
Sensitization of Resistant Cells with a BET Bromodomain Inhibitor in a Cell Culture Model of Deep Intrinsic Resistance in Breast Cancer
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Drugs
2.2. JQ1 Treatment of Resistant Cancer Cells
2.3. Assays of Relative Resistance of Cells to Paclitaxel and Doxorubicin
2.4. Western Blotting
3. Results
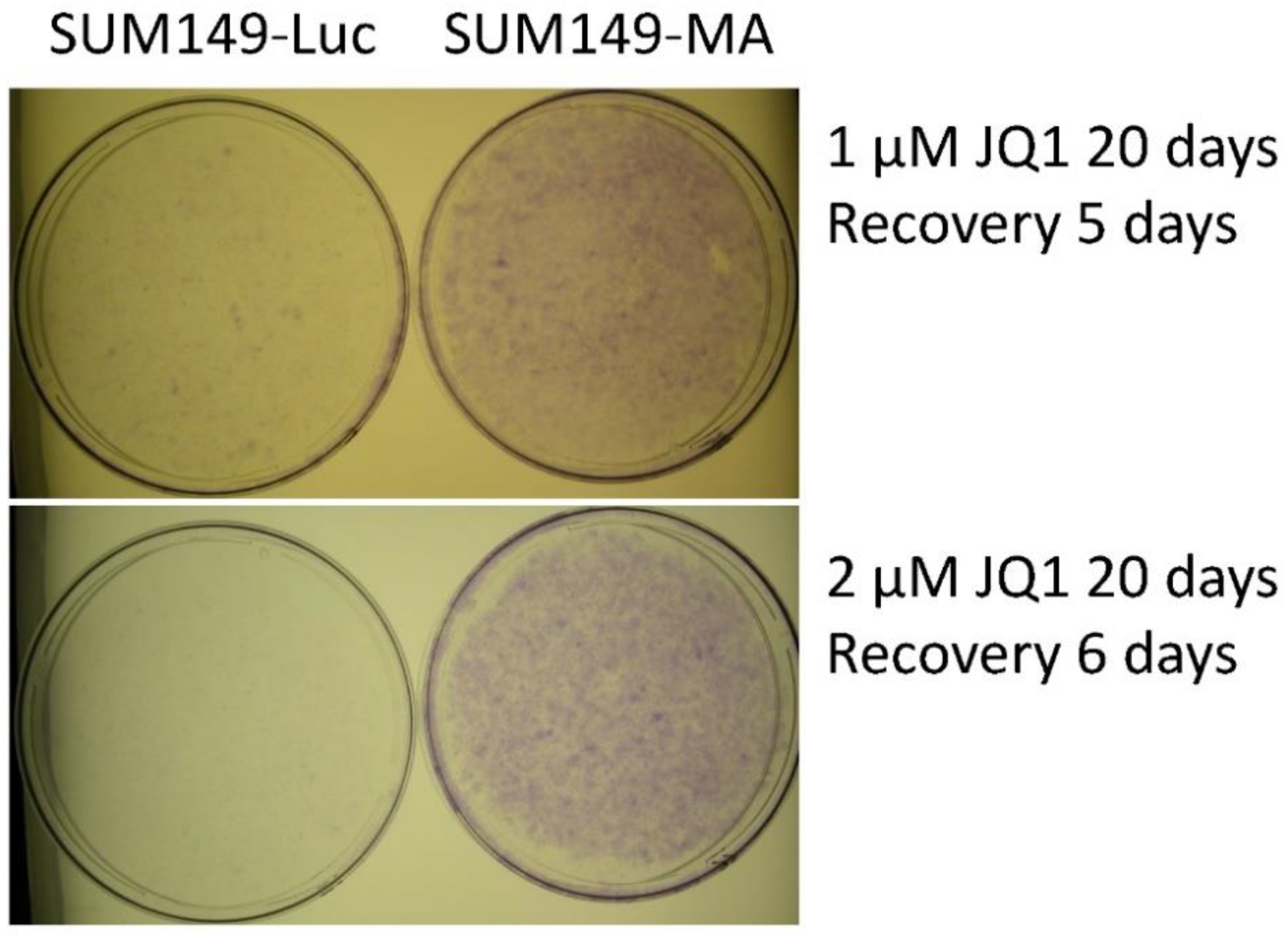
3.1. JQ1 Resistance of SUM149-MA Cells
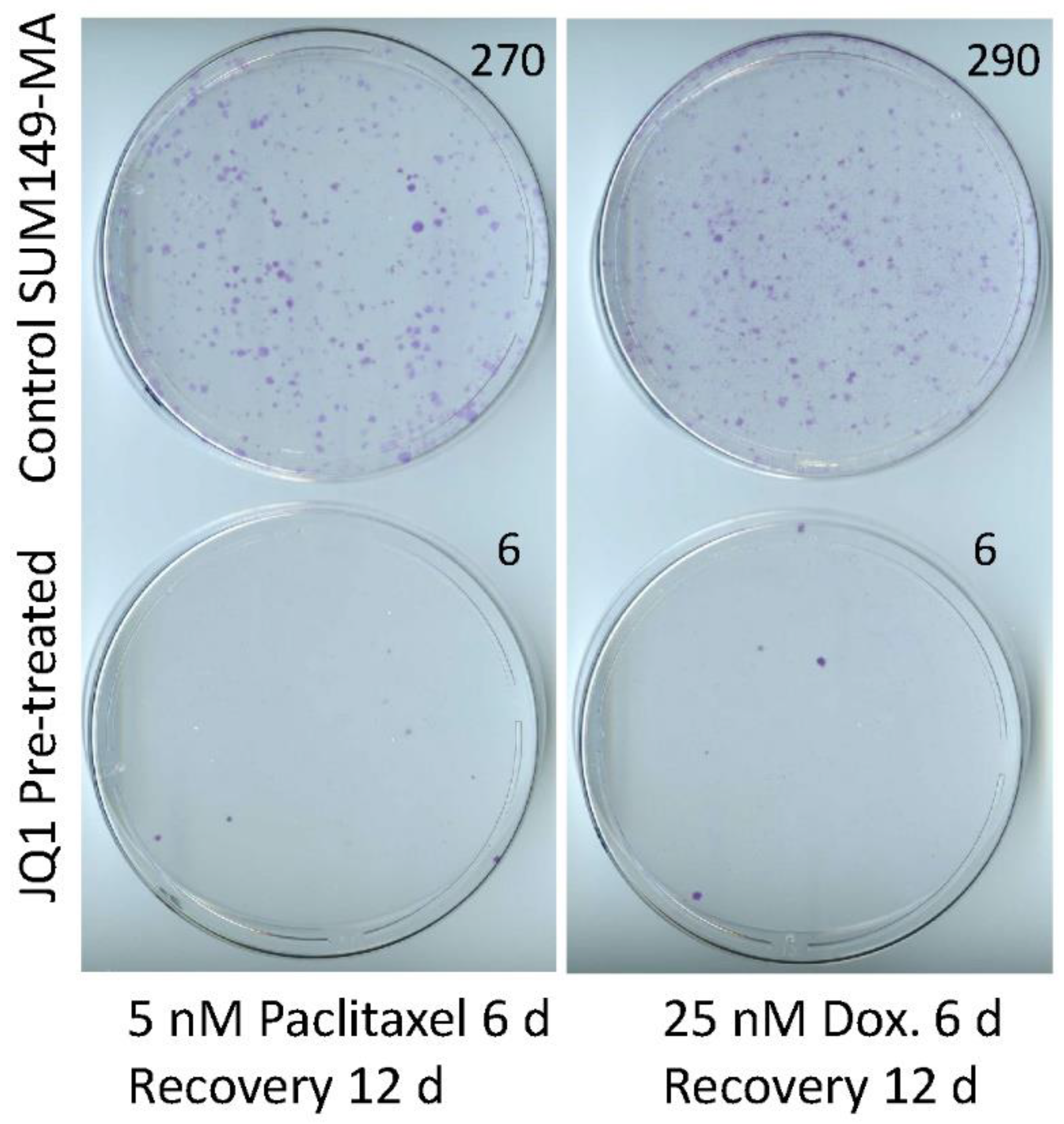
3.2. Sensitization of SUM149-MA Cells to Chemotherapeutic Drugs by Treatment with JQ1
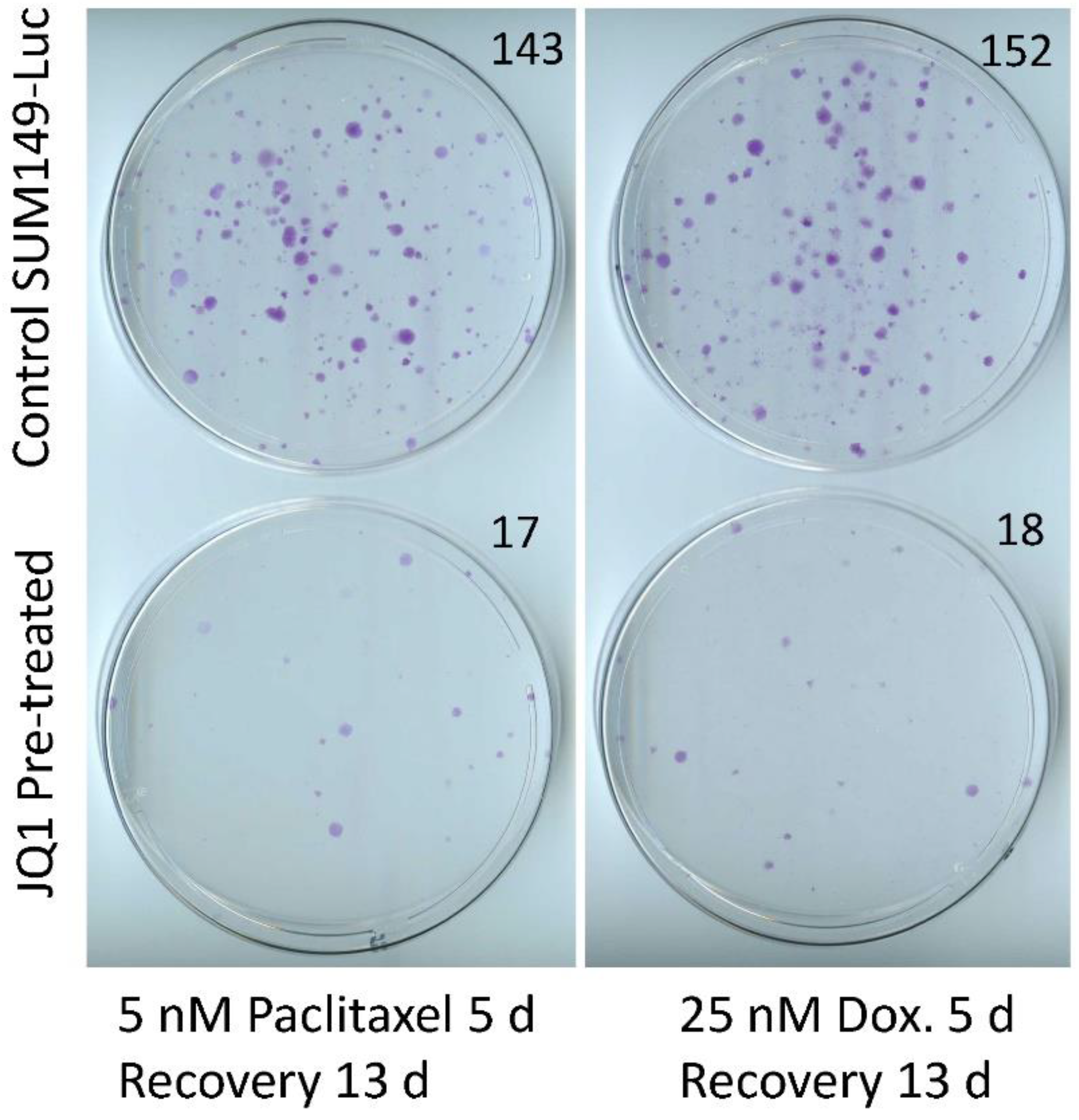
3.3. Sensitization of SUM149-Luc Cells to Chemotherapeutic Drugs by Treatment with JQ1
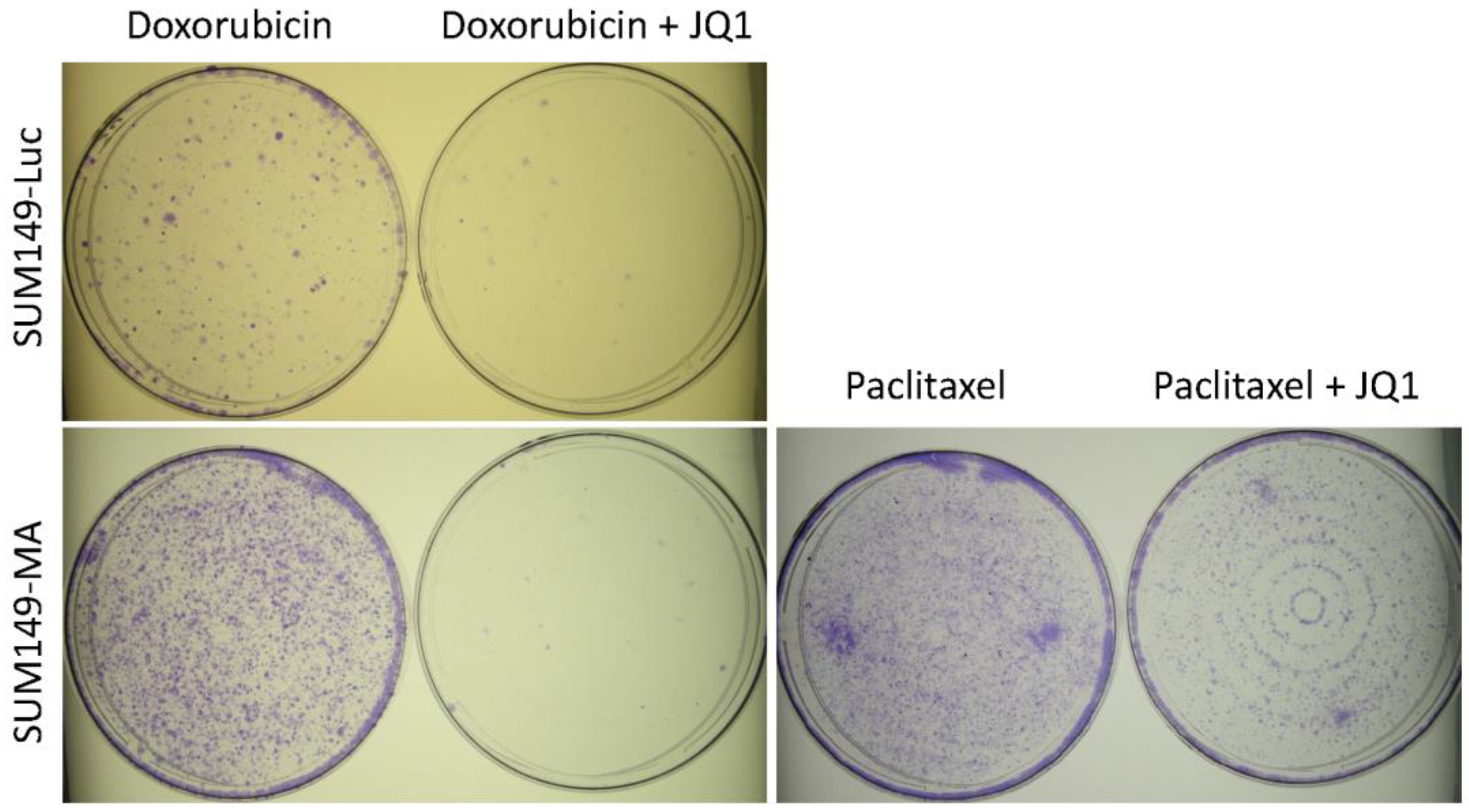
3.4. Combination with JQ1 Enhanced Efficacy of Chemotherapeutic Drugs against Resistant Cancer Cells
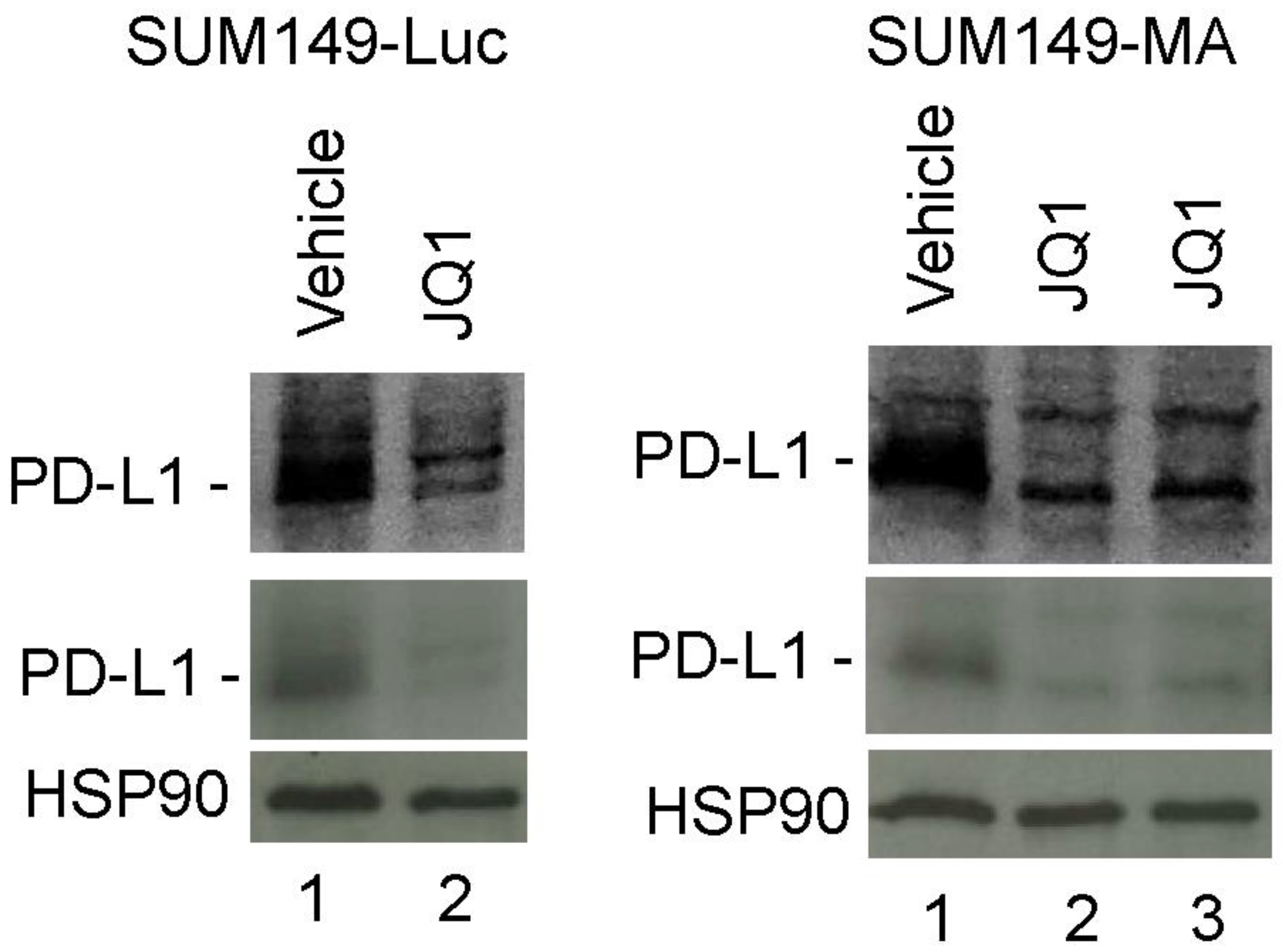
3.5. Decreased PD-L1 Expression in Cancer Cells upon Treatment with JQ1
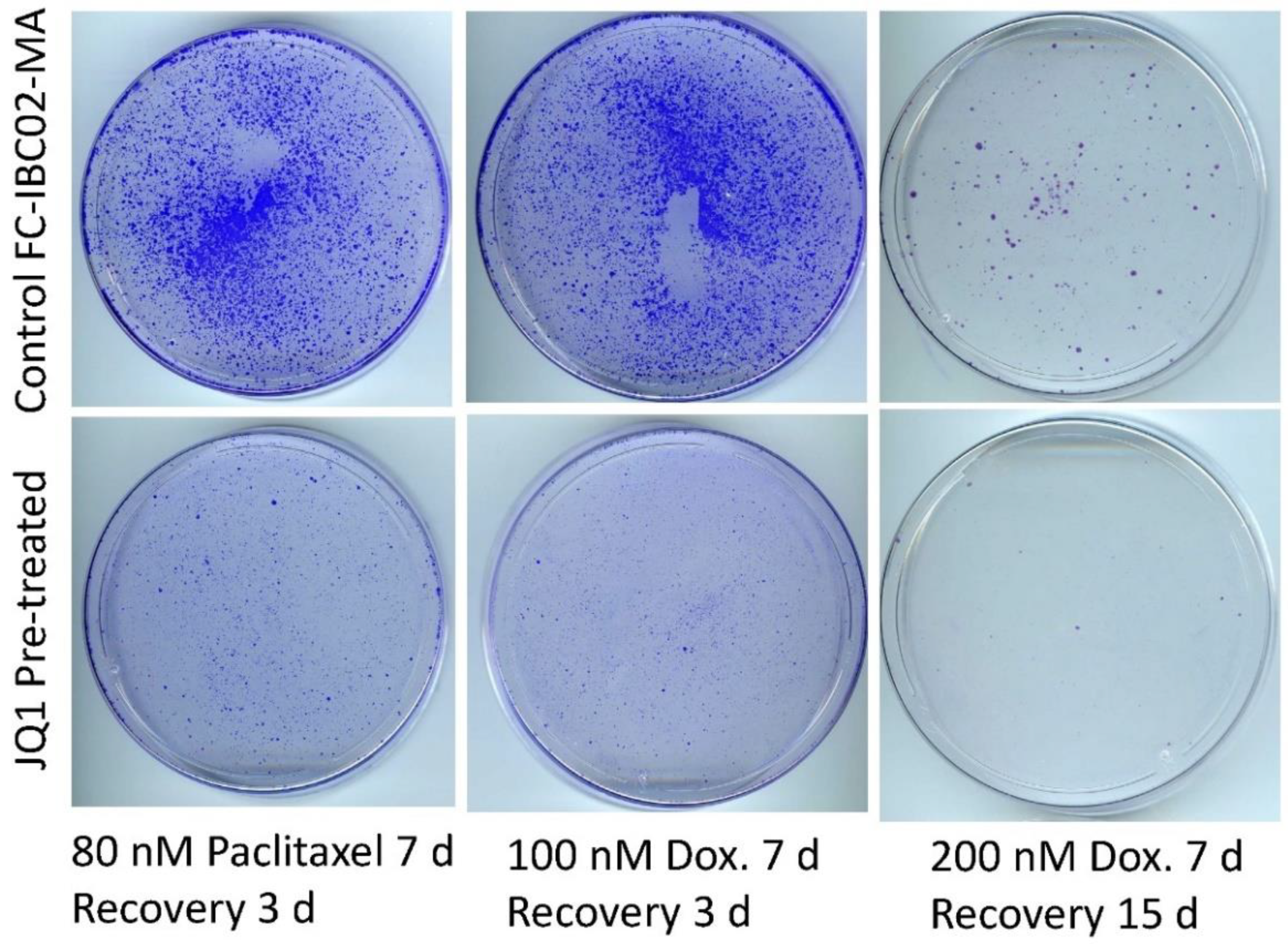
3.6. Sensitization of FC-IBC02-MA Cells to Chemotherapeutic Drugs by Treatment with JQ1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kummel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef]
