
New Approaches to Targeting Epigenetic Regulation in Bladder Cancer

1
Department of Surgery, Austin Health, The University of Melbourne, Heidelberg, VIC 3084, Australia
2
Division of Cancer Surgery, Peter MacCallum Cancer Centre, The University of Melbourne, Melbourne, VIC 3000, Australia
3
Department of Urology, The Royal Melbourne Hospital, Melbourne, VIC 3050, Australia
4
EJ Whitten Prostate Cancer Research Centre at Epworth Healthcare, Melbourne, VC 3121, Australia
5
Olivia Newton-John Cancer and Wellness Centre, Austin Health, Melbourne, VIC 3084, Australia
*
Author to whom correspondence should be addressed.
Cancers 2023, 15(6), 1856; https://doi.org/10.3390/cancers15061856
Submission received: 28 February 2023
/
Revised: 14 March 2023
/
Accepted: 17 March 2023
/
Published: 20 March 2023
(This article belongs to the Topic Novel Approaches in Bladder Cancer Treatment)
Abstract
:Simple Summary
Epigenetic changes occur in parts of the genome other than in nucleotides. They are considered reversible and are therefore important targets for cancer therapy. Epigenetic changes have been observed in urological cancers, including urothelial carcinoma, and in recent years have been a topic of investigation for the treatment of metastatic bladder cancer that has failed traditional therapy. We performed a review of the current literature to assess the evidence and role for targeted epigenetic therapy in bladder cancer. While we found 25 clinical trials investigating this topic, there have been no phase 3 human clinical trials to date. This is an emerging topic in urology, and future directions involve further research into bladder cancer-specific epigenetic changes, as well as the development of novel agents to target these mutations.
Abstract
Epigenetics is a growing field and in bladder cancer, it is of particular interest in advanced or metastatic disease. As opposed to genetic mutations in which the nucleotide sequence itself is altered, epigenetic alterations refer to changes to the genome that do not involve nucleotides. This is of great interest in cancer research because epigenetic alterations are reversible, making them a promising target for pharmacological agents. While chemoimmunotherapy is the mainstay for metastatic disease, there are few alternatives for patients who have progressed on first- or second-line treatment. By targeting reversible epigenetic alterations, novel epigenetic therapies are important potential treatment options for these patients. A search of clinical registries was performed in order to identify and collate epigenetic therapies currently in human trials. A literature search was also performed to identify therapies that are currently in preclinical stages, whether this be in vivo or in vitro models. Twenty-five clinical trials were identified that investigated the use of epigenetic inhibitors in patients with bladder cancer, often in combination with another agent, such as platinum-based chemotherapy or pembrolizumab. The main classes of epigenetic inhibitors studied include DNA-methyltransferase (DNMT) inhibitors, histone deacetylase (HDAC) inhibitors, and histone methyltransferase (HMT) inhibitors. At present, no phase 3 clinical trials have been registered. Few trials have published results, though DNMT inhibitors have shown the most promise thus far. Many patients with advanced or metastatic bladder cancer have limited treatment options, particularly when first- or second-line chemoimmunotherapy fails. Epigenetic alterations, which are common in bladder cancer, are potential targets for drug therapies, and these epigenetic agents are already in use for many cancers. While they have shown promise in pre-clinical trials for bladder cancer, more research is needed to assess their benefit in clinical settings.
1. Introduction
Bladder cancer is a common urological malignancy with approximately 573,000 new diagnoses and 213,000 deaths reported worldwide in 2020, and among men, it ranks 6th for new cancer diagnoses and 9th for cancer deaths [1]. The 5-year survival rate for bladder cancer is reported as 54%, and even after curative-intent treatment with radical cystectomy, disease recurrence ensues in up to 30% [2,3]. Advanced or metastatic urothelial cancer of the bladder carries an even poorer prognosis with an overall survival of just over one year, even with first line chemotherapy and immunotherapy [4].
The current standard of care for patients with metastatic urothelial cancer depends on fitness for platinum. Platinum-fit patients typically receive cisplatin- or carboplatin-containing chemotherapy first-line, whereas platinum-unfit patients receive checkpoint inhibitors, such as pembrolizumab [5]. Patients who fail chemoimmunotherapy, however, have limited treatment options. For this reason, several novel agents are currently being studied in both pre-clinical and clinical trials. These include epigenetic inhibitors, which are drugs that target epigenetic alterations found in multiple human cancers. Epigenetic therapy is already established in the treatment of haematological malignancies and is increasingly being studied in urological cancers. In this review, we aim to summarise the current status of epigenetics and potential epigenetic treatments for bladder cancer.
2. Epigenetics and Bladder Cancer
Epigenetics is a term that was first used in 1940 to broadly describe anything relating to the process by which a genotype is expressed as a phenotype [6]. Today epigenetic alterations refer to acquired or heritable changes in a gene’s function that do not involve disruption of the nucleotide sequence [7]. This is in contrast to genetic mutations, in which the nucleotide sequences themselves are affected. Epigenetic alterations have been found in a variety of human malignancies and are thought to be reversible, making them promising targets for novel cancer therapies [8]. This is of particular interest in advanced or metastatic malignancies for which limited treatment options exist.
The enzymes responsible for epigenetic alterations can be categorised into three broad groups: writers, erasers, and readers. ‘Writers’ are proteins that add modifications, such as methyl or acetyl groups, to DNA or histones [9]. Common writers implicated in human cancers include DNA methyltransferases (DNMTs), histone-lysine N-methyltransferases (HMTs), and histone acetyltransferases (HATs). ‘Erasers’ directly oppose the action of writers by catalysing the removal of acetyl or methyl groups and include histone deacetylases (HDACs) [9]. The final group, ‘readers’, are proteins that are responsible for enacting the alterations made by writers and erasers [9].
2.1. Writers
2.1.1. DNA Methyltransferases (DNMTs)
DNA methylation, carried out by DNMTs, involves the addition of a methyl group to CpG sites, which is the region of DNA in which a cytosine nucleotide is followed by a guanine nucleotide (Figure 1) [10]. There are four types of DNMTs found in mammals, of which DNMT1 is the most abundant [11]. DNA methylation is essential for normal cell growth and development, imprinting, and X-chromosome inactivation [8]. Given that up to 80% of human CpG sites are methylated [10], any deviation from the normal process can have profound effects on gene expression. Aberrant methylation, whether it be hyper- or hypomethylation, has been observed in many cancers, particularly when it occurs in gene promoters, oncogenes, or tumour suppressor genes [8].
Patients with urothelial carcinoma of the bladder have been shown to have hypermethylation in multiple genes, with several studies even finding that the detection of hypermethylation in urine samples has higher sensitivity for diagnosing bladder cancer than traditional cytology [12,13]. There is also evidence to suggest that the degree of hypermethylation correlates with the aggressiveness of the cancer [14,15,16]. Hypermethylation in certain promoters has also been linked to tobacco smoking, a known risk factor for bladder cancer [15]. Tissue studies, such as that performed by Liu et al., have shown that DNMT1 is upregulated in bladder cancer samples compared to levels in the normal urothelium [17]. Here, the authors showed that silencing DNMT1 inhibited the growth and migration of tumour cells, whereas increasing DNMT1 expression had the opposite effect, confirming its role in bladder cancer [17].
