Senolytic Flavonoids Enhance Type-I and Type-II Cell Death in Human Radioresistant Colon Cancer Cells through AMPK/MAPK Pathway





Abstract
:Simple Summary
Abstract
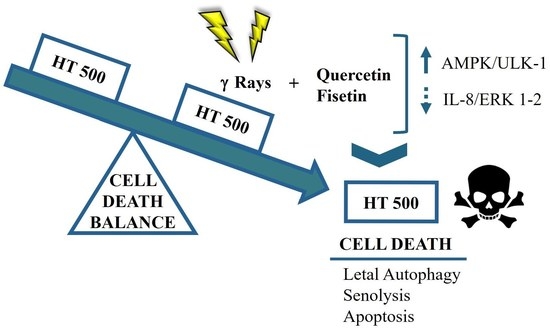
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
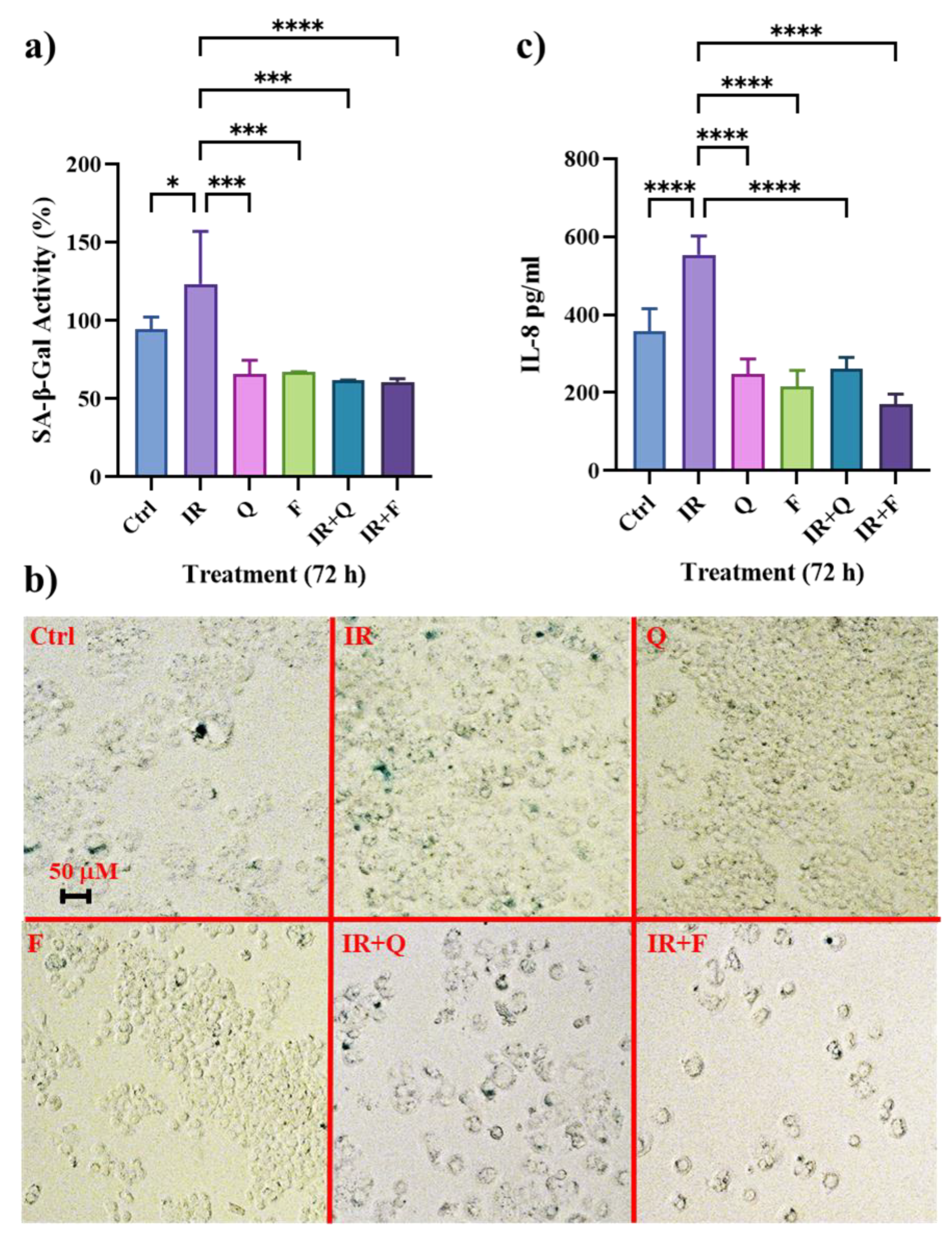
2.2. Evaluation of Senescence
2.3. Analysis of Cytokine Production
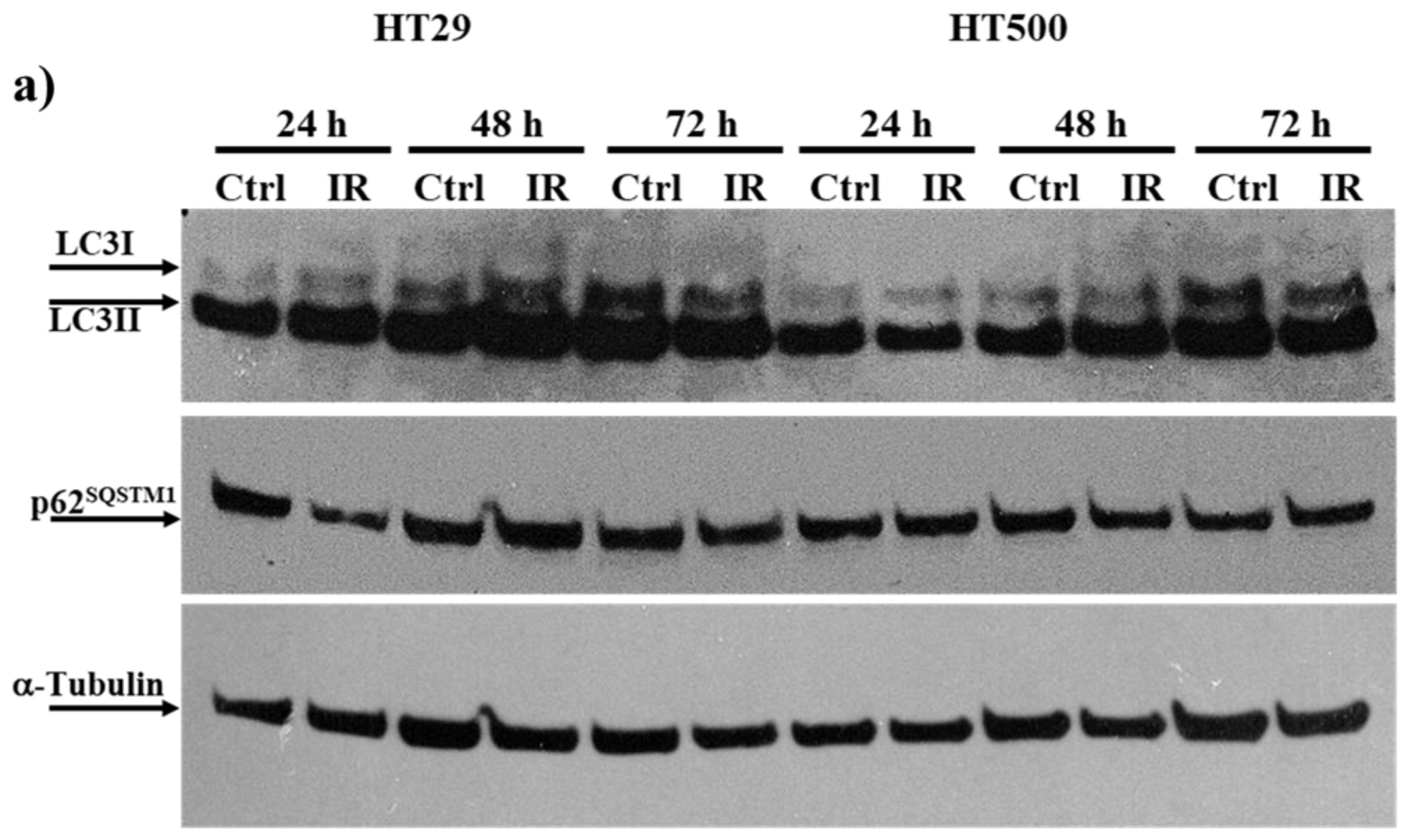
2.4. Measurement of Autophagy
2.5. Measurement of Apoptosis
2.6. Immunoblotting
2.7. Statistical Analysis
3. Results
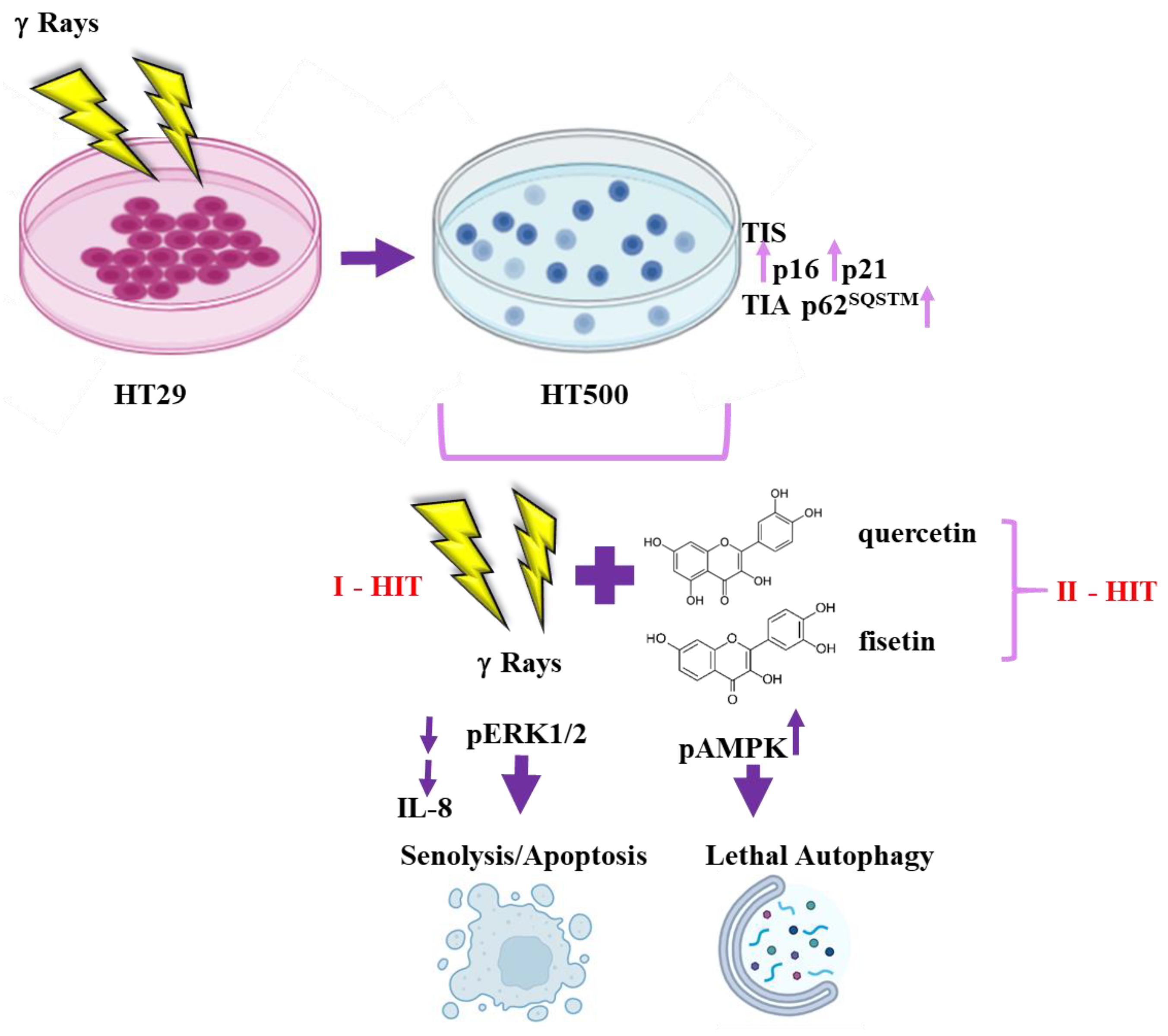
3.1. Comparison of Cellular and Biochemical Markers in HT29 and HT500 Cell Lines
3.2. Quercetin and Fisetin Show Senolytic and Anti-SASP Activity in Radioresistant HT500 Cells
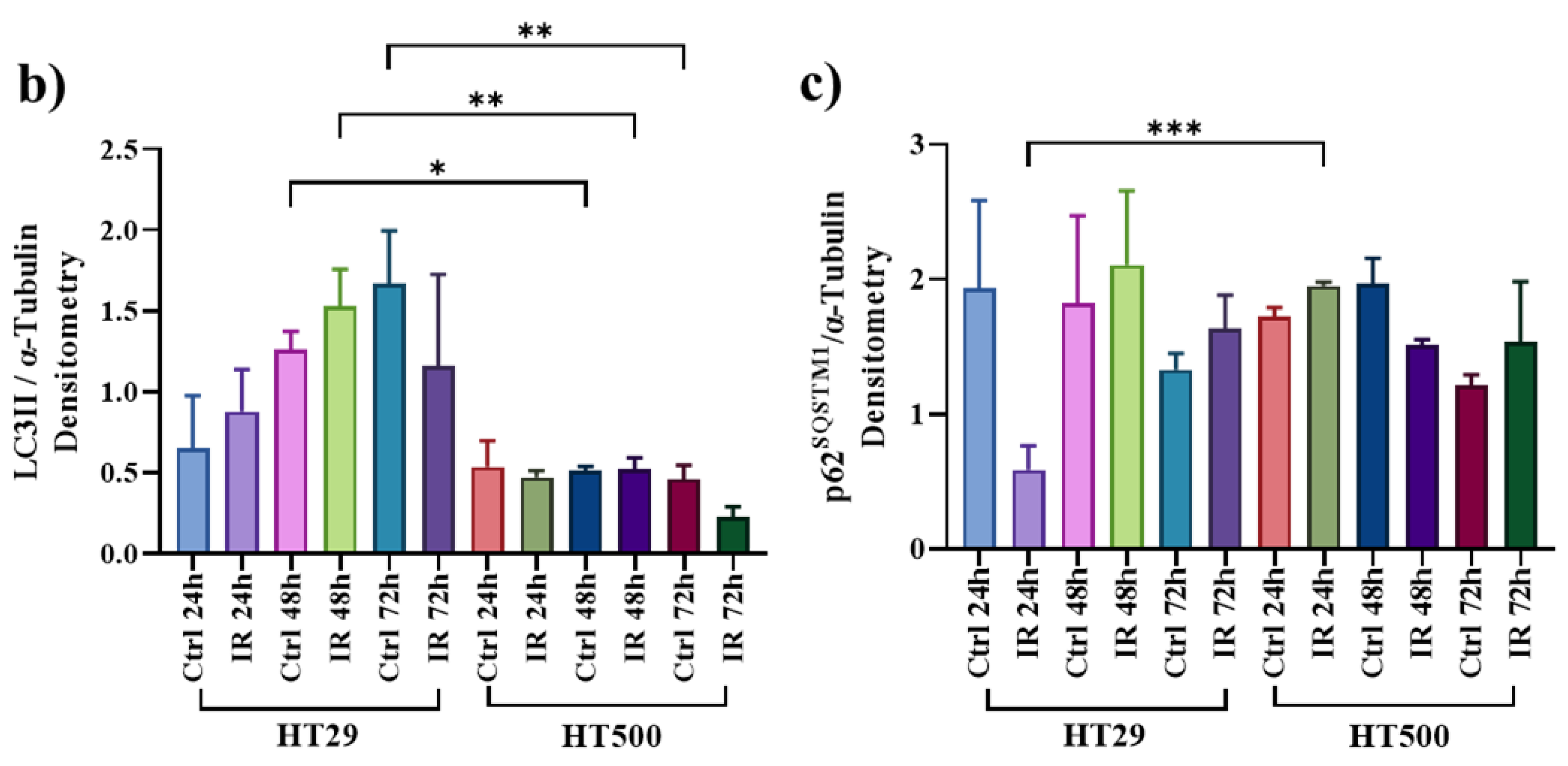
3.3. IR Differently Modulates the Autophagic Flux in HT29 and HT500 Cells
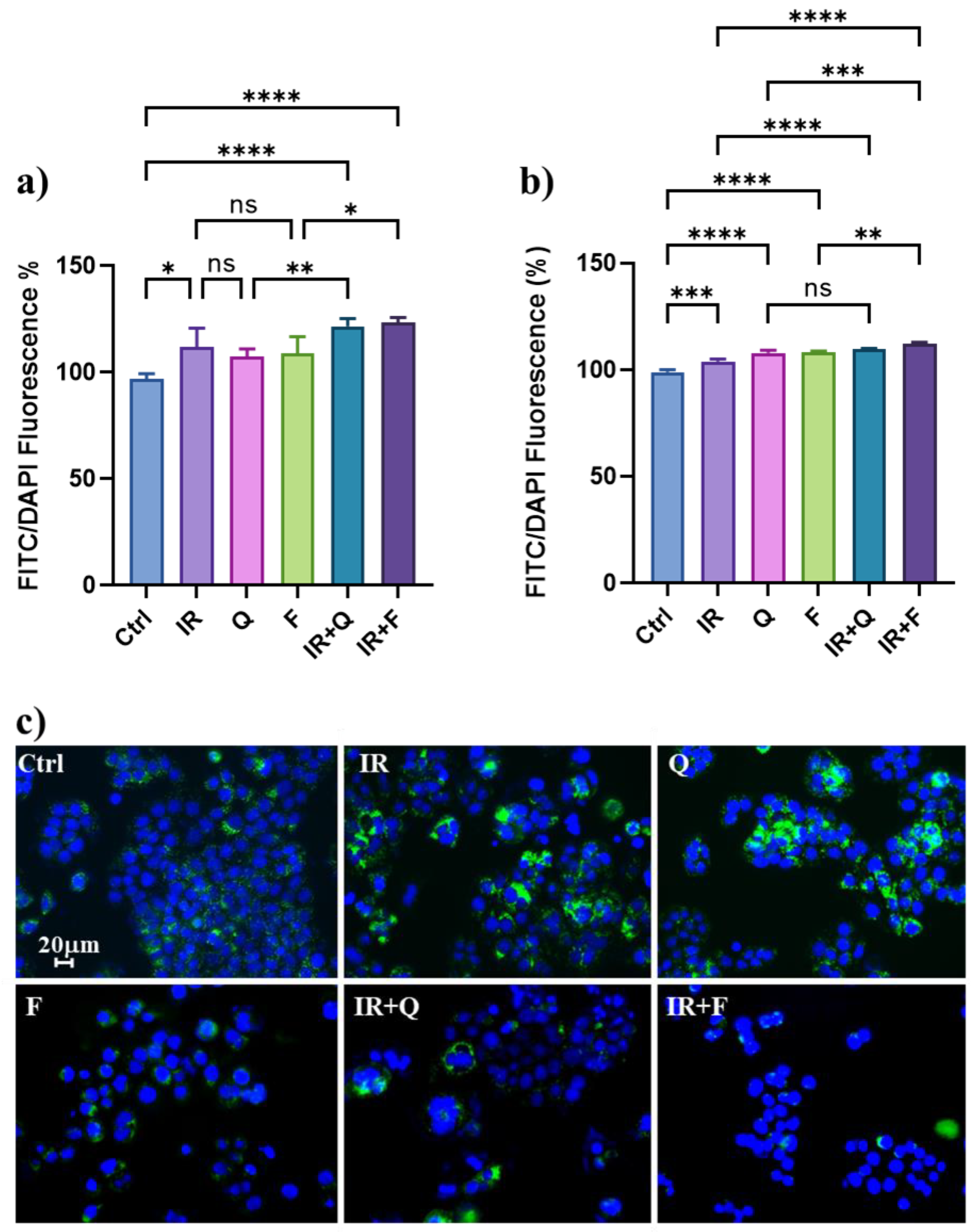
3.4. Quercetin and Fisetin Modulate Autophagic Flux in HT500 Cells
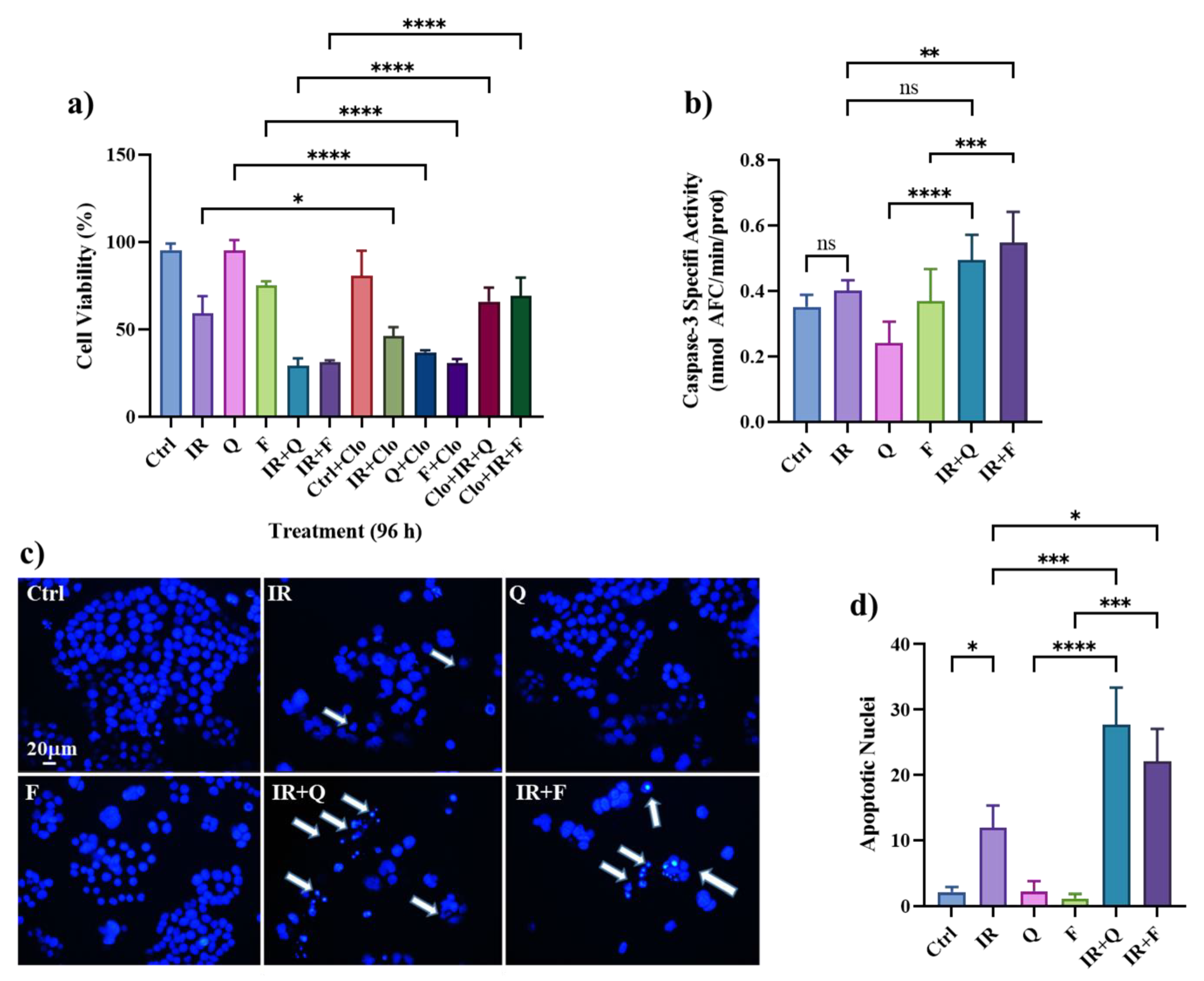
3.5. Quercetin and Fisetin Enhance Type I (Apoptosis) and Type II (Autophagic) Cell Death in HT500 Cells
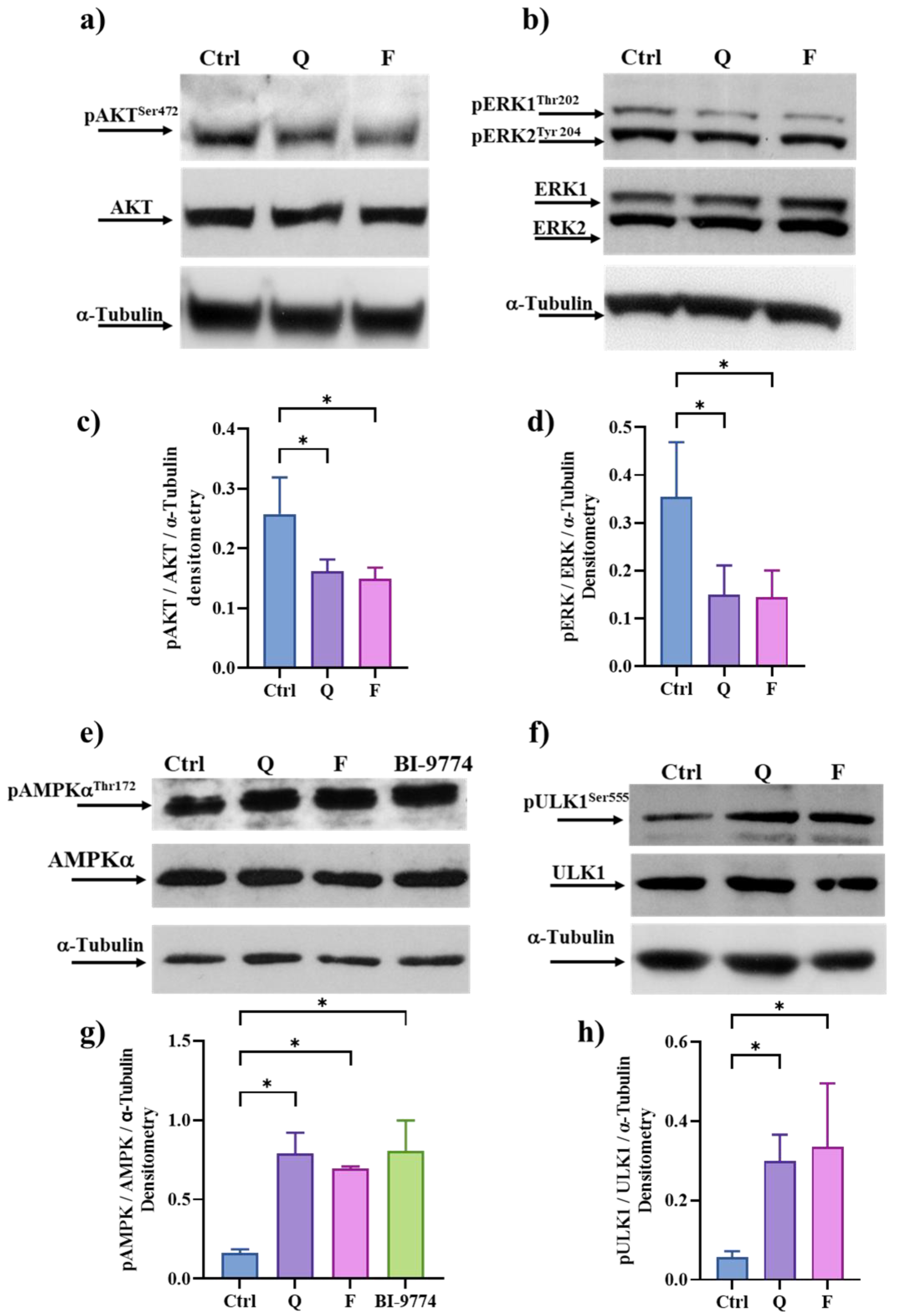
3.6. Quercetin and Fisetin Target Different Signaling Pathways Associated to the Senolytic Effects and Autophagy Induction in HT500 Cell Line
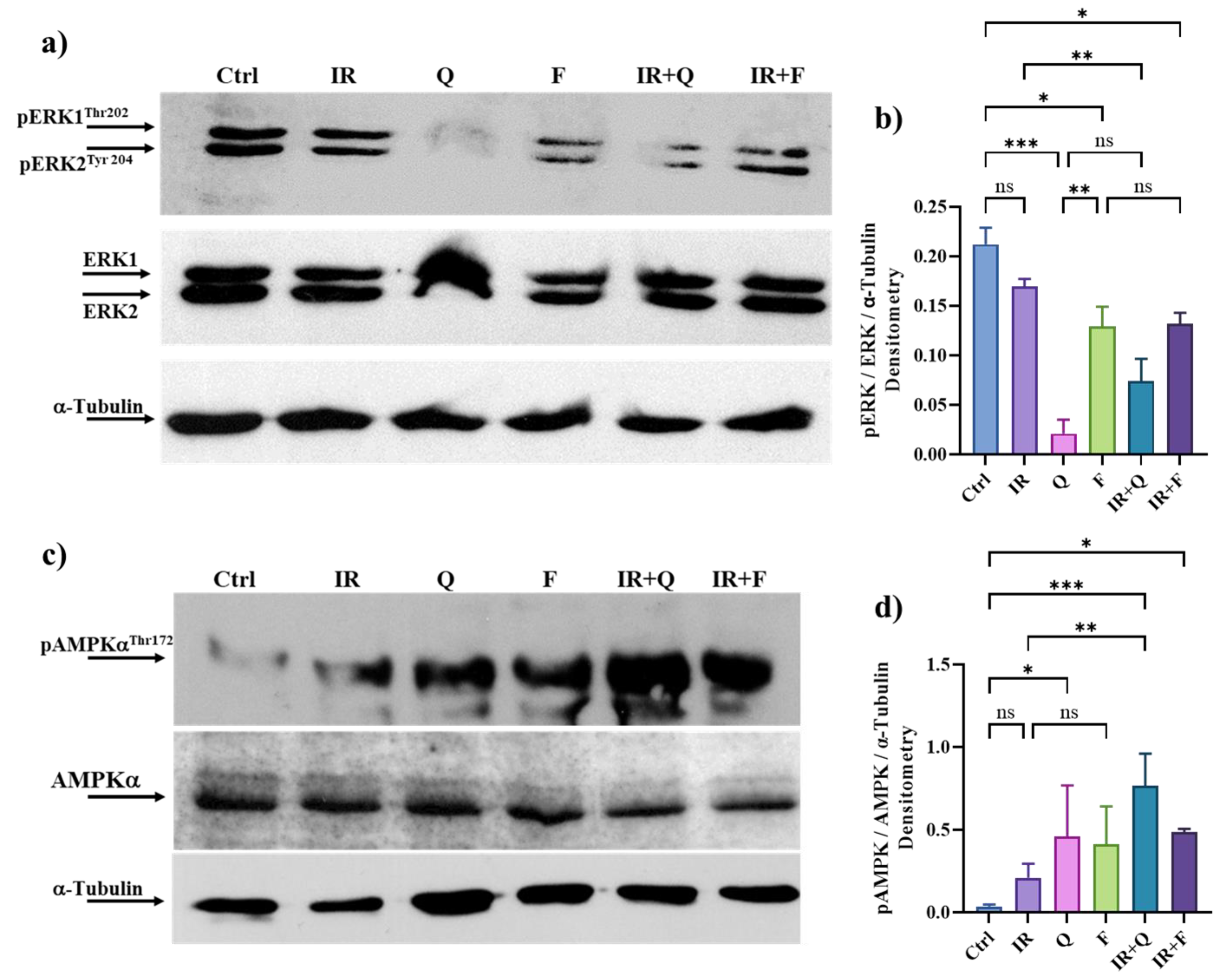
3.7. Quercetin and Fisetin in Association with γ-Ray Downregulate ERK/MAPK and Activate AMPK Kinases
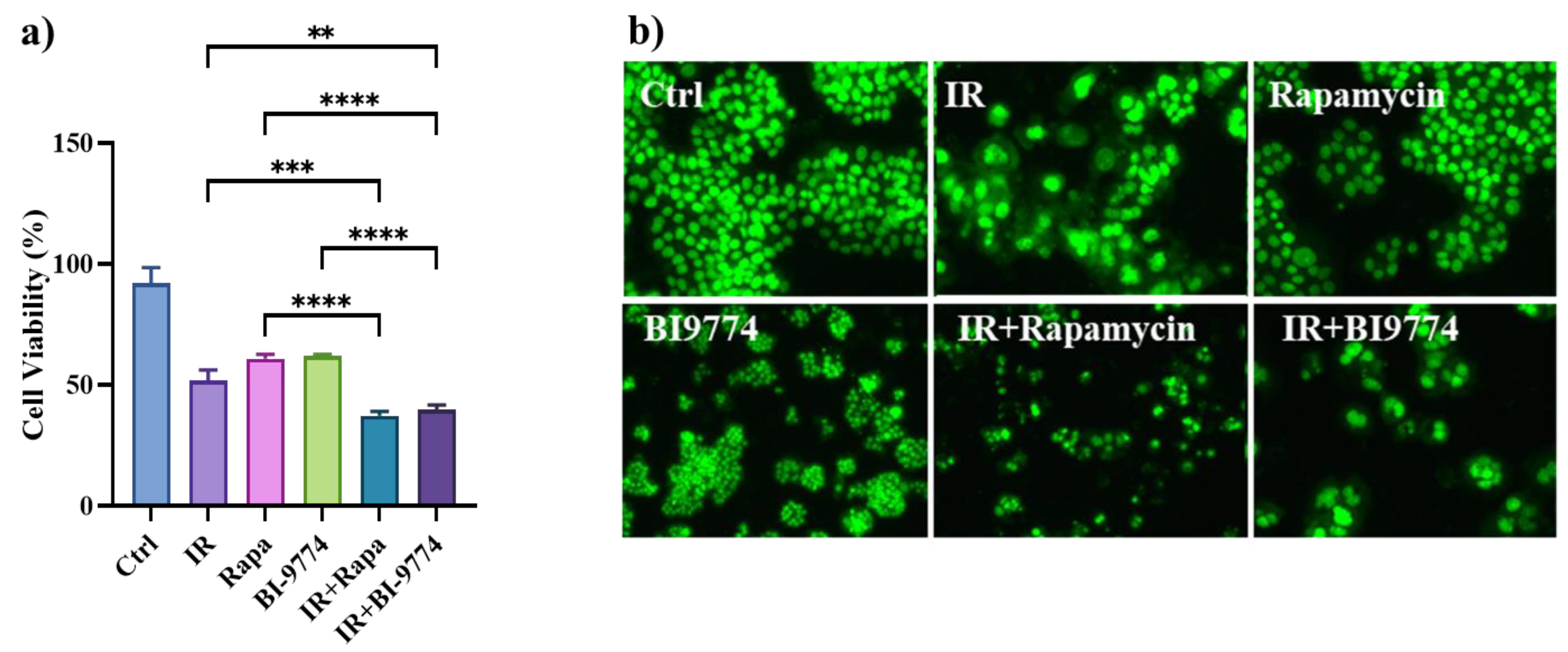
3.8. Pharmacological Modulation of mTOR and AMPK Kinases Confirm the Role of Autophagy in Sensitizing Radioresistant HT500 Cells
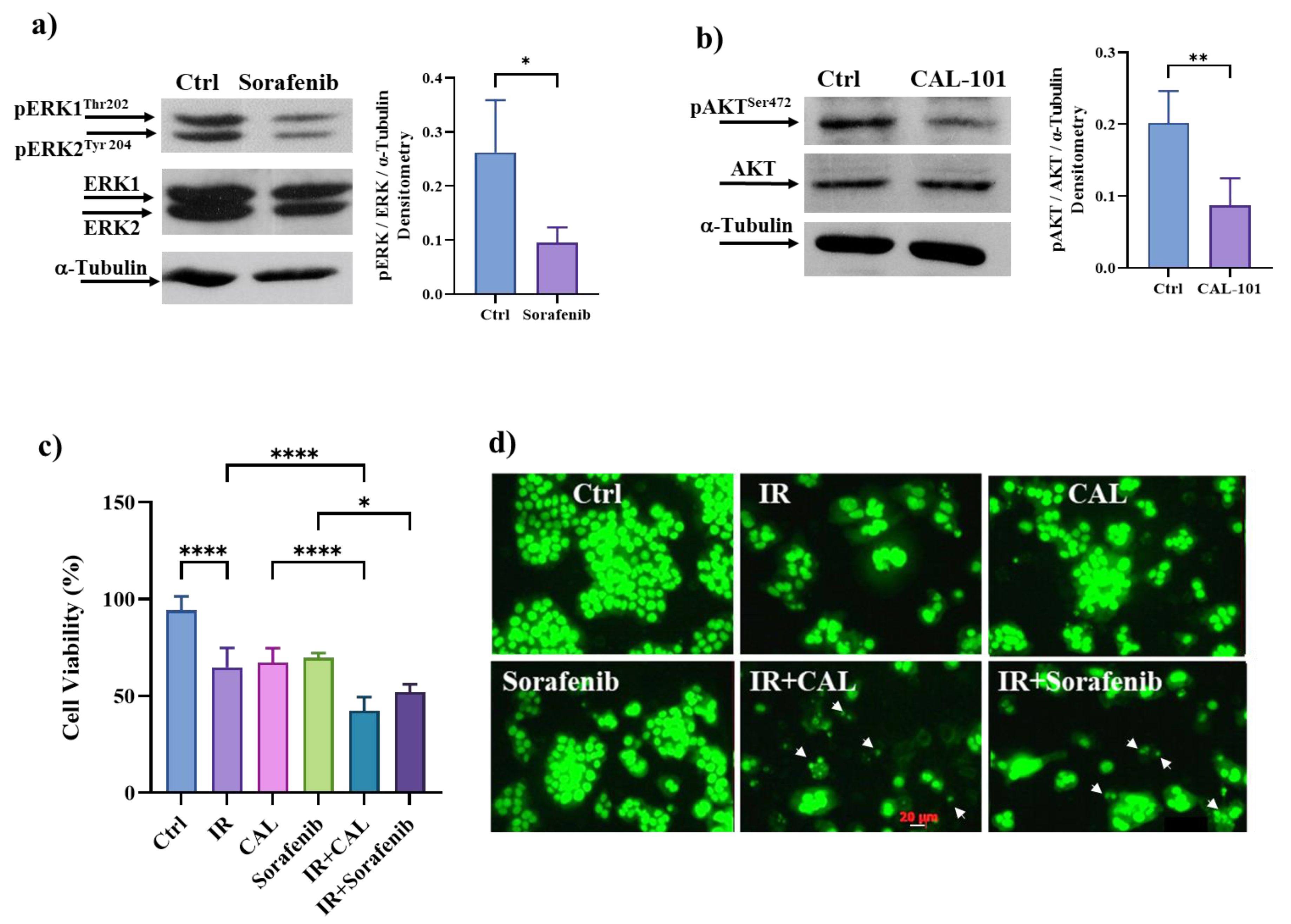
3.9. Pharmacological Inhibition of ERK/MAPKs or PI3K/AKT Signalling Pathways Induces Cell Death in Radioresistant Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Global Burden of Disease Cancer Collaboration. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived with Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2017: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2019, 5, 1749–1768. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Kroemer, G. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Russo, G.L.; Spagnuolo, C.; Russo, M.; Tedesco, I.; Moccia, S.; Cervellera, C. Mechanisms of aging and potential role of selected polyphenols in extending healthspan. Biochem. Pharmacol. 2020, 173, 113719. [Google Scholar] [CrossRef]
- Patel, N.H.; Bloukh, S.; Alwohosh, E.; Alhesa, A.; Saleh, T.; Gewirtz, D.A. Autophagy and senescence in cancer therapy. Adv. Cancer Res. 2021, 150, 1–74. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.H.; Sohal, S.S.; Manjili, M.H.; Harrell, J.C.; Gewirtz, D.A. The Roles of Autophagy and Senescence in the Tumor Cell Response to Radiation. Radiat. Res. 2020, 194, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B. Autophagy in Human Diseases. N. Engl. J. Med. 2020, 383, 1564–1576. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. The ATG conjugation systems in autophagy. Curr. Opin. Cell Biol. 2020, 63, 1–10. [Google Scholar] [CrossRef]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef]
- Russo, G.L.; Russo, M.; Ungaro, P. AMP-activated protein kinase: A target for old drugs against diabetes and cancer. Biochem. Pharmacol. 2013, 86, 339–350. [Google Scholar] [CrossRef] [PubMed]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, Metabolism, and Cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef] [PubMed]
- Poillet-Perez, L.; Despouy, G.; Delage-Mourroux, R.; Boyer-Guittaut, M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 2015, 4, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Reddel, R.R. Senescence: An antiviral defense that is tumor suppressive? Carcinogenesis 2010, 31, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.L.; Sierra, F.; Howcroft, K.; Singer, D.S.; Sharpless, N.; Hodes, R.J.; Wilder, E.L.; Anderson, J.M. A Blueprint for Characterizing Senescence. Cell 2020, 183, 1143–1146. [Google Scholar] [CrossRef]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef]