- Braun, S.; Vogl, F.D.; Naume, B.; Janni, W.; Osborne, M.P.; Coombes, R.C.; Schlimok, G.; Diel, I.J.; Gerber, B.; Gebauer, G.; et al. A pooled analysis of bone marrow micrometastasis in breast cancer. N. Engl. J. Med. 2005, 353, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Gianni, L.; Huang, C.S.; Egle, D.; Bermejo, B.; Zamagni, C.; Thill, M.; Anton, A.; Zambelli, S.; Bianchini, G.; Russo, S.; et al. Pathologic complete response (pCR) to neoadjuvant treatment with or without atezolizumab in triple-negative, early high-risk and locally advanced breast cancer: NeoTRIP Michelangelo randomized study. Ann. Oncol. 2022, 33, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Janni, W.J.; Rack, B.; Terstappen, L.W.; Pierga, J.Y.; Taran, F.A.; Fehm, T.; Hall, C.; de Groot, M.R.; Bidard, F.C.; Friedl, T.W.; et al. Pooled Analysis of the Prognostic Relevance of Circulating Tumor Cells in Primary Breast Cancer. Clin. Cancer Res. 2016, 22, 2583–2593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavrommati, I.; Johnson, F.; Echeverria, G.V.; Natrajan, R. Subclonal heterogeneity and evolution in breast cancer. NPJ Breast Cancer 2021, 7, 155. [Google Scholar] [CrossRef]