2.1.2. Histone-Lysine N-Methyltransferases (HMTs)
HMTs are enzymes that catalyse the addition of methyl groups to histones, which are the proteins around which DNA is wound into nucleosomes [18]. Like DNA methylation, histone methylation also plays a crucial role in normal development and is involved in processes, including DNA replication and repair and gene transcription [19]. Aberrant histone methylation has been observed in many human cancers, and the most well-known aberrations occur in enhancer of zeste 2 (EZH2), which is thought to be the main enzyme involved in histone modification [20].
In urological oncology EZH2 is best known for its role in castrate-resistant prostate cancer (CRPC), in which its mutated form can activate genes involved in the androgen receptor pathway [21]. It has also been implicated in bladder cancer, with one study finding a correlation between EZH2 overexpression and non-muscle invasive bladder cancer (NMIBC) in both mouse models and humans, as well as an increased likelihood of disease recurrence in those with EZH2 overexpression [22]. Warrick et al. found that EZH2 expression was highest in bladder CIS, followed in descending order by muscle invasive bladder cancer (MIBC), NMIBC, and the normal epithelium, though they did not find an association with the oncological outcome [23].
Similar findings were observed by Chen et al., leading authors to conclude that not only is EZH2 associated with bladder cancer, but that its degree of expression correlates with disease severity [24]. EZH2 has also been found in urine through RNA released from cancer cells, and Zhang et al. report that its measurement can distinguish between MIBC and NMIBC and is a more sensitive diagnostic test than urine cytology [25].
2.1.3. Histone Acetyltransferases (HATs)
HATs also act on histones but catalyse the addition of acetyl groups rather than methyl groups. They are a diverse group of enzymes that play an important role in DNA repair and overall gene stability [26]. One protein complex demonstrating HAT activity, though not technically classified as a HAT, is the CREB-binding protein (CBP)/p300 coactivator family. CBP/p300 acetylates various oncoproteins and tumour-suppressor proteins, and thus, it is via these relationships that CBP/p300 abnormalities are associated with cancer [26].
While CBP/p300 mutations are more closely associated in haematological malignancies, they have also been found in many solid tumours, including colorectal, breast, ovarian, and pancreatic cancers [27]. Its impact on the androgen receptor pathway and subsequent association with prostate cancer is well known [28,29], though its role in bladder cancer is less clear. A 2011 study, however, noted that p300 is under-expressed in doxorubicin-resistant bladder cancer cells, generating interest in this class as a potential therapeutic target for chemotherapy-resistant cancers [30].
2.2. Erasers
2.2.1. Histone Deacetylases (HDACs)
HDACs are a group of enzymes that oppose the actions of HATs by catalysing the removal of acetyl groups from histones. This is a large group comprising 18 subtypes in humans, which are further divided into four classes [31].
Class I enzymes, which include HDAC1, 2, 3, and 8, are expressed in all cells, whereas the other three classes are more tissue-specific [32]. Just like their HAT counterparts, HDACs act on histones to regulate gene expression, apoptosis, and cell proliferation [31]. Inappropriate expression of these enzymes has been identified in haematological malignancies, such as acute myeloid leukaemia (AML) and non-Hodgkin’s lymphoma, as well in a range of solid tumours, including breast, cervical, colon, and prostate cancers [33].
More is known about the relationship between bladder cancer and class I HDACs, though enzymes from other classes have been studied as well [32]. Mutations in multiple HDAC genes have been identified in urothelial bladder cancers, leading to the overexpression of HDAC1 [34], HDAC2, and HDAC8, as well as the under-expression of HDAC5 and HDAC7 [35]. HDAC4’s association with bladder cancer is somewhat unclear, with one study finding it under-expressed in urothelial cancer [35] and another showing it as both overexpressed and associated with more severe disease [36]. Aberrations in HDAC1, in particular, are associated with poorer prognosis compared to other HDAC types [37].
HDAC3 microRNA in urine has also been shown to be significantly higher in patients with urothelial bladder cancer than in control patients [38]. HDACs have also been studied in non-urothelial bladder cancer, with one study finding HDAC4, HDAC7, and HDAC9 to be overexpressed in basal-squamous bladder cancer [39].
2.2.2. DNA Demethylases
DNA demethylases counteract DNMTs by removing a methyl group from the CpG sites. DNA hypomethylation was actually the first epigenetic alteration implicated in tumour development, though it was quickly overlooked as researchers shifted their focus to hypermethylation. Nonetheless, hypomethylation is a feature of several cancers, including ovarian epithelial carcinoma, metastatic prostate cancer, hepatocellular carcinoma, and colon adenocarcinoma [40]. In bladder cancer, hypomethylation has not been studied as thoroughly as hypermethylation, and most current research focuses on inducing hypomethylation to slow or reverse tumour growth. However, the upregulation of DNA demethylase has been noted in bladder cancer, although it appears to be linked to low-grade tumours, whereas hypermethylation is more common in high grade tumours [41].
2.2.3. Histone Demethylases (HDMs)
The addition of methyl groups to histones by methyltransferases was historically thought to be an irreversible process, and it was not until 2004 that the discovery of the first histone demethylase demonstrated that methyl groups could in fact be removed [42]. Histone demethylases (HDMs) are grouped by their mechanism of action and comprise two families: lysine-specific demethylases (LSDs) and Jumonji C-domain-containing demethylases (JMJDs), both of which are overexpressed in certain malignancies, such as prostate cancer [43]. While JMJDs appear protective against bladder cancer, LSD1 has been reported at higher levels in urothelial cancer compared to those in the normal urothelium [44]. Evidence on LSD1’s prognostic value is mixed, however, with some studies finding it linked to low grade but not high grade tumours [44], and others finding it significantly overexpressed in high-grade or metastatic cancers [45].
2.3. Readers
2.3.1. Bromodomain and Extraterminal Domain (BET)
The bromodomain and extraterminal domain (BET) family is a group of proteins that includes BRD2, BRD3, BRD4, and BRDT. While each protein performs a different function, they are all involved in the recognition of acetylated lysine residues on histones [46]. By ‘reading’ acetyl groups put down by HATs, BET proteins can regulate the activity and function of enzymes and proteins, thereby affecting gene transcription and chromatin remodelling [47].
One such type of gene directly regulated by BET proteins is the Myc family, which includes regulator genes and proto-oncogenes. Abnormal BET activity can result in the amplification or overexpression of Myc genes, resulting in tumorigenesis [48]. Myc genes are major players in haematological malignancies, such as Burkitt lymphoma, but are also important in many solid tumours, including neuroblastoma, melanoma, and breast cancer [48].