- Collado, M.; Gil, J.; Efeyan, A.; Guerra, C.; Schuhmacher, A.J.; Barradas, M.; Benguria, A.; Zaballos, A.; Flores, J.M.; Barbacid, M.; et al. Tumour biology: Senescence in premalignant tumours. Nature 2005, 436, 642. [Google Scholar] [CrossRef]
- Russo, G.L.; Moccia, S.; Russo, M.; Spagnuolo, C. Redox regulation by carotenoids: Evidence and conflicts for their application in cancer. Biochem. Pharmacol. 2021, 194, 114838. [Google Scholar] [CrossRef]
- Musial, C.; Siedlecka-Kroplewska, K.; Kmiec, Z.; Gorska-Ponikowska, M. Modulation of Autophagy in Cancer Cells by Dietary Polyphenols. Antioxidants 2021, 10, 123. [Google Scholar] [CrossRef]
- Russo, M.; Russo, G.L. Autophagy inducers in cancer. Biochem. Pharmacol. 2018, 153, 51–61. [Google Scholar] [CrossRef]
- Pang, X.; Zhang, X.; Jiang, Y.; Su, Q.; Li, Q.; Li, Z. Autophagy: Mechanisms and Therapeutic Potential of Flavonoids in Cancer. Biomolecules 2021, 11, 135. [Google Scholar] [CrossRef]
- Sellares, J.; Rojas, M. Quercetin in Idiopathic Pulmonary Fibrosis: Another Brick in the Senolytic Wall. Am. J. Respir. Cell Mol. Biol. 2019, 60, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Wang, B.; Shi, Y.; Xie, C.; Huang, C.; Chen, B.; Zhang, H.; Zeng, G.; Liang, H.; Wu, Y.; et al. Senolytic agent Quercetin ameliorates intervertebral disc degeneration via the Nrf2/NF-κB axis. Osteoarthr. Cartil. 2021, 29, 413–422. [Google Scholar] [CrossRef]
- Short, S.; Fielder, E.; Miwa, S.; von Zglinicki, T. Senolytics and senostatics as adjuvant tumour therapy. eBioMedicine 2019, 41, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Spagnuolo, C.; Tedesco, I.; Bilotto, S.; Russo, G.L. The flavonoid quercetin in disease prevention and therapy: Facts and fancies. Biochem. Pharmacol. 2012, 83, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Lall, R.K.; Adhami, V.M.; Mukhtar, H. Dietary flavonoid fisetin for cancer prevention and treatment. Mol. Nutr. Food Res. 2016, 60, 1396–1405. [Google Scholar] [CrossRef]
- Russo, M.; Spagnuolo, C.; Tedesco, I.; Russo, G.L. Phytochemicals in cancer prevention and therapy: Truth or dare? Toxins 2010, 2, 517–551. [Google Scholar] [CrossRef]