- Minussi, D.C.; Nicholson, M.D.; Ye, H.; Davis, A.; Wang, K.; Baker, T.; Tarabichi, M.; Sei, E.; Du, H.; Rabbani, M.; et al. Breast tumours maintain a reservoir of subclonal diversity during expansion. Nature 2021, 592, 302–308. [Google Scholar] [CrossRef]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Somarelli, J.A.; Gardner, H.; Cannataro, V.L.; Gunady, E.F.; Boddy, A.M.; Johnson, N.A.; Fisk, J.N.; Gaffney, S.G.; Chuang, J.H.; Li, S.; et al. Molecular Biology and Evolution of Cancer: From Discovery to Action. Mol. Biol. Evol. 2020, 37, 320–326. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Wolman, S.R.; Fidler, I.J. Evidence for the clonal origin of spontaneous metastases. Science 1982, 217, 361–363. [Google Scholar] [CrossRef]
- Singh, B.; Sarli, V.N.; Washburn, L.J.; Raythatha, M.R.; Lucci, A. A usable model of “decathlon winner” cancer cells in triple-negative breast cancer: Survival of resistant cancer cells in quiescence. Oncotarget 2018, 9, 11071–11082. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.; Tai, K.; Madan, S.; Raythatha, M.R.; Cady, A.M.; Braunlin, M.; Irving, L.R.; Bajaj, A.; Lucci, A. Selection of metastatic breast cancer cells based on adaptability of their metabolic state. PLoS ONE 2012, 7, e36510. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.; Shamsnia, A.; Raythatha, M.R.; Milligan, R.D.; Cady, A.M.; Madan, S.; Lucci, A. Highly adaptable triple-negative breast cancer cells as a functional model for testing anticancer agents. PLoS ONE 2014, 9, e109487. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Kinne, H.E.; Milligan, R.D.; Washburn, L.J.; Olsen, M.; Lucci, A. Important Role of FTO in the Survival of Rare Panresistant Triple-Negative Inflammatory Breast Cancer Cells Facing a Severe Metabolic Challenge. PLoS ONE 2016, 11, e0159072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, B.; Sarli, V.N.; Kinne, H.E.; Shamsnia, A.; Lucci, A. Evaluation of 6-mercaptopurine in a cell culture model of adaptable triple-negative breast cancer with metastatic potential. Oncotarget 2019, 10, 3681–3693. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.; Sarli, V.N.; Lucci, A. Inhibition of resistant triple-negative breast cancer cells with low-dose 6-mercaptopurine and 5-azacitidine. Oncotarget 2021, 12, 626–637. [Google Scholar] [CrossRef] [PubMed]
- Asangani, I.A.; Dommeti, V.L.; Wang, X.; Malik, R.; Cieslik, M.; Yang, R.; Escara-Wilke, J.; Wilder-Romans, K.; Dhanireddy, S.; Engelke, C.; et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014, 510, 278–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Z.; Gong, Y.; Ma, Y.; Lu, K.; Lu, X.; Pierce, L.A.; Thompson, R.C.; Muller, S.; Knapp, S.; Wang, J. Inhibition of BET bromodomain targets genetically diverse glioblastoma. Clin. Cancer Res. 2013, 19, 1748–1759. [Google Scholar] [CrossRef] [Green Version]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [Green Version]