BRD4 is particularly relevant in urothelial bladder cancer and has been shown to be overexpressed in cancerous cells compared to levels in the normal urothelium, likely causing tumorigenesis through the Myc pathway [49]. Higher BRD4 expression has also been linked to a higher histological grade, as well as lymph node and distant metastasis [50].
2.3.2. Methyl-CpG-Binding Domain (MBD) Proteins
MBD proteins are a group of readers that are involved in the readout of DNA methylation, though they may also influence chromatin remodelling and histone methylation [51]. They are most well-known for their role in Rett syndrome, a neurodegenerative disorder caused by an X-linked mutation of the MBD gene MECP2, in which the gene loses its function [52]. Loss-of-function mutations in MBD protein genes have also been reported in human malignancies, including breast, lung, pancreas, colon, and prostate cancers [51]. Because MBD proteins help regulate DNA methylation, the absence of these proteins can cause DNA methylation to occur unchecked, which can be cancer-inducing as discussed previously. MBD2, a type of MBD protein, is thought to have a protective role against bladder cancer as demonstrated by Zhu et al., whose 2004 study reported that high expression of MBD2 is associated with a reduced risk of urothelial carcinoma [53].
3. Pre-Clinical and Clinical Trials
3.1. DNA Methyltransferases (DNMTs)
DNMT inhibitors are one of the few classes of epigenetic inhibitors approved by the American Food and Drug Association (FDA) for use in malignancy. These approved agents, azacytidine and decitabine, are currently only approved for use in myelodysplastic syndrome, though they have also been trialled for other malignancies, including breast and prostate [54]. Azacytidine acts on all DNMT classes, whereas decitabine acts preferentially on DNMT1.
To date, eight clinical trials have been performed using DNMT inhibitors in patients with advanced or metastatic urothelial bladder cancer, often in combination with platinum-based chemotherapy or pembrolizumab (Table 1). Three of these studies were phase I dose-finding trials, reporting that azacytidine and the novel agent guadecitabine were well-tolerated [55,56,57]. Only one phase 2 trial has been completed, and although it was terminated early due to slow accrual, progression-free survival was better than expected in urothelial cancer patients treated with the DNMT inhibitor 5-fluoro-2′-deoxycytidine [58]. There is currently one active phase 2 trial investigating guadecitabine in combination with atezolizumab, and it was expected to be completed in 2022 (NCT03179943). A phase 1/2 trial using NTX-301, an inhibitor targeting DNMT1 specifically, in combination with platinum-based chemotherapy, is currently recruiting (NCT04851834). A further studying using azacytidine was terminated early by sponsors, though further details were not provided (NCT02959437).
While human clinical trials have thus far administered azacytidine as a subcutaneous or intravenous injection, intravesical instillation has recently been investigated in a 2021 study using rat models, which reported a reduction in tumour burden using this method [61]. Other DNMT inhibitors that have not yet progressed to clinical trials include chromobox 7 (CBX7), which has been shown to cause tumour suppression in vitro [62], and CM-272, which can cause tumour and metastasis regression in in vivo transgenic mouse models [63].
3.2. Histone Methyltransferases (HMTs)
Inhibition of Enhancer of Zeste 2 (EZH2), a type of HMT, has been proven effective for metastatic or locally advanced epithelioid sarcoma and it is for this indication that the EZH2 inhibitor tazemetostat has been approved by the FDA [54]. Other EZH2 inhibitors are undergoing clinical trials to assess efficacy in myeloma, non-Hodgkin’s and B-cell lymphoma, and solid tumours, including breast, lung, and prostate, though none have progressed to phase 3 trials [64].
Few clinical trials have been conducted using EZH2 inhibitors in bladder cancer (Table 1), and the single completed trial has not reported results (NCT03525795). A phase 1/2 trial investigating tazemetostat in combination with pembrolizumab is currently recruiting patients with locally advanced or metastatic bladder cancer who have progressed on first-line chemotherapy (NCT03854474).
Despite the paucity of human clinical trials, there have been multiple studies published on EZH2 inhibitors using in vitro cell lines or animal models. The EZH2 inhibitor EPZ011989 has been reported to cause cell cycle arrest in in vitro assays and decreased tumour volume in xenograft mouse models [65]. UNC1999 is another EZH2 inhibitor that is observed to have an anti-tumorigenic effect on human bladder cancer cells [24]. EZH2 is also thought to have a direct relationship with lysine-specific demethylase 6A (LSD-6A), which is a histone demethylase and therefore performs the opposite function of EZH2. Several studies have reported that bladder cancers showing mutations in KDM6A, the gene coding for LSD-6A, are especially susceptible to EZH2 inhibition [65,66]. This is an interesting prospect given the rise of personal genomics as it suggests that particular genetic mutations may be more amenable to one kind of treatment than another.
3.3. Histone Acetyltransferases (HATs)
The inhibition of HAT-like CBP/p300 in cancer is less well-studied than other epigenetic targets with currently no approved agents in use. Clinical trials are also lacking with only three active currently and none completed. CCS1477 is a CBP/p300 inhibitor with phase 1/2 trials currently recruiting patients with haematological malignancy (NCT04068597), as well as advanced or metastatic solid tumours, including breast, lung, and prostate (NCT03568656). A phase 1 clinical trial using another CBP/p300 inhibitor, FT-7051, is also currently recruiting and is focusing on patients with metastatic CRPC (NCT04575766). To date, there are no past or current trials using CBP/p300 inhibitors in bladder cancer patients.
There is also a paucity of pre-clinical data investigating the use of CBP/p300 inhibitors in bladder cancer. A 2019 in vitro study had promising results, finding that CBP/p300 inhibition in bladder cancer cells led to decreased Myc expression, thereby increasing apoptosis and reducing the proliferation of malignant cells [67]. However, CBP/p300 inhibition in this study was performed using the process of transfection rather than via the use of an epigenetic agent. Thus, further studies are needed to identify CBP/p300-inhibiting drugs and to test their effects on urothelial cancer.
3.4. Histone Deacetylases (HDACs)
HDAC inhibitors are a well-studied group and make up three of the seven currently approved epigenetic agents, all of which are in use for T-cell lymphoma [54]. Vorinostat, also known as suberoylanilide hydroxamic acid or SAHA, was the first epigenetic agent approved by the FDA and is used mainly for refractory cutaneous T-cell lymphoma (CTCL) [54]. It also has shown an effect in clinical trials investigating lymphoma, lung cancer, breast cancer, head and neck carcinoma, and colorectal and prostate cancers [66]. As an HDAC inhibitor, its primary effect is on acetylation, but in vitro studies have shown that it may also have some action on methylation and miRNA expression in certain cancers [66].
Thus far, vorinostat has not yielded particularly promising results in human bladder cancer trials. A phase 1 trial combining vorinostat with docetaxel was terminated in 2008 due to toxicity (NCT00565227), and a phase 2 trial using it as a single agent was terminated due to both a lack of demonstrable efficacy, as well as intolerable side effects [59]. One trial using vorinostat as a single agent ran to completion in 2008, though results have not been published (NCT00045006). There is currently one active trial investigating vorinostat in combination with pembrolizumab in patients with metastatic bladder cancer who have failed chemotherapy, which was expected to finish in 2022 (NCT02619253).
