
Roles of the PARP Inhibitor in BRCA1 and BRCA2 Pathogenic Mutated Metastatic Prostate Cancer: Direct Functions and Modification of the Tumor Microenvironment

Abstract
:Simple Summary
Abstract
1. Introduction
2. Adenosine Diphosphate-Ribosylation
3. Poly (ADP-Ribosyl) Polymerase-1 (PARP-1)
4. PARP Inhibitors
5. Characteristics of Prostate Cancer in Cases with BRCA1/BRCA2 Pathogenic Variants
6. The PROfound Study and Olaparib (Lynparza®)
7. Olaparib (Lynparza®) and Genetic Testing
8. Background of Successful Cases of Olaparib (Lynparza®)
9. Mechanisms of Resistance to PARP Inhibitors
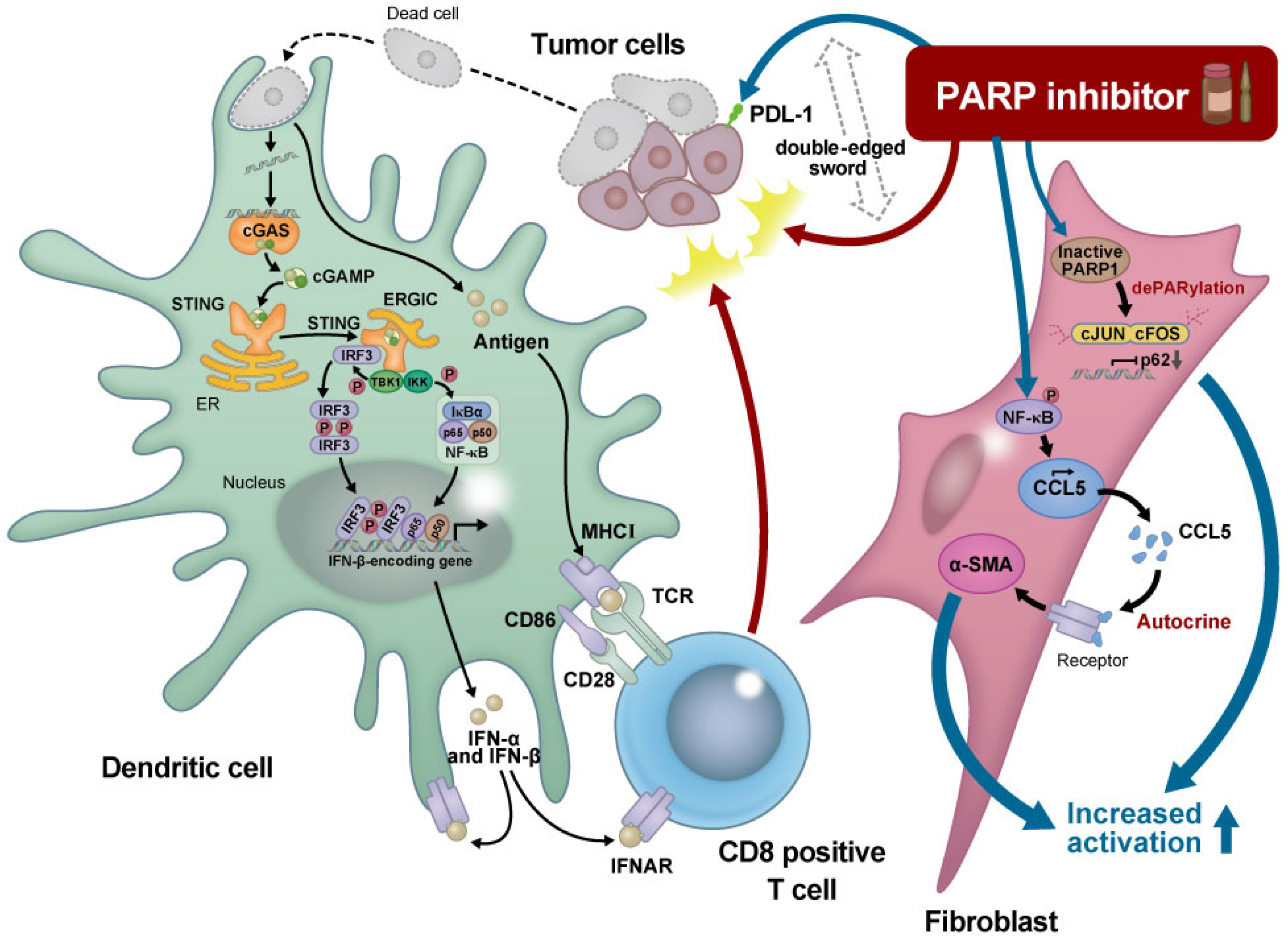
10. Modulation of the Tumor Microenvironment by PARP Inhibitors
11. Future Application of PARP Inhibitors for Prostate Cancer
12. Conclusions
13. Future Directions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gradishar, W.J.; Moran, M.S.; Abraham, J.; Aft, R.; Agnese, D.; Allison, K.H.; Anderson, B.; Burstein, H.J.; Chew, H.; Dang, C.; et al. Breast Cancer, Version 3.2022, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2022, 20, 691–722. [Google Scholar] [CrossRef]
- Ettinger, D.S.; Wood, D.E.; Aisner, D.L.; Akerley, W.; Bauman, J.R.; Bharat, A.; Bruno, D.S.; Chang, J.Y.; Chirieac, L.R.; D’Amico, T.A.; et al. Non-Small Cell Lung Cancer, Version 3.2022, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2022, 20, 497–530. [Google Scholar] [CrossRef]
- Schaeffer, E.M.; Srinivas, S.; Adra, N.; An, Y.; Barocas, D.; Bitting, R.; Bryce, A.; Chapin, B.; Cheng, H.H.; D’Amico, A.V.; et al. NCCN Guidelines® Insights: Prostate Cancer, Version 1.2023. J. Natl. Compr. Cancer Netw. 2022, 20, 1288–1298. [Google Scholar]
- De Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Groelly, F.J.; Fawkes, M.; Dagg, R.A.; Blackford, A.N.; Tarsounas, M. Targeting DNA damage response pathways in cancer. Nat. Rev. Cancer 2023, 23, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Rimar, K.J.; Tran, P.T.; Matulewicz, R.S.; Hussain, M.; Meeks, J.J. The emerging role of homologous recombination repair and PARP inhibitors in genitourinary malignancies. Cancer 2017, 123, 1912–1924. [Google Scholar] [CrossRef] [PubMed]
