in glioblastoma: Therapeutic opportunities and challenges. Front. Mol. Biosci. 2022, 9, 974156. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Lei, C.; Wang, Y.; Guo, D.; Zhang, S.; Wang, X.; Zhang, Z.; Wang, Y.; Ma, W. Prognostic Prediction Model for Glioblastoma: A Ferroptosis-Related Gene Prediction Model and Independent External Validation. J. Clin. Med. 2023, 12, 1341. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Ye, Z.; Hu, Y.; Ye, L.; Gao, L.; Wang, Y.; Sun, Q.; Tong, S.; Zhang, S.; Wu, L. Fatostatin induces ferroptosis through inhibition of the AKT/mTORC1/GPX4 signaling pathway in glioblastoma. Cell Death Dis. 2023, 14, 211. [Google Scholar] [CrossRef]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, P.; Lombardi, F.; Augello, F.R.; Giusti, I.; Dolo, V.; Leocata, P.; Cifone, M.G.; Cinque, B. Biological effects of selective COX-2 inhibitor NS398 on human glioblastoma cell lines. Cancer Cell Int. 2020, 20, 167. [Google Scholar] [CrossRef]
- Shono, T.; Tofilon, P.J.; Bruner, J.M.; Owolabi, O.; Lang, F.F. Cyclooxygenase-2 expression in human gliomas: Prognostic significance and molecular correlations. Cancer Res. 2001, 61, 4375–4381. [Google Scholar]
- Payner, T.; Leaver, H.A.; Knapp, B.; Whittle, I.R.; Trifan, O.C.; Miller, S.; Rizzo, M.T. Microsomal prostaglandin E synthase-1 regulates human glioma cell growth via prostaglandin E2–dependent activation of type II protein kinase A. Mol. Cancer Ther. 2006, 5, 1817–1826. [Google Scholar] [CrossRef]
- Tsai, Y.-T.; Lo, W.-L.; Chen, P.-Y.; Ko, C.-Y.; Chuang, J.-Y.; Kao, T.-J.; Yang, W.-B.; Chang, K.-Y.; Hung, C.-Y.; Kikkawa, U. Reprogramming of arachidonate metabolism confers temozolomide resistance to glioblastoma through enhancing mitochondrial activity in fatty acid oxidation. J. Biomed. Sci. 2022, 29, 21. [Google Scholar] [CrossRef] [PubMed]
- Lalier, L.; Cartron, P.; Pedelaborde, F.; Olivier, C.; Loussouarn, D.; Martin, S.; Meflah, K.; Menanteau, J.; Vallette, F. Increase in PGE2 biosynthesis induces a Bax dependent apoptosis correlated to patients’ survival in glioblastoma multiforme. Oncogene 2007, 26, 4999–5009. [Google Scholar] [CrossRef] [PubMed]
- Hari, A.; Vegi, N.; Das, U. Arachidonic and eicosapentaenoic acids induce oxidative stress to suppress proliferation of human glioma cells. Arch. Med. Sci. 2020, 16, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Tabuchi, S. The autotaxin-lysophosphatidic acid–lysophosphatidic acid receptor cascade: Proposal of a novel potential therapeutic target for treating glioblastoma multiforme. Lipids Health Dis. 2015, 14, 56. [Google Scholar] [CrossRef] [PubMed]
- Kishi, Y.; Okudaira, S.; Tanaka, M.; Hama, K.; Shida, D.; Kitayama, J.; Yamori, T.; Aoki, J.; Fujimaki, T.; Arai, H. Autotaxin is overexpressed in glioblastoma multiforme and contributes to cell motility of glioblastoma by converting lysophosphatidylcholine to lysophosphatidic acid. J. Biol. Chem. 2006, 281, 17492–17500. [Google Scholar] [CrossRef] [PubMed]
- Bhave, S.R.; Dadey, D.Y.; Karvas, R.M.; Ferraro, D.J.; Kotipatruni, R.P.; Jaboin, J.J.; Hallahan, A.N.; DeWees, T.A.; Linkous, A.G.; Hallahan, D.E. Autotaxin inhibition with PF-8380 enhances the radiosensitivity of human and murine glioblastoma cell lines. Front. Oncol. 2013, 3, 236. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, C.; Capasso, A. The endocannabinoid system in the cancer therapy: An overview. Curr. Med. Chem. 2011, 18, 1575–1583. [Google Scholar] [CrossRef]
- Velasco, G.; Carracedo, A.; Blázquez, C.; Lorente, M.; Aguado, T.; Haro, A.; Sánchez, C.; Galve-Roperh, I.; Guzmán, M. Cannabinoids and gliomas. Mol. Neurobiol. 2007, 36, 60–67. [Google Scholar] [CrossRef]