- Cervellera, C.; Russo, M.; Dotolo, S.; Facchiano, A.; Russo, G.L. STL1, a New AKT Inhibitor, Synergizes with Flavonoid Quercetin in Enhancing Cell Death in A Chronic Lymphocytic Leukemia Cell Line. Molecules 2021, 26, 5810. [Google Scholar] [CrossRef]
- Granato, M.; Rizzello, C.; Romeo, M.A.; Yadav, S.; Santarelli, R.; D’Orazi, G.; Faggioni, A.; Cirone, M. Concomitant reduction of c-Myc expression and PI3K/AKT/mTOR signaling by quercetin induces a strong cytotoxic effect against Burkitt’s lymphoma. Int. J. Biochem. Cell Biol. 2016, 79, 393–400. [Google Scholar] [CrossRef]
- Lewinska, A.; Adamczyk-Grochala, J.; Bloniarz, D.; Olszowka, J.; Kulpa-Greszta, M.; Litwinienko, G.; Tomaszewska, A.; Wnuk, M.; Pazik, R. AMPK-mediated senolytic and senostatic activity of quercetin surface functionalized Fe3O4 nanoparticles during oxidant-induced senescence in human fibroblasts. Redox Biol. 2020, 28, 101337. [Google Scholar] [CrossRef]
- Khan, N.; Jajeh, F.; Eberhardt, E.L.; Miller, D.D.; Albrecht, D.M.; Van Doorn, R.; Hruby, M.D.; Maresh, M.E.; Clipson, L.; Mukhtar, H.; et al. Fisetin and 5-fluorouracil: Effective combination for PIK3CA-mutant colorectal cancer. Int. J. Cancer 2019, 145, 3022–3032. [Google Scholar] [CrossRef] [PubMed]
- Chamcheu, J.C.; Esnault, S.; Adhami, V.M.; Noll, A.L.; Banang-Mbeumi, S.; Roy, T.; Singh, S.S.; Huang, S.; Kousoulas, K.G.; Mukhtar, H. Fisetin, a 3,7,3′,4′-Tetrahydroxyflavone Inhibits the PI3K/Akt/mTOR and MAPK Pathways and Ameliorates Psoriasis Pathology in 2D and 3D Organotypic Human Inflammatory Skin Models. Cells 2019, 8, 1089. [Google Scholar] [CrossRef]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. eBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef]
- Hohmann, M.S.; Habiel, D.M.; Coelho, A.L.; Verri, W.A., Jr.; Hogaboam, C.M. Quercetin Enhances Ligand-induced Apoptosis in Senescent Idiopathic Pulmonary Fibrosis Fibroblasts and Reduces Lung Fibrosis In Vivo. Am. J. Respir. Cell Mol. Biol. 2019, 60, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. eBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T. Senolytic drugs: From discovery to translation. J. Intern. Med. 2020, 288, 518–536. [Google Scholar] [CrossRef]