- Schuller, M.; Butler, R.E.; Ariza, A.; Tromans-Coia, C.; Jankevicius, G.; Claridge, T.D.W.; Kendall, S.L.; Goh, S.; Stewart, G.R.; Ahel, I. Molecular basis for DarT ADP-ribosylation of a DNA base. Nature 2021, 596, 597–602. [Google Scholar] [CrossRef]
- Li, P.; Lei, Y.; Qi, J.; Liu, W.; Yao, K. Functional roles of ADP-ribosylation writers, readers and erasers. Front. Cell. Dev. Biol. 2022, 10, 941356. [Google Scholar] [CrossRef]
- Pandey, N.; Black, B.E. Rapid Detection and Signaling of DNA Damage by PARP-1. Trends Biochem. Sci. 2021, 46, 744–757. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Snyder, L.A.; Damle, R.; Patel, S.; Bohrer, J.; Fiorella, A.; Driscoll, J.; Hawkins, R.; Stratton, C.F.; Manning, C.D.; Tatikola, K.; et al. Niraparib Shows Superior Tissue Distribution and Efficacy in a Prostate Cancer Bone Metastasis Model Compared with Other PARP Inhibitors. Mol. Cancer Ther. 2022, 21, 1115–1124. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Chu, Y.Y.; Yam, C.; Yamaguchi, H.; Hung, M.C. Biomarkers beyond BRCA: Promising combinatorial treatment strategies in overcoming resistance to PARP inhibitors. J. Biomed. Sci. 2022, 29, 86. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; de Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef]
- Abida, W.; Armenia, J.; Gopalan, A.; Brennan, R.; Walsh, M.; Barron, D.; Danila, D.; Rathkopf, D.; Morris, M.; Slovin, S.; et al. Prospective Genomic Profiling of Prostate Cancer Across Disease States Reveals Germline and Somatic Alterations That May Affect Clinical Decision Making. JCO Precis. Oncol. 2017, 2017, 1–26. [Google Scholar] [CrossRef]
- Network, C.G.A.R. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015, 163, 1011–1025. [Google Scholar] [CrossRef]
- Page, E.C.; Bancroft, E.K.; Brook, M.N.; Assel, M.; Hassan Al Battat, M.; Thomas, S.; Taylor, N.; Chamberlain, A.; Pope, J.; Raghallaigh, H.N.; et al. Interim Results from the IMPACT Study: Evidence for Prostate-specific Antigen Screening in BRCA2 Mutation Carriers. Eur. Urol. 2019, 76, 831–842. [Google Scholar] [CrossRef]