- Leonard, B.; Brand, T.M.; O’Keefe, R.A.; Lee, E.D.; Zeng, Y.; Kemmer, J.D.; Li, H.; Grandis, J.R.; Bhola, N.E. BET Inhibition Overcomes Receptor Tyrosine Kinase-Mediated Cetuximab Resistance in HNSCC. Cancer Res. 2018, 78, 4331–4343. [Google Scholar] [CrossRef] [Green Version]
- Qi, J. Bromodomain and extraterminal domain inhibitors (BETi) for cancer therapy: Chemical modulation of chromatin structure. Cold Spring Harb. Perspect. Biol. 2014, 6, a018663. [Google Scholar] [CrossRef]
- Shimamura, T.; Chen, Z.; Soucheray, M.; Carretero, J.; Kikuchi, E.; Tchaicha, J.H.; Gao, Y.; Cheng, K.A.; Cohoon, T.J.; Qi, J.; et al. Efficacy of BET bromodomain inhibition in Kras-mutant non-small cell lung cancer. Clin. Cancer Res. 2013, 19, 6183–6192. [Google Scholar] [CrossRef] [Green Version]
- Feng, Q.; Zhang, Z.; Shea, M.J.; Creighton, C.J.; Coarfa, C.; Hilsenbeck, S.G.; Lanz, R.; He, B.; Wang, L.; Fu, X.; et al. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res. 2014, 24, 809–819. [Google Scholar] [CrossRef] [Green Version]