- Russo, M.; Spagnuolo, C.; Moccia, S.; Tedesco, I.; Lauria, F.; Russo, G.L. Biochemical and Cellular Characterization of New Radio-Resistant Cell Lines Reveals a Role of Natural Flavonoids to Bypass Senescence. Int. J. Mol. Sci. 2021, 23, 301. [Google Scholar] [CrossRef]
- Von Kleist, S.; Chany, E.; Burtin, P.; King, M.; Fogh, J. Immunohistology of the antigenic pattern of a continuous cell line from a human colon tumor. J. Natl. Cancer Inst. 1975, 55, 555–560. [Google Scholar] [CrossRef]
- Feoktistova, M.; Geserick, P.; Leverkus, M. Crystal Violet Assay for Determining Viability of Cultured Cells. Cold Spring Harb. Protoc. 2016, 2016, pdb.prot087379. [Google Scholar] [CrossRef] [PubMed]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef]
- Russo, M.; Moccia, S.; Bilotto, S.; Spagnuolo, C.; Durante, M.; Lenucci, M.S.; Mita, G.; Volpe, M.G.; Aquino, R.P.; Russo, G.L. A Carotenoid Extract from a Southern Italian Cultivar of Pumpkin Triggers Nonprotective Autophagy in Malignant Cells. Oxidative Med. Cell. Longev. 2017, 2017, 7468538. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.; Abdel-Aziz, A.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Moccia, S.; Spagnuolo, C.; Tedesco, I.; Russo, G.L. Carotenoid-Enriched Nanoemulsions and gamma-Rays Synergistically Induce Cell Death in a Novel Radioresistant Osteosarcoma Cell Line. Int. J. Mol. Sci. 2022, 23, 15959. [Google Scholar] [CrossRef] [PubMed]
- Gewirtz, D.A. The four faces of autophagy: Implications for cancer therapy. Cancer Res. 2014, 74, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Milito, A.; Spagnuolo, C.; Carbone, V.; Rosen, A.; Minasi, P.; Lauria, F.; Russo, G.L. CK2 and PI3K are direct molecular targets of quercetin in chronic lymphocytic leukaemia. Oncotarget 2017, 8, 42571–42587. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Zhang, Y.; Liu, Y.; Zhou, M.; Lu, Y.; Yuan, L.; Zhang, C.; Hong, M.; Wang, S.; Li, X. MiR-106b induces cell radioresistance via the PTEN/PI3K/AKT pathways and p21 in colorectal cancer. J. Transl. Med. 2015, 13, 252. [Google Scholar] [CrossRef] [PubMed]