- Nyberg, T.; Frost, D.; Barrowdale, D.; Evans, D.G.; Bancroft, E.; Adlard, J.; Ahmed, M.; Barwell, J.; Brady, A.F.; Brewer, C.; et al. Prostate Cancer Risks for Male BRCA1 and BRCA2 Mutation Carriers: A Prospective Cohort Study. Eur. Urol. 2020, 77, 24–35. [Google Scholar] [CrossRef]
- Momozawa, Y.; Iwasaki, Y.; Hirata, M.; Liu, X.; Kamatani, Y.; Takahashi, A.; Sugano, K.; Yoshida, T.; Murakami, Y.; Matsuda, K.; et al. Germline Pathogenic Variants in 7636 Japanese Patients with Prostate Cancer and 12,366 Controls. J. Natl. Cancer Inst. 2020, 112, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Momozawa, Y.; Sasai, R.; Usui, Y.; Shiraishi, K.; Iwasaki, Y.; Taniyama, Y.; Parsons, M.T.; Mizukami, K.; Sekine, Y.; Hirata, M.; et al. Expansion of Cancer Risk Profile for BRCA1 and BRCA2 Pathogenic Variants. JAMA Oncol. 2022, 8, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Castro, E.; Romero-Laorden, N.; del Pozo, A.; Lozano, R.; Medina, A.; Puente, J.; Piulats, J.M.; Lorente, D.; Saez, M.I.; Morales-Barrera, R.; et al. PROREPAIR-B: A Prospective Cohort Study of the Impact of Germline DNA Repair Mutations on the Outcomes of Patients with Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2019, 37, 490–503. [Google Scholar] [CrossRef]
- Carter, H.B.; Helfand, B.; Mamawala, M.; Wu, Y.; Landis, P.; Yu, H.; Wiley, K.; Na, R.; Shi, Z.; Petkewicz, J.; et al. Germline Mutations in ATM and BRCA1/2 are Associated with Grade Reclassification in Men on Active Surveillance for Prostate Cancer. Eur. Urol. 2019, 75, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 383, 2345–2357. [Google Scholar] [CrossRef]
- Matsubara, N.; Nishimura, K.; Kawakami, S.; Joung, J.Y.; Uemura, H.; Goto, T.; Kwon, T.G.; Sugimoto, M.; Kato, M.; Wang, S.S.; et al. Olaparib in patients with mCRPC with homologous recombination repair gene alterations: PROfound Asian subset analysis. Jpn. J. Clin. Oncol. 2022, 52, 441–448. [Google Scholar] [CrossRef]
- EAU Guidelines. Edn. Presented at the EAU Annual Congress Milan 2023 Prostate Cancer. Available online: https://uroweb.org/guidelines/prostate-cancer (accessed on 1 May 2023).