- Shu, S.; Lin, C.Y.; He, H.H.; Witwicki, R.M.; Tabassum, D.P.; Roberts, J.M.; Janiszewska, M.; Huh, S.J.; Liang, Y.; Ryan, J.; et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 2016, 529, 413–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risom, T.; Langer, E.M.; Chapman, M.P.; Rantala, J.; Fields, A.J.; Boniface, C.; Alvarez, M.J.; Kendsersky, N.D.; Pelz, C.R.; Johnson-Camacho, K.; et al. Differentiation-state plasticity is a targetable resistance mechanism in basal-like breast cancer. Nat. Commun. 2018, 9, 3815. [Google Scholar] [CrossRef] [Green Version]
- Jing, X.; Shao, S.; Zhang, Y.; Luo, A.; Zhao, L.; Zhang, L.; Gu, S.; Zhao, X. BRD4 inhibition suppresses PD-L1 expression in triple-negative breast cancer. Exp. Cell Res. 2020, 392, 112034. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Bengsch, F.; Svoronos, N.; Rutkowski, M.R.; Bitler, B.G.; Allegrezza, M.J.; Yokoyama, Y.; Kossenkov, A.V.; Bradner, J.E.; Conejo-Garcia, J.R.; et al. BET Bromodomain Inhibition Promotes Anti-tumor Immunity by Suppressing PD-L1 Expression. Cell Rep. 2016, 16, 2829–2837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, B.; Cook, K.R.; Martin, C.; Huang, E.H.; Mosalpuria, K.; Krishnamurthy, S.; Cristofanilli, M.; Lucci, A. Evaluation of a CXCR4 antagonist in a xenograft mouse model of inflammatory breast cancer. Clin. Exp. Metastasis 2010, 27, 233–240. [Google Scholar] [CrossRef]