- Ngoi, N.Y.; Liew, A.Q.; Chong, S.J.F.; Davids, M.S.; Clement, M.V.; Pervaiz, S. The redox-senescence axis and its therapeutic targeting. Redox Biol. 2021, 45, 102032. [Google Scholar] [CrossRef]
- Reyes-Farias, M.; Carrasco-Pozo, C. The Anti-Cancer Effect of Quercetin: Molecular Implications in Cancer Metabolism. Int. J. Mol. Sci. 2019, 20, 3177. [Google Scholar] [CrossRef]
- Shui, L.; Wang, W.; Xie, M.; Ye, B.; Li, X.; Liu, Y.; Zheng, M. Isoquercitrin induces apoptosis and autophagy in hepatocellular carcinoma cells via AMPK/mTOR/p70S6K signaling pathway. Aging 2020, 12, 24318–24332. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cao, L.; Xu, G.; He, H.; Zhao, H.; Liu, T. Co-delivery of sorafenib and metformin from amphiphilic polypeptide-based micelles for colon cancer treatment. Front. Med. 2022, 9, 1009496. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhu, Z.; Zhang, X.; Zhang, N.; Yao, Z. Idelalisib induces PUMA-dependent apoptosis in colon cancer cells. Oncotarget 2017, 8, 6102–6113. [Google Scholar] [CrossRef]
- Braithwaite, D.; Anton, S.; Mohile, S.; DeGregori, J.; Gillis, N.; Zhou, D.; Bloodworth, S.; Pahor, M.; Licht, J. Cancer and aging: A call to action. Aging Cancer 2022, 3, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Nam, H.Y.; Kang, H.B.; Lee, W.H.; Lee, G.H.; Sung, G.J.; Han, M.W.; Cho, K.J.; Chang, E.J.; Choi, K.C.; et al. Epigenetic regulation of p62/SQSTM1 overcomes the radioresistance of head and neck cancer cells via autophagy-dependent senescence induction. Cell Death Dis. 2021, 12, 250. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, P.G.; Citrin, D.E.; Hildesheim, J.; Ahmed, M.M.; Venkatachalam, S.; Riscuta, G.; Xi, D.; Zheng, G.; van Deursen, J.; Goronzy, J.; et al. Therapy-Induced Senescence: Opportunities to Improve Anti-Cancer Therapy. J. Natl. Cancer Inst. 2021, 113, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Gewirtz, D.A. An autophagic switch in the response of tumor cells to radiation and chemotherapy. Biochem. Pharmacol. 