Belinostat, or PXD101, is another FDA-approved HDAC inhibitor that is used for peripheral T-cell lymphoma (PTCL) [54]. It has also progressed to phase 2 trials for ovarian cancer [68], hepatocellular carcinoma [69], and AML [70]. In vivo urothelial cancer trials using animal models have reported promising results, including a reduced tumour volume, less haematuria, and slower disease progression, in mice treated with belinostat [71,72]. There have been three completed clinical trials using belinostat in patients with refractory bladder cancer, two of which have not reported results (NCT00413075, NCT00413322). The third, which combined belinostat with carboplatin or paclitaxel, has not published results but preliminary outcomes show at least a partial response in four of fifteen enrolled patients (NCT00421889). A further phase 1 trial investigating belinostat in combination with tremelimumab and durvalumab is currently recruiting (NCT05154994).
The third HDAC inhibitor that has been granted FDA approval is FK-228, or romidepsin, used for CTCL [54]. Romidepsin was initially isolated in 1994 from Chromobacterium violaceum, and although it displays minimal antibacterial properties, it was noted to have cytotoxic effects selectively on malignant cells [73]. In 2007, Karam et al. reported that in vitro treatment of bladder cancer cells with FK-228 caused tumour regression and also observed a similar finding in xenograft mice given intravenous doses of FK-228 [74]. To date, there have been two registered clinical trials using romidepsin in bladder cancer patients, one of which was terminated in 2006 due to poor accrual (NCT00087295). The other is currently active in phase 1 but has published dose-finding results, which showed that romidepsin is generally tolerable and has a similar toxicity profile to that of other HDAC inhibitors [60].
Valproic acid, a drug that is typically used to treat epilepsy and bipolar disorder, also has an inhibitory effect on HDACs. Given that valproate has been widely used for the management of neurological conditions, subsequent work on its HDAC-inhibiting properties focused on neurodegenerative disorders, such as Alzheimer’s disease, Huntington’s disease, and Parkinson’s disease [75]. It has also garnered attention in oncological settings, particularly in advanced cervical cancer where a double-blinded randomised controlled trial reported a significant improvement in PFS [76].
In urothelial cancer, multiple in vitro studies have shown that valproate is cytotoxic against human bladder cancer cells [77,78,79]. Intravesical instillation into rat bladder cancer models has also shown anti-tumorigenic properties, particularly when used in synergy with chemotherapy [79]. There have been two phase 1 clinical trials investigating valproate in bladder cancer patients, one of which is completed but without reported results (NCT01738815) and the other which is currently active (NCT01552434).
The last HDAC inhibitor involved in clinical trials for bladder cancer is entinostat. A 2020 in vitro study by Wang et al. showed that when used in combination with decitabine, a DNMT inhibitor, entinostat, exhibited a cytotoxic effect on chemoresistant bladder cancer cells without damaging normal urothelial cells [80]. A phase 2 trial of entinostat paired with pembrolizumab is currently underway, with results expected in late 2023 (NCT03978624).
3.5. Histone Methylation Readers
Several small-molecule BET inhibitors, including OTX015 and TEN-010, have progressed to clinical trials for haematological malignancy and select solid tumours, such as breast and prostate cancer [81]. Toxicity has been a major concern, however, and this is thought to be due to relatively poor selectivity. Further work into BET inhibition has yielded newer agents that target specific components of the bromodomain, such as ABBV-744, which acts primarily on bromodomain 2 [81]. While there have so far been no clinical trials using BET inhibitors in bladder cancer, a 2019 in vitro study by Li et al. reported the suppression of urothelial cancer cells by the BET inhibitor JQ1, an analogue of TEN-010 [82]. It has challenging pharmacokinetics due to its short half-life and poor bioavailability, however, which is perhaps a reason why it and so many other BET inhibitors have not progressed as far as other types of epigenetic agents.
4. Conclusions
There are few treatment options for patients with advanced or metastatic bladder cancer who have failed first-line chemoimmunotherapy, making this an important area for future research. Epigenetic agents, long in use for haematological malignancies, have also shown promise for the treatment of advanced solid tumours. In vitro and in vivo studies have repeatedly shown that epigenetic agents can be of benefit for bladder cancer, even with chemoresistant cells, but so far, these results have not been convincingly replicated in human clinical trials.
A recurrent issue limiting the clinical applicability of epigenetic therapies has been a lack of selectivity for urothelial cancer cells, leading to high toxicity and a lack of demonstrable efficacy. Combining these agents with established first- or second-line therapy, such as platin-based chemotherapy or pembrolizumab, is a common strategy to improve the side-effect profile as it usually allows for a lower dose of epigenetic inhibitors to be given. This may also increase efficacy as many epigenetic inhibitors act in synergy with chemoimmunotherapy. However, most epigenetic drugs trialled for bladder cancer were originally developed for other diseases or malignancies, such as lymphoma, and greater selectivity for malignant urothelial cells remains the goal of treatment at this time. Although further work is required to identify novel epigenetic targets and agents specifically for urothelial cancer, it is inevitable that we will see progress in this domain and likely the greater applicability of epigenetic therapies to urothelial cancer of the bladder in the future. Urologists should be aware of this class of agent and understand their derivation and potential to improve bladder cancer-specific survival in the future.
Author Contributions
Conceptualisation, N.L. and D.B.; Methodology, D.T. and D.B.; Validation, D.T.; Investigation, D.T.; Resources, N.L. and D.B.; Data Curation, D.T.; Writing—Original Draft Preparation, D.T.; Writing—Review & Editing, N.L. and D.B.; Visualisation, D.B.; Supervision, N.L. and D.B.; Project Administration, D.B.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors would like to acknowledge the Department of Urology at Austin Health, Melbourne Australia.
Conflicts of Interest
The authors report no conflict of interest.
Abbreviations
| BET | bromodomain and extraterminal domain |
| CBP | CREB-binding protein |
| DNMT | DNA methyltransferase |
| EZH2 | enhancer of zeste 2 |
| HAT | histone acetyltransferase |
| HDAC | histone deacetylase |
| HMT | histone-lysine N-methyltransferase |
| MBD | methyl-CpG-binding domain |
| MIBC | muscle invasive bladder cancer |
| NMIBC | non-muscle invasive bladder cancer |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Ca, N. 5-Year Relative Survival. Natl. Cancer Control Indic. Available online: https://ncci.canceraustralia.gov.au/outcomes/relative-survival-rate/5-year-relative-survival-diagnosis (accessed on 25 July 2022).