- Fernandez, S.V.; Robertson, F.M.; Pei, J.; Aburto-Chumpitaz, L.; Mu, Z.; Chu, K.; Alpaugh, R.K.; Huang, Y.; Cao, Y.; Ye, Z.; et al. Inflammatory breast cancer (IBC): Clues for targeted therapies. Breast Cancer Res. Treat. 2013, 140, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.; Berry, J.A.; Shoher, A.; Ayers, G.D.; Wei, C.; Lucci, A. COX-2 involvement in breast cancer metastasis to bone. Oncogene 2007, 26, 3789–3796. [Google Scholar] [CrossRef] [Green Version]
- Dongre, A.; Rashidian, M.; Reinhardt, F.; Bagnato, A.; Keckesova, Z.; Ploegh, H.L.; Weinberg, R.A. Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res. 2017, 77, 3982–3989. [Google Scholar] [CrossRef] [Green Version]
- Horn, L.A.; Fousek, K.; Palena, C. Tumor Plasticity and Resistance to Immunotherapy. Trends Cancer 2020, 6, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Baldominos, P.; Barbera-Mourelle, A.; Barreiro, O.; Huang, Y.; Wight, A.; Cho, J.W.; Zhao, X.; Estivill, G.; Adam, I.; Sanchez, X.; et al. Quiescent cancer cells resist T cell attack by forming an immunosuppressive niche. Cell 2022, 185, 1694–1708.e19. [Google Scholar] [CrossRef] [PubMed]