2014, 90, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Gewirtz, D.A. The Switch between Protective and Nonprotective Autophagy; Implications for Autophagy Inhibition as a Therapeutic Strategy in Cancer. Biology 2020, 9, 12. [Google Scholar] [CrossRef]
- Yu, S.H.; Yang, P.M.; Peng, C.W.; Yu, Y.C.; Chiu, S.J. Securin depletion sensitizes human colon cancer cells to fisetin-induced apoptosis. Cancer Lett. 2011, 300, 96–104. [Google Scholar] [CrossRef]
- Leu, J.D.; Wang, B.S.; Chiu, S.J.; Chang, C.Y.; Chen, C.C.; Chen, F.D.; Avirmed, S.; Lee, Y.J. Combining fisetin and ionizing radiation suppresses the growth of mammalian colorectal cancers in xenograft tumor models. Oncol. Lett. 2016, 12, 4975–4982. [Google Scholar] [CrossRef]
- Suh, Y.; Afaq, F.; Johnson, J.J.; Mukhtar, H. A plant flavonoid fisetin induces apoptosis in colon cancer cells by inhibition of COX2 and Wnt/EGFR/NF-κB-signaling pathways. Carcinogenesis 2009, 30, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Syed, D.N.; Adhami, V.M.; Khan, N.; Khan, M.I.; Mukhtar, H. Exploring the molecular targets of dietary flavonoid fisetin in cancer. Semin. Cancer Biol. 2016, 40–41, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Adhami, V.M.; Syed, D.N.; Khan, N.; Mukhtar, H. Dietary flavonoid fisetin: A novel dual inhibitor of PI3K/Akt and mTOR for prostate cancer management. Biochem. Pharmacol. 2012, 84, 1277–1281. [Google Scholar] [CrossRef] [PubMed]
- Russo, G.L.; Russo, M.; Spagnuolo, C.; Tedesco, I.; Bilotto, S.; Iannitti, R.; Palumbo, R. Quercetin: A pleiotropic kinase inhibitor against cancer. Cancer Treat. Res. 2014, 159, 185–205. [Google Scholar] [CrossRef]
- Yuan, J.; Dong, X.; Yap, J.; Hu, J. The MAPK and AMPK signalings: Interplay and implication in targeted cancer therapy. J. Hematol. Oncol. 2020, 13, 113. [Google Scholar] [CrossRef]