- Stein, J.P.; Lieskovsky, G.; Cote, R.; Groshen, S.; Feng, A.C.; Boyd, S.; Skinner, E.; Bochner, B.; Thangathurai, D.; Mikhail, M.; et al. Radical cystectomy in the treatment of invasive bladder cancer: Long-term results in 1054 patients. J. Clin. Oncol. 2001, 19, 666–675. [Google Scholar] [CrossRef]
- Von der Maase, H.; Sengelov, L.; Roberts, J.T.; Ricci, S.; Dogliotti, L.; Oliver, T.; Moore, M.J.; Zimmermann, A.; Arning, M. Long-term survival results of a randomized trial comparing gemcitabine plus cisplatin, with methotrexate, vinblastine, doxorubicin, plus cisplatin in patients with bladder cancer. J. Clin. Oncol. 2005, 23, 4602–4608. [Google Scholar] [CrossRef] [PubMed]
- Stenzl, A.; Cowan, N.C.; De Santis, M.; Jakse, G.; Kuczyk, M.A.; Merseburger, A.S.; Ribal, M.J.; Sherif, A.; Witjes, J.A. The updated EAU guidelines on muscle-invasive and metastatic bladder cancer. Eur. Urol. 2009, 55, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Waddington, C.H. The epigenotype. Endeavour 1942, 1, 18–20. [Google Scholar] [CrossRef] [Green Version]
- Dupont, C.; Armant, D.R.; Brenner, C.A. Epigenetics: Definition, mechanisms and clinical perspective. Semin. Reprod. Med. 2009, 27, 351–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadikovic, B.; Al-Romaih, K.; Squire, J.; Zielenska, M. Cause and consequences of genetic and epigenetic alterations in human cancer. Curr. Genom. 2008, 9, 394–408. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.; Rao, C.M. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur. J. Pharm. 2018, 837, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Jabbari, K.; Bernardi, G. Cytosine methylation and cpg, tpg (cpa) and tpa frequencies. Gene 2004, 333, 143–149. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, Y. Role of mammalian DNA methyltransferases in development. Annu. Rev. Biochem. 2020, 89, 135–158. [Google Scholar] [CrossRef]
- Chan, M.W.; Chan, L.W.; Tang, N.L.; Tong, J.H.; Lo, K.W.; Lee, T.L.; Cheung, H.Y.; Wong, W.S.; Chan, P.S.; Lai, F.M. Hypermethylation of multiple genes in tumor tissues and voided urine in urinary bladder cancer patients. Clin. Cancer Res. 2002, 8, 464–470. [Google Scholar]
- Dulaimi, E.; Uzzo, R.G.; Greenberg, R.E.; Al-Saleem, T.; Cairns, P. Detection of bladder cancer in urine by a tumor suppressor gene hypermethylation panel. Clin. Cancer Res. 2004, 10, 1887–1893. [Google Scholar] [CrossRef] [Green Version]
- Yates, D.R.; Rehman, I.; Abbod, M.F.; Meuth, M.; Cross, S.S.; Linkens, D.A.; Hamdy, F.C.; Catto, J.W. Promoter hypermethylation identifies progression risk in bladder cancer. Clin. Cancer Res. 2007, 13, 2046–2053. [Google Scholar] [CrossRef] [Green Version]
- Marsit, C.J.; Karagas, M.R.; Danaee, H.; Liu, M.; Andrew, A.; Schned, A.; Nelson, H.H.; Kelsey, K.T. Carcinogen exposure and gene promoter hypermethylation in bladder cancer. Carcinogenesis 2005, 27, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Ellinger, J.; El Kassem, N.; Heukamp, L.C.; Matthews, S.; Cubukluoz, F.; Kahl, P.; Perabo, F.G.; Müller, S.C.; von Ruecker, A.; Bastian, P.J. Hypermethylation of cell-free serum DNA indicates worse outcome in patients with bladder cancer. J. Urol. 2008, 179, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wu, L.; Chand, H.; Li, C.; Hu, X.; Li, Y. Silencing of miR-152 contributes to DNMT1-mediated CpG methylation of the PTEN promoter in bladder cancer. Life Sci. 2020, 261, 118311. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, R.D.; Lorch, Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 1999, 98, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Sawan, C.; Herceg, Z. Histone modifications and cancer. Adv. Genet. 2010, 70, 57–85. [Google Scholar] [PubMed]
- Michalak, E.M.; Burr, M.L.; Bannister, A.J.; Dawson, M.A. The roles of DNA, RNA and histone methylation in ageing and cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Wu, Z.J.; Groner, A.C.; He, H.H.; Cai, C.; Lis, R.T.; Wu, X.; Stack, E.C.; Loda, M.; Liu, T. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 2012, 338, 1465–1469. [Google Scholar] [CrossRef] [Green Version]
- Santos, M.; Martínez-Fernández, M.; Duenas, M.; García-Escudero, R.; Alfaya, B.; Villacampa, F.; Saiz-Ladera, C.; Costa, C.; Oteo, M.; Duarte, J. In Vivo Disruption of an Rb–E2F–Ezh2 Signaling Loop Causes Bladder Cancer. Cancer Res. 2014, 74, 6565–6577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warrick, J.I.; Raman, J.D.; Kaag, M.; Bruggeman, T.; Cates, J.; Clark, P.; DeGraff, D.J. Enhancer of zeste homolog 2 (EZH2) expression in bladder cancer. Urol. Oncol. Semin. Orig. Investig. 2016, 34, 258.e251–258.e256. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Du, Y.; Liu, X.; Chen, H.; Weng, X.; Guo, J.; Wang, M.; Wang, X.; Wang, L. EZH2 inhibition suppresses bladder cancer cell growth and metastasis via the JAK2/STAT3 signaling pathway. Oncol. Lett. 2019, 18, 907–915. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, Y.; Liu, X.; Liu, T.; Li, P.; Du, L.; Yang, Y.; Wang, L.; Wang, C. Nested quantitative PCR approach for urinary cell-free EZH2 mRNA and its potential clinical application in bladder cancer. Int. J. Cancer 2016, 139, 1830–1838. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.K.; Workman, J.L. Histone acetyltransferase complexes: One size doesn’t fit all. Nat. Rev. Mol. Cell Biol. 2007, 8, 284–295. [Google Scholar] [CrossRef]