- Winer, E.P.; Lipatov, O.; Im, S.A.; Goncalves, A.; Munoz-Couselo, E.; Lee, K.S.; Schmid, P.; Tamura, K.; Testa, L.; Witzel, I.; et al. Pembrolizumab versus investigator-choice chemotherapy for metastatic triple-negative breast cancer (KEYNOTE-119): A randomised, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 499–511. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, B.; Sarli, V.N.; Milligan, R.D.; Kinne, H.E.; Shamsnia, A.; Washburn, L.J.; Addanki, S.; Lucci, A. Sensitization of Resistant Cells with a BET Bromodomain Inhibitor in a Cell Culture Model of Deep Intrinsic Resistance in Breast Cancer. Cancers 2023, 15, 2036. https://doi.org/10.3390/cancers15072036
Singh B, Sarli VN, Milligan RD, Kinne HE, Shamsnia A, Washburn LJ, Addanki S, Lucci A. Sensitization of Resistant Cells with a BET Bromodomain Inhibitor in a Cell Culture Model of Deep Intrinsic Resistance in Breast Cancer. Cancers. 2023; 15(7):2036. https://doi.org/10.3390/cancers15072036
Chicago/Turabian StyleSingh, Balraj, Vanessa N. Sarli, Ryan D. Milligan, Hannah E. Kinne, Anna Shamsnia, Laura J. Washburn, Sridevi Addanki, and Anthony Lucci. 2023. "Sensitization of Resistant Cells with a BET Bromodomain Inhibitor in a Cell Culture Model of Deep Intrinsic Resistance in Breast Cancer" Cancers 15, no. 7: 2036. https://doi.org/10.3390/cancers15072036