- Ning, Y.; Lenz, H.J. Targeting IL-8 in colorectal cancer. Expert Opin. Ther. Targets 2012, 16, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jeon, M.; Lee, J.E.; Nam, S.J. MEK activity controls IL-8 expression in tamoxifen-resistant MCF-7 breast cancer cells. Oncol. Rep. 2016, 35, 2398–2404. [Google Scholar] [CrossRef]
- Walters, H.E.; Deneka-Hannemann, S.; Cox, L.S. Reversal of phenotypes of cellular senescence by pan-mTOR inhibition. Aging 2016, 8, 231–244. [Google Scholar] [CrossRef]
- Steinberg, G.R.; Hardie, D.G. New insights into activation and function of the AMPK. Nat. Rev. Mol. Cell Biol. 2022, 24, 255–272. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Z.; Huang, Y.; Zhou, Y.; Sheng, X.; Jiang, Q.; Wang, Y.; Luo, P.; Luo, M.; Shi, C. Senolytics (DQ) Mitigates Radiation Ulcers by Removing Senescent Cells. Front. Oncol. 2019, 9, 1576. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cellular/Biochemical Markers | HT29 | HT500 |
|---|---|---|
| EC50 IR (72 h) a | 21 Gy | 58 Gy *** |
| SA-βGAL basal activity b | 1716.8 ± 8.6 | 3540 ± 59 *** |
| Fisetin/Quercetin radio-sensitizing effect c | Not present | Synergic |
| (%) Intracellular peroxide after IR (5 min) d | >226% ± 92 | >119% ± 10 *** |
| (%) Intracellular GSH after IR (2 h) e | No increase | >24% ± 5 ** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russo, M.; Moccia, S.; Luongo, D.; Russo, G.L. Senolytic Flavonoids Enhance Type-I and Type-II Cell Death in Human Radioresistant Colon Cancer Cells through AMPK/MAPK Pathway. Cancers 2023, 15, 2660. https://doi.org/10.3390/cancers15092660
Russo M, Moccia S, Luongo D, Russo GL. Senolytic Flavonoids Enhance Type-I and Type-II Cell Death in Human Radioresistant Colon Cancer Cells through AMPK/MAPK Pathway. Cancers. 2023; 15(9):2660. https://doi.org/10.3390/cancers15092660
Chicago/Turabian StyleRusso, Maria, Stefania Moccia, Diomira Luongo, and Gian Luigi Russo. 2023. "Senolytic Flavonoids Enhance Type-I and Type-II Cell Death in Human Radioresistant Colon Cancer Cells through AMPK/MAPK Pathway" Cancers 15, no. 9: 2660. https://doi.org/10.3390/cancers15092660