- Iyer, N.G.; Özdag, H.; Caldas, C. p300/CBP and cancer. Oncogene 2004, 23, 4225–4231. [Google Scholar] [CrossRef] [Green Version]
- Waddell, A.R.; Huang, H.; Liao, D. CBP/p300: Critical Co-Activators for Nuclear Steroid Hormone Receptors and Emerging Therapeutic Targets in Prostate and Breast Cancers. Cancers 2021, 13, 2872. [Google Scholar] [CrossRef]
- Hong, Z.; Xiang, Z.; Zhang, P.; Wu, Q.; Xu, C.; Wang, X.; Shi, G.; Wu, D. Histone acetyltransferase 1 upregulates androgen receptor expression to modulate CRPC cell resistance to enzalutamide. Clin. Transl. Med. 2021, 11, e495. [Google Scholar] [CrossRef]
- Takeuchi, A.; Shiota, M.; Tatsugami, K.; Yokomizo, A.; Tanaka, S.; Kuroiwa, K.; Eto, M.; Naito, S. p300 mediates cellular resistance to doxorubicin in bladder cancer. Mol. Med. Rep. 2012, 5, 173–176. [Google Scholar] [PubMed] [Green Version]
- Xu, W.; Parmigiani, R.; Marks, P. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef] [Green Version]
- Giannopoulou, A.F.; Velentzas, A.D.; Konstantakou, E.G.; Avgeris, M.; Katarachia, S.A.; Papandreou, N.C.; Kalavros, N.I.; Mpakou, V.E.; Iconomidou, V.; Anastasiadou, E. Revisiting histone deacetylases in human tumorigenesis: The paradigm of urothelial bladder cancer. Int. J. Mol. Sci. 2019, 20, 1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Ezzeldin, H.H.; Diasio, R.B. Histone Deacetylase Inhibitors. Drugs 2009, 69, 1911–1934. [Google Scholar] [CrossRef]
- Alivand, M.; Soufi, R.; Madani, A.; Esmaeili, S.; Vaziri, H.; Sohani, M.; Rafati, M.; Hamami, P.; Ajamian, F. Histonedeacetylase 1 mRNA has elevated expression in clinical specimen of bladder cancer. Bratisl. Med. J. 2018, 119, 12–16. [Google Scholar] [CrossRef] [Green Version]
- Niegisch, G.; Knievel, J.; Koch, A.; Hader, C.; Fischer, U.; Albers, P.; Schulz, W.A. Changes in histone deacetylase (HDAC) expression patterns and activity of HDAC inhibitors in urothelial cancers. Urol. Oncol. Semin. Orig. Investig. 2013, 31, 1770–1779. [Google Scholar] [CrossRef]
- Xu, X.S.; Wang, L.; Abrams, J.; Wang, G. Histone deacetylases (HDACs) in XPC gene silencing and bladder cancer. J. Hematol. Oncol. 2011, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Poyet, C.; Jentsch, B.; Hermanns, T.; Schweckendiek, D.; Seifert, H.-H.; Schmidtpeter, M.; Sulser, T.; Moch, H.; Wild, P.J.; Kristiansen, G. Expression of histone deacetylases 1, 2 and 3 in urothelial bladder cancer. BMC Clin. Pathol. 2014, 14, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucca, I.; Hofbauer, S.L.; Haitel, A.; Susani, M.; Shariat, S.F.; Klatte, T.; De Martino, M. Urinary expression of genes involved in DNA methylation and histone modification for diagnosis of bladder cancer in patients with asymptomatic microscopic haematuria. Oncol. Lett. 2019, 18, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Buckwalter, J.M.; Chan, W.; Shuman, L.; Wildermuth, T.; Ellis-Mohl, J.; Walter, V.; Warrick, J.I.; Wu, X.-R.; Kaag, M.; Raman, J.D. Characterization of histone deacetylase expression within in vitro and in vivo bladder cancer model systems. Int. J. Mol. Sci. 2019, 20, 2599. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef] [Green Version]
- Besaratinia, A.; Cockburn, M.; Tommasi, S. Alterations of DNA methylome in human bladder cancer. Epigenetics 2013, 8, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Liu, J.; Lin, Q. Histone demethylase KDM2A: Biological functions and clinical values (Review). Exp. Ther. Med. 2021, 22, 723. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, E.C.; Robinson, B.D.; Downes, M.J.; Powell, L.G.; Lee, M.M.; Scherr, D.S.; Gudas, L.J.; Mongan, N.P. Role of androgen receptor and associated lysine-demethylase coregulators, LSD1 and JMJD2A, in localized and advanced human bladder cancer. Mol. Carcinog. 2011, 50, 931–944. [Google Scholar] [CrossRef] [Green Version]
- Xie, Q.; Tang, T.; Pang, J.; Xu, J.; Yang, X.; Wang, L.; Huang, Y.; Huang, Z.; Liu, G.; Tong, D.; et al. LSD1 Promotes Bladder Cancer Progression by Upregulating LEF1 and Enhancing EMT. Front. Oncol. 2020, 10, 1234. [Google Scholar] [CrossRef] [PubMed]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.-M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Sanchez, R.; Zhou, M.-M. Scaling the druggability landscape of human bromodomains, a new class of drug targets. J. Med. Chem. 2012, 55, 7342–7345. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.-l.; Tian, M.; Li, X.; Li, J.-j.; Huang, J.; Ouyang, L.; Zhang, Y.; Liu, B. Inhibition of BET bromodomains as a therapeutic strategy for cancer drug discovery. Oncotarget 2015, 6, 5501. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Liu, D.; Tao, D.; Xiang, W.; Xiao, X.; Wang, M.; Wang, L.; Luo, G.; Li, Y.; Zeng, F. BRD4 Regulates EZH2 Transcription through Upregulation of C-MYC and Represents a Novel Therapeutic Target in Bladder CancerBRD4 Regulates EZH2 Transcription in Bladder Cancer. Mol. Cancer Ther. 2016, 15, 1029–1042. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Yang, F.-Q.; Zhang, H.-M.; Li, J.; Li, W.; Wang, G.-C.; Che, J.-P.; Zheng, J.-H.; Liu, M. Bromodomain 4 protein is a predictor of survival for urothelial carcinoma of bladder. Int. J. Clin. Exp. Pathol. 2014, 7, 4231. [Google Scholar]