- Chi, K.N.; Barnicle, A.; Sibilla, C.; Lai, Z.; Corcoran, C.; Barrett, J.C.; Adelman, C.A.; Qiu, P.; Easter, A.; Dearden, S.; et al. Detection of BRCA1, BRCA2, and ATM Alterations in Matched Tumor Tissue and Circulating Tumor DNA in Patients with Prostate Cancer Screened in PROfound. Clin. Cancer Res. 2023, 29, 81–91. [Google Scholar] [CrossRef]
- Gunderson, C.C.; Moore, K.N. BRACAnalysis CDx as a companion diagnostic tool for Lynparza. Expert Rev. Mol. Diagn. 2015, 15, 1111–1116. [Google Scholar] [CrossRef]
- Kage, H.; Shinozaki-Ushiku, A.; Ishigaki, K.; Sato, Y.; Tanabe, M.; Tanaka, S.; Tanikawa, M.; Watanabe, K.; Kato, S.; Akagi, K.; et al. Clinical utility of Todai OncoPanel in the setting of approved comprehensive cancer genomic profiling tests in Japan. Cancer Sci. 2023, 114, 1710–1717. [Google Scholar] [CrossRef]
- Takeda, M.; Takahama, T.; Sakai, K.; Shimizu, S.; Watanabe, S.; Kawakami, H.; Tanaka, K.; Sato, C.; Hayashi, H.; Nonagase, Y.; et al. Clinical Application of the FoundationOne CDx Assay to Therapeutic Decision-Making for Patients with Advanced Solid Tumors. Oncologist 2021, 26, e588–e596. [Google Scholar] [CrossRef]
- Taza, F.; Holler, A.E.; Fu, W.; Wang, H.; Adra, N.; Albany, C.; Ashkar, R.; Cheng, H.H.; Sokolova, A.O.; Agarwal, N.; et al. Differential Activity of PARP Inhibitors in BRCA1-Versus BRCA2-Altered Metastatic Castration-Resistant Prostate Cancer. JCO Precis. Oncol. 2021, 5, 1200–1220. [Google Scholar] [CrossRef] [PubMed]
- Carreira, S.; Porta, N.; Arce-Gallego, S.; Seed, G.; Llop-Guevara, A.; Bianchini, D.; Rescigno, P.; Paschalis, A.; Bertan, C.; Baker, C.; et al. Biomarkers Associating with PARP Inhibitor Benefit in Prostate Cancer in the TOPARP-B Trial. Cancer Discov. 2021, 11, 2812–2827. [Google Scholar] [CrossRef]
- Loehr, A.; Hussain, A.; Patnaik, A.; Bryce, A.H.; Castellano, D.; Font, A.; Shapiro, J.; Zhang, J.; Sautois, B.; Vogelzang, N.J.; et al. Emergence of BRCA Reversion Mutations in Patients with Metastatic Castration-resistant Prostate Cancer After Treatment with Rucaparib. Eur. Urol. 2023, 83, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Ter Brugge, P.; Kristel, P.; van der Burg, E.; Boon, U.; de Maaker, M.; Lips, E.; Mulder, L.; de Ruiter, J.; Moutinho, C.; Gevensleben, H.; et al. Mechanisms of Therapy Resistance in Patient-Derived Xenograft Models of BRCA1-Deficient Breast Cancer. J. Natl. Cancer Inst. 2016, 108, 1075. [Google Scholar] [CrossRef] [PubMed]
- Mekonnen, N.; Yang, H.; Shin, Y.K. Homologous Recombination Deficiency in Ovarian, Breast, Colorectal, Pancreatic, Non-Small Cell Lung and Prostate Cancers, and the Mechanisms of Resistance to PARP Inhibitors. Front. Oncol. 2022, 12, 880643. [Google Scholar] [CrossRef]
- Du, Y.; Yamaguchi, H.; Wei, Y.; Hsu, J.L.; Wang, H.L.; Hsu, Y.H.; Lin, W.C.; Yu, W.H.; Leonard, P.G.; Lee, G.R.; et al. Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat. Med. 2016, 22, 194–201. [Google Scholar] [CrossRef]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef]