- Guzman, M.; Duarte, M.; Blazquez, C.; Ravina, J.; Rosa, M.; Galve-Roperh, I.; Sánchez, C.; Velasco, G.; González-Feria, L. A pilot clinical study of Δ9-tetrahydrocannabinol in patients with recurrent glioblastoma multiforme. Br. J. Cancer 2006, 95, 197–203. [Google Scholar] [CrossRef]
- Hernan Perez de la Ossa, D.; Lorente, M.; Gil-Alegre, M.E.; Torres, S.; Garcia-Taboada, E.; Aberturas, M.d.R.; Molpeceres, J.; Velasco, G.; Torres-Suarez, A.I. Local delivery of cannabinoid-loaded microparticles inhibits tumor growth in a murine xenograft model of glioblastoma multiforme. PLoS ONE 2013, 8, e54795. [Google Scholar] [CrossRef]
- Torres, S.; Lorente, M.; Rodríguez-Fornés, F.; Hernández-Tiedra, S.; Salazar, M.; García-Taboada, E.; Barcia, J.; Guzmán, M.; Velasco, G. A combined preclinical therapy of cannabinoids and temozolomide against glioma. Mol. Cancer Ther. 2011, 10, 90–103. [Google Scholar] [CrossRef]
- Carracedo, A.; Lorente, M.; Egia, A.; Blázquez, C.; García, S.; Giroux, V.; Malicet, C.; Villuendas, R.; Gironella, M.; González-Feria, L. The stress-regulated protein p8 mediates cannabinoid-induced apoptosis of tumor cells. Cancer Cell 2006, 9, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Tiedra, S.; Fabrias, G.; Dávila, D.; Salanueva, I.J.; Casas, J.; Montes, L.R.; Antón, Z.; Garcia-Taboada, E.; Salazar-Roa, M.; Lorente, M. Dihydroceramide accumulation mediates cytotoxic autophagy of cancer cells via autolysosome destabilization. Autophagy 2016, 12, 2213–2229. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.; Carracedo, A.; Salanueva, Í.J.; Hernández-Tiedra, S.; Lorente, M.; Egia, A.; Vázquez, P.; Blázquez, C.; Torres, S.; García, S. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J. Clin. Investig. 2009, 119, 1359–1372. [Google Scholar] [CrossRef] [PubMed]
- Likar, R.; Koestenberger, M.; Stutschnig, M.; Nahler, G. Cannabidiol Μay prolong survival in patients with Glioblastoma Multiforme. Cancer Diagn. Progn. 2021, 1, 77–82. [Google Scholar] [CrossRef]
- Twelves, C.; Sabel, M.; Checketts, D.; Miller, S.; Tayo, B.; Jove, M.; Brazil, L.; Short, S.C.; McBain, C.; Haylock, B.; et al. A phase 1b randomised, placebo-controlled trial of nabiximols cannabinoid oromucosal spray with temozolomide in patients with recurrent glioblastoma. Br. J. Cancer 2021, 124, 1379–1387. [Google Scholar] [CrossRef]
- Cadas, H.; di Tomaso, E.; Piomelli, D. Occurrence and biosynthesis of endogenous cannabinoid precursor, N-arachidonoyl phosphatidylethanolamine, in rat brain. J. Neurosci. 1997, 17, 1226–1242. [Google Scholar] [CrossRef]
- Liu, J.; Wang, L.; Harvey-White, J.; Osei-Hyiaman, D.; Razdan, R.; Gong, Q.; Chan, A.C.; Zhou, Z.; Huang, B.X.; Kim, H.-Y. A biosynthetic pathway for anandamide. Proc. Natl. Acad. Sci. USA 2006, 103, 13345–13350. [Google Scholar] [CrossRef]
- Liu, J.; Wang, L.; Harvey-White, J.; Huang, B.X.; Kim, H.-Y.; Luquet, S.; Palmiter, R.D.; Krystal, G.; Rai, R.; Mahadevan, A. Multiple pathways involved in the biosynthesis of anandamide. Neuropharmacology 2008, 54, 1–7. [Google Scholar] [CrossRef]
- Simon, G.M.; Cravatt, B.F. Anandamide biosynthesis catalyzed by the phosphodiesterase GDE1 and detection of glycerophospho-N-acyl ethanolamine precursors in mouse brain. J. Biol. Chem. 2008, 283, 9341–9349. [Google Scholar] [CrossRef]
- Cravatt, B.F.; Giang, D.K.; Mayfield, S.P.; Boger, D.L.; Lerner, R.A.; Gilula, N.B. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 1996, 384, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Zuzarte-Augustin, M.L.; Schmid, H. Properties of rat liver N-acylethanolamine amidohydrolase. J. Biol. Chem. 1985, 260, 14145–14149. [Google Scholar] [CrossRef] [PubMed]
- Ueda, N.; Yamamoto, K.; Yamamoto, S.; Tokunaga, T.; Shirakawa, E.; Shinkai, H.; Ogawa, M.; Sato, T.; Kudo, I.; Inoue, K. Lipoxygenase-catalyzed oxygenation of arachidonylethanolamide, a cannabinoid receptor agonist. Biochim. Biophys. Acta (BBA)-Lipids Lipid Metab. 1995, 1254, 127–134. [Google Scholar] [CrossRef]