- Du, Q.; Luu, P.L.; Stirzaker, C.; Clark, S.J. Methyl-CpG-binding domain proteins: Readers of the epigenome. Epigenomics 2015, 7, 1051–1073. [Google Scholar] [CrossRef]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Spitz, M.R.; Zhang, H.; Grossman, H.B.; Frazier, M.L.; Wu, X. Methyl-CpG-binding domain 2: A protective role in bladder carcinoma. Cancer 2004, 100, 1853–1858. [Google Scholar] [CrossRef] [PubMed]
- Nepali, K.; Liou, J.P. Recent developments in epigenetic cancer therapeutics: Clinical advancement and emerging trends. J. Biomed. Sci. 2021, 28, 27. [Google Scholar] [CrossRef] [PubMed]
- Crabb, S.J.; Danson, S.; Catto, J.; McDowell, C.; Dunkley, D.; Huddart, R.A.; Griffiths, G.; Group, S.T.M. Phase I Trial of DNA Methyltransferase Inhibitor Guadecitabine Combined with Cisplatin and Gem-citabine for Solid Malignancies Including Urothelial Carcinoma (SPIRE). Clin. Cancer Res. 2021, 27, 1882–1892. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.; Rasco, D.; Bendell, J.; Sachdev, J.; Ramanathan, R.; Weiss, G.; Munster, P.; Edenfield, W.J.; Liu, K.; Blackwood-Chirchir, A. Abstract A120: A Phase Ib study of CC-486 (Oral Azacitidine) as a priming agent for carboplatin or NAB-paclitaxel in subjects with relapsed and refractory solid tumors. Mol. Cancer Ther. 2013, 12, A120. [Google Scholar] [CrossRef]
- Lin, J.; Gilbert, J.; Rudek, M.A.; Zwiebel, J.A.; Gore, S.; Jiemjit, A.; Zhao, M.; Baker, S.D.; Ambinder, R.F.; Herman, J.G. A phase I dose-finding study of 5-azacytidine in combination with sodium phenylbutyrate in patients with refractory solid tumors. Clin. Cancer Res. 2009, 15, 6241–6249. [Google Scholar] [CrossRef] [Green Version]
- Coyne, G.O.S.; Wang, L.; Zlott, J.; Juwara, L.; Covey, J.M.; Beumer, J.H.; Cristea, M.C.; Newman, E.M.; Koehler, S.; Nieva, J.J. Intravenous 5-fluoro-2′-deoxycytidine administered with tetrahydrouridine increases the proportion of p16-expressing circulating tumor cells in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2020, 85, 979–993. [Google Scholar] [CrossRef]
- Quinn, D.I.; Tsao-Wei, D.D.; Twardowski, P.; Aparicio, A.M.; Frankel, P.; Chatta, G.; Wright, J.J.; Groshen, S.G.; Khoo, S.; Lenz, H.-J. Phase II study of the histone deacetylase inhibitor vorinostat (Suberoylanilide Hydroxamic Acid; SAHA) in recurrent or metastatic transitional cell carcinoma of the urothelium–an NCI-CTEP sponsored: California Cancer Consortium trial, NCI 6879. Investig. New Drugs 2021, 39, 812–820. [Google Scholar] [CrossRef]
- Grant, C.; Rahman, F.; Piekarz, R.; Peer, C.; Frye, R.; Robey, R.W.; Gardner, E.R.; Figg, W.D.; Bates, S.E. Romidepsin: A new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors. Expert Rev. Anticancer Ther. 2010, 10, 997–1008. [Google Scholar] [CrossRef]
- Wang, S.-C.; Chang, Y.-C.; Wu, M.-Y.; Yu, C.-Y.; Chen, S.-L.; Sung, W.-W. Intravesical Instillation of Azacitidine Suppresses Tumor Formation through TNF-R1 and TRAIL-R2 Signaling in Genotoxic Carcinogen-Induced Bladder Cancer. Cancers 2021, 13, 3933. [Google Scholar] [CrossRef]
- Huang, Z.; Yan, Y.; Zhu, Z.; Liu, J.; He, X.; Dalangood, S.; Li, M.; Tan, M.; Cai, J.; Tang, P. CBX7 suppresses urinary bladder cancer progression via modulating AKR1B10–ERK signaling. Cell Death Dis. 2021, 12, 537. [Google Scholar] [CrossRef] [PubMed]
- Segovia, C.; José-Enériz, S.; Munera-Maravilla, E.; Martinez-Fernandez, M.; Garate, L.; Miranda, E.; Vilas-Zornoza, A.; Lodewijk, I.; Rubio, C.; Segrelles, C. Inhibition of a G9a/DNMT network triggers immune-mediated bladder cancer regression. Nat. Med. 2019, 25, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, Y.; Gong, Y.; Zhang, T.; Huang, J.; Tan, Z.; Xue, L. Finding an easy way to harmonize: A review of advances in clinical research and combination strategies of EZH2 inhibitors. Clin. Epigenet. 2021, 13, 62. [Google Scholar] [CrossRef]
- Ramakrishnan, S.; Granger, V.; Rak, M.; Hu, Q.; Attwood, K.; Aquila, L.; Krishnan, N.; Osiecki, R.; Azabdaftari, G.; Guru, K. Inhibition of EZH2 induces NK cell-mediated differentiation and death in muscle-invasive bladder cancer. Cell Death Differ. 2019, 26, 2100–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ler, L.D.; Ghosh, S.; Chai, X.; Thike, A.A.; Heng, H.L.; Siew, E.Y.; Dey, S.; Koh, L.K.; Lim, J.Q.; Lim, W.K. Loss of tumor suppressor KDM6A amplifies PRC2-regulated transcriptional repression in bladder cancer and can be targeted through inhibition of EZH2. Sci. Transl. Med. 2017, 9, eaai8312. [Google Scholar] [CrossRef]