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.; Zander, S.A.; Derksen, P.W.; de Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084. [Google Scholar] [CrossRef]
- Gogola, E.; Duarte, A.A.; de Ruiter, J.R.; Wiegant, W.W.; Schmid, J.A.; de Bruijn, R.; James, D.I.; Guerrero Llobet, S.; Vis, D.J.; Annunziato, S.; et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell. 2018, 33, 1078–1093.e12. [Google Scholar] [CrossRef]
- Cahuzac, M.; Péant, B.; Mes-Masson, A.M.; Saad, F. Development of Olaparib-Resistance Prostate Cancer Cell Lines to Identify Mechanisms Associated with Acquired Resistance. Cancers 2022, 14, 3877. [Google Scholar] [CrossRef]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Pantelidou, C.; Sonzogni, O.; de Oliveria Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP Inhibitor Efficacy Depends on CD8. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Corte, C.M.D.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef]
- Juncheng, P.; Joseph, A.; Lafarge, A.; Martins, I.; Obrist, F.; Pol, J.; Saavedra, E.; Li, S.; Sauvat, A.; Cerrato, G.; et al. Cancer cell-autonomous overactivation of PARP1 compromises immunosurveillance in non-small cell lung cancer. J. Immunother. Cancer 2022, 10, e004280. [Google Scholar] [CrossRef]
- Samstein, R.M.; Krishna, C.; Ma, X.; Pei, X.; Lee, K.W.; Makarov, V.; Kuo, F.; Chung, J.; Srivastava, R.M.; Purohit, T.A.; et al. Mutations in BRCA1 and BRCA2 differentially affect the tumor microenvironment and response to checkpoint blockade immunotherapy. Nat. Cancer 2021, 1, 1188–1203. [Google Scholar] [CrossRef]
- Trigos, A.S.; Pasam, A.; Banks, P.; Wallace, R.; Guo, C.; Keam, S.; Thorne, H.; Mitchell, C.; Lade, S.; Clouston, D.; et al. Tumor immune microenvironment of primary prostate cancer with and without germline mutations in homologous recombination repair genes. J. Immunother. Cancer 2022, 10, e003744. [Google Scholar] [CrossRef]
- Li, X.; Fang, T.; Xu, S.; Jin, P.; Zhou, D.; Wang, Z.; Li, H.; Yang, Z.; Chen, G.; Zheng, X.; et al. PARP inhibitors promote stromal fibroblast activation by enhancing CCL5 autocrine signaling in ovarian cancer. NPJ Precis. Oncol. 2021, 5, 49. [Google Scholar] [CrossRef]
- Linares, J.F.; Cid-Diaz, T.; Duran, A.; Osrodek, M.; Martinez-Ordoñez, A.; Reina-Campos, M.; Kuo, H.H.; Elemento, O.; Martin, M.L.; Cordes, T.; et al. The lactate-NAD+ axis activates cancer-associated fibroblasts by downregulating p62. Cell. Rep. 2022, 39, 110792. [Google Scholar] [CrossRef] [PubMed]
- Schiewer, M.J.; Goodwin, J.F.; Han, S.; Brenner, J.C.; Augello, M.A.; Dean, J.L.; Liu, F.; Planck, J.L.; Ravindranathan, P.; Chinnaiyan, A.M.; et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012, 2, 1134–1149. [Google Scholar] [CrossRef]
- Clarke, N.W.; Armstrong, A.J.; Thiery-Vuillemin, A.; Oya, M.; Shore, N.; Loredo, E.; Procopio, G.; de Menezes, J.; Girotto, G.; Arslan, C.; et al. Abiraterone and Olaparib for Metastatic Castration-Resistant Prostate Cancer. NEJM Evid. 2022, 1, EVIDoa2200043. [Google Scholar] [CrossRef]