- Yu, M.; Ives, D.; Ramesha, C.S. Synthesis of prostaglandin E2 ethanolamide from anandamide by cyclooxygenase-2. J. Biol. Chem. 1997, 272, 21181–21186. [Google Scholar] [CrossRef]
- Petersen, G.; Moesgaard, B.; Schmid, P.C.; Schmid, H.H.; Broholm, H.; Kosteljanetz, M.; Hansen, H.S. Endocannabinoid metabolism in human glioblastomas and meningiomas compared to human non-tumour brain tissue. J. Neurochem. 2005, 93, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Han, L.; Zhang, X.; Li, L.; Jiang, C.; Qiu, Y.; Huang, R.; Xie, B.; Lin, Z.; Ren, J. Alteration of endocannabinoid system in human gliomas. J. Neurochem. 2012, 120, 842–849. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Melck, D.; Palmisano, A.; Bisogno, T.; Laezza, C.; Bifulco, M.; Di Marzo, V. The endogenous cannabinoid anandamide inhibits human breast cancer cell proliferation. Proc. Natl. Acad. Sci. USA 1998, 95, 8375–8380. [Google Scholar] [CrossRef]
- Maccarrone, M.; Lorenzon, T.; Bari, M.; Melino, G.; Finazzi-Agro, A. Anandamide induces apoptosis in human cells via vanilloid receptors: Evidence for a protective role of cannabinoid receptors. J. Biol. Chem. 2000, 275, 31938–31945. [Google Scholar] [CrossRef]
- Jacobsson, S.O.; Wallin, T.; Fowler, C.J. Inhibition of rat C6 glioma cell proliferation by endogenous and synthetic cannabinoids. Relative involvement of cannabinoid and vanilloid receptors. J. Pharmacol. Exp. Ther. 2001, 299, 951–959. [Google Scholar]
- Ramer, R.; Brune, K.; Pahl, A.; Hinz, B. R (+)-methanandamide induces cyclooxygenase-2 expression in human neuroglioma cells via a non-cannabinoid receptor-mediated mechanism. Biochem. Biophys. Res. Commun. 2001, 286, 1144–1152. [Google Scholar] [CrossRef]
- Adinolfi, B.; Romanini, A.; Vanni, A.; Martinotti, E.; Chicca, A.; Fogli, S.; Nieri, P. Anticancer activity of anandamide in human cutaneous melanoma cells. Eur. J. Pharmacol. 2013, 718, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Ravi, J.; Sneh, A.; Shilo, K.; Nasser, M.W.; Ganju, R.K. FAAH inhibition enhances anandamide mediated anti-tumorigenic effects in non-small cell lung cancer by downregulating the EGF/EGFR pathway. Oncotarget 2014, 5, 2475–2486. [Google Scholar] [CrossRef] [PubMed]
- Soliman, E.; Van Dross, R. Anandamide-induced endoplasmic reticulum stress and apoptosis are mediated by oxidative stress in non-melanoma skin cancer: Receptor-independent endocannabinoid signaling. Mol. Carcinog. 2016, 55, 1807–1821. [Google Scholar] [CrossRef] [PubMed]
- Kesari, S.; Schiff, D.; Henson, J.W.; Muzikansky, A.; Gigas, D.C.; Doherty, L.; Batchelor, T.T.; Longtine, J.A.; Ligon, K.L.; Weaver, S. Phase II study of temozolomide, thalidomide, and celecoxib for newly diagnosed glioblastoma in adults. Neuro-Oncology 2008, 10, 300–308. [Google Scholar] [CrossRef]
- Altwairgi, A.K.; Alghareeb, W.A.; AlNajjar, F.H.; Alhussain, H.; Alsaeed, E.; Balbaid, A.A.O.; Aldanan, S.; Orz, Y.; Alsharm, A.A. Atorvastatin in combination with radiotherapy and temozolomide for glioblastoma: A prospective phase II study. Investig. New Drugs 2021, 39, 226–231. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Darwish, A.; Pammer, M.; Gallyas, F., Jr.; Vígh, L.; Balogi, Z.; Juhász, K. Emerging Lipid Targets in Glioblastoma. Cancers 2024, 16, 397. https://doi.org/10.3390/cancers16020397
Darwish A, Pammer M, Gallyas F Jr., Vígh L, Balogi Z, Juhász K. Emerging Lipid Targets in Glioblastoma. Cancers. 2024; 16(2):397. https://doi.org/10.3390/cancers16020397
Chicago/Turabian StyleDarwish, Ammar, Milán Pammer, Ferenc Gallyas, Jr., László Vígh, Zsolt Balogi, and Kata Juhász. 2024. "Emerging Lipid Targets in Glioblastoma" Cancers 16, no. 2: 397. https://doi.org/10.3390/cancers16020397