- Saad, F.; Armstrong, A.J.; Thiery-Vuillemin, A.; Oya, M.; Loredo, E.; Procopio, G.; Janoski de Menezes, J.; Girotto, G.C.; Arslan, C.; Mehra, N.; et al. PROpel: Phase III trial of olaparib (ola) and abiraterone (abi) versus placebo (pbo) and abi as first-line (1L) therapy for patients (pts) with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2022, 40, 11. [Google Scholar] [CrossRef]
- Agarwal, N.; Azad, A.; Shore, N.D.; Carles, J.; Fay, A.P.; Dunshee, C.; Karsh, L.I.; Paccagnella, M.L.; Santo, N.D.; Elmeliegy, M.; et al. Talazoparib plus enzalutamide in metastatic castration-resistant prostate cancer: TALAPRO-2 phase III study design. Future Oncol. 2022, 18, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.M.; Nguyen, H.D.; Ho, C.K.; Todorova Kwan, T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef]


{kind=link}
| Trial | PROfound | PROpel | TALAPRO2 | |||||
|---|---|---|---|---|---|---|---|---|
| Treatment | Olaparib | Abiraterone/ Enzalutamide | Olaparib + Abiraterone | Placecbo + Abiraterone | Talazoparib + Enzalutamide | Placebo + Enzalutamide | ||
| Patient eligibility | mCRPC refractory against docetaxel and either abiraterone/enzalutamide | mCRPC without abiraterone and other second-generation AR inhibitor treatment (docetaxel was allowed as a neoadjuvant or adjuvant treatment after localized disease or as a first-line treatment for mHSPC) | mCRPC without second generation AR inhibitors treatment (enzalutamide, apalutamide, darolutamide); docetaxel and abiraterone was allowed as a treatment for mHSPC | |||||
| Genetic background | BRCA1/2, ATM | 12 HRR-related genes other than BRCA1/2, ATM | BRCA1/2, ATM | 12 HRR-related genes other than BRCA1/2, ATM | All comers | All comers | ||
| Number of patients | 162 | 94 | 83 | 48 | 399 | 397 | 402 | 403 |
| Primary endpoint | rPFS of patients with BRCA1/2 or ATM mutations | rPFS | rPFS | |||||
| 7.4 ms | - | 3.6 ms | - | 24.8 ms | 16.6 ms | NR | 21.9 ms | |
| HR, 0.34; (CI, 0.25–0.47) p < 0.001 | HR, 0.66; (CI, 0.55–0.81) p < 0.0001 | HR, 0.63; (CI, 0.51–0.78) p < 0.001 | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inoue, T.; Sekito, S.; Kageyama, T.; Sugino, Y.; Sasaki, T. Roles of the PARP Inhibitor in BRCA1 and BRCA2 Pathogenic Mutated Metastatic Prostate Cancer: Direct Functions and Modification of the Tumor Microenvironment. Cancers 2023, 15, 2662. https://doi.org/10.3390/cancers15092662
Inoue T, Sekito S, Kageyama T, Sugino Y, Sasaki T. Roles of the PARP Inhibitor in BRCA1 and BRCA2 Pathogenic Mutated Metastatic Prostate Cancer: Direct Functions and Modification of the Tumor Microenvironment. Cancers. 2023; 15(9):2662. https://doi.org/10.3390/cancers15092662
Chicago/Turabian StyleInoue, Takahiro, Sho Sekito, Takumi Kageyama, Yusuke Sugino, and Takeshi Sasaki. 2023. "Roles of the PARP Inhibitor in BRCA1 and BRCA2 Pathogenic Mutated Metastatic Prostate Cancer: Direct Functions and Modification of the Tumor Microenvironment" Cancers 15, no. 9: 2662. https://doi.org/10.3390/cancers15092662