- Li, J.; Huang, C.; Xiong, T.; Zhuang, C.; Zhuang, C.; Li, Y.; Ye, J.; Gui, Y. A CRISPR Interference of CBP and p300 Selectively Induced Synthetic Lethality in Bladder Cancer Cells In Vitro. Int. J. Biol. Sci. 2019, 15, 1276. [Google Scholar] [CrossRef] [Green Version]
- Mackay, H.J.; Hirte, H.; Colgan, T.; Covens, A.; MacAlpine, K.; Grenci, P.; Wang, L.; Mason, J.; Pham, P.-A.; Tsao, M.-S. Phase II trial of the histone deacetylase inhibitor belinostat in women with platinum resistant epithelial ovarian cancer and micropapillary (LMP) ovarian tumours. Eur. J. Cancer 2010, 46, 1573–1579. [Google Scholar] [CrossRef] [Green Version]
- Yeo, W.; Chung, H.C.; Chan, S.L.; Wang, L.Z.; Lim, R.; Picus, J.; Boyer, M.; Mo, F.K.; Koh, J.; Rha, S.Y. Epigenetic therapy using belinostat for patients with unresectable hepatocellular carcinoma: A multicenter phase I/II study with biomarker and pharmacokinetic analysis of tumors from patients in the Mayo Phase II Consortium and the Cancer Therapeutics Research Group. J. Clin. Oncol. 2012, 30, 3361. [Google Scholar]
- Kirschbaum, M.H.; Foon, K.A.; Frankel, P.; Ruel, C.; Pulone, B.; Tuscano, J.M.; Newman, E.M. A phase 2 study of belinostat (PXD101) in patients with relapsed or refractory acute myeloid leukemia or patients over the age of 60 with newly diagnosed acute myeloid leukemia: A California Cancer Consortium Study. Leuk. Lymphoma 2014, 55, 2301–2304. [Google Scholar] [CrossRef]
- Buckley, M.T.; Yoon, J.; Yee, H.; Chiriboga, L.; Liebes, L.; Ara, G.; Qian, X.; Bajorin, D.F.; Sun, T.-T.; Wu, X.-R. The histone deacetylase inhibitor belinostat (PXD101) suppresses bladder cancer cell growth in vitro and in vivo. J. Transl. Med. 2007, 5, 49. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.T.; Hoimes, C.J.; Kaimakliotis, H.Z.; Cheng, C.J.; Zhang, K.; Liu, J.; Wheeler, M.A.; Kelly, W.K.; Tew, G.N.; Saltzman, W.M. Nanoparticles for urothelium penetration and delivery of the histone deacetylase inhibitor belinostat for treatment of bladder cancer. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 1124–1134. [Google Scholar] [CrossRef] [Green Version]
- Ueda, H.; Nakajima, H.; Hori, Y.; Fujita, T.; Nishimura, M.; Goto, T.; Okuhara, M. FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968 I. Taxonomy, fermentation, isolation, physico-chemical and biological properties, and antitumor activity. J. Antibiot. 1994, 47, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Karam, J.A.; Fan, J.; Stanfield, J.; Richer, E.; Benaim, E.A.; Frenkel, E.; Antich, P.; Sagalowsky, A.I.; Mason, R.P.; Hsieh, J.T. The use of histone deacetylase inhibitor FK228 and DNA hypomethylation agent 5-azacytidine in human bladder cancer therapy. Int. J. Cancer 2007, 120, 1795–1802. [Google Scholar] [CrossRef] [PubMed]
- Nalivaeva, N.N.; Belyaev, N.D.; Turner, A.J. Sodium valproate: An old drug with new roles. Trends Pharmacol. Sci. 2009, 30, 509–514. [Google Scholar] [CrossRef]
- Coronel, J.; Cetina, L.; Pacheco, I.; Trejo-Becerril, C.; González-Fierro, A.; De La Cruz-Hernandez, E.; Perez-Cardenas, E.; Taja-Chayeb, L.; Arias-Bofill, D.; Candelaria, M. A double-blind, placebo-controlled, randomized phase III trial of chemotherapy plus epigenetic therapy with hydralazine valproate for advanced cervical cancer. Preliminary results. Med. Oncol. 2011, 28, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Vallo, S.; Xi, W.; Hudak, L.; Juengel, E.; Tsaur, I.; Wiesner, C.; Haferkamp, A.; Blaheta, R.A. HDAC inhibition delays cell cycle progression of human bladder cancer cells in vitro. Anti-Cancer Drugs 2011, 22, 1002–1009. [Google Scholar] [CrossRef]
- Byler, T.K.; Leocadio, D.; Shapiro, O.; Bratslavsky, G.; Stodgell, C.J.; Wood, R.W.; Messing, E.M.; Reeder, J.E. Valproic acid decreases urothelial cancer cell proliferation and induces thrombospondin-1 expression. BMC Urol. 2012, 12, 21. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Jing, Y.; Ouyang, S.; Liu, B.; Zhu, T.; Niu, H.; Tian, Y. Inhibitory effect of valproic acid on bladder cancer in combination with chemotherapeutic agents in vitro and in vivo. Oncol. Lett. 2013, 6, 1492–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Hamacher, A.; Petzsch, P.; Köhrer, K.; Niegisch, G.; Hoffmann, M.J.; Schulz, W.A.; Kassack, M.U. Combination of decitabine and entinostat synergistically inhibits urothelial bladder cancer cells via activation of FoxO1. Cancers 2020, 12, 337. [Google Scholar] [CrossRef] [Green Version]
- Shorstova, T.; Foulkes, W.D.; Witcher, M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br. J. Cancer 2021, 124, 1478–1490. [Google Scholar] [CrossRef]
- Li, F.; Yang, C.; Zhang, H.B.; Ma, J.; Jia, J.; Tang, X.; Zeng, J.; Chong, T.; Wang, X.; He, D. BET inhibitor JQ1 suppresses cell proliferation via inducing autophagy and activating LKB1/AMPK in bladder cancer cells. Cancer Med. 2019, 8, 4792–4805. [Google Scholar] [CrossRef] [PubMed]
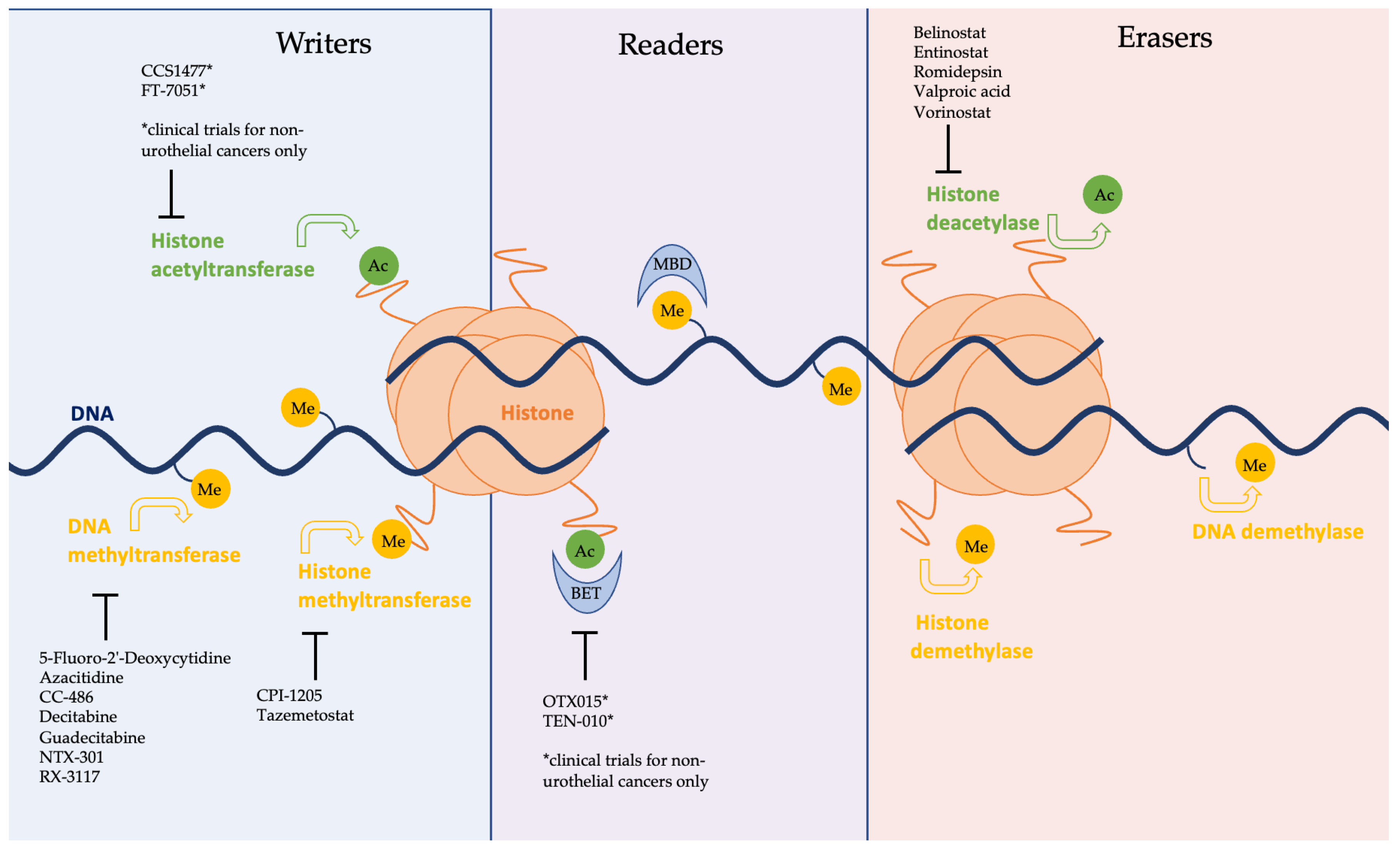
Figure 1.
Schematic representation of the epigenome with enzymes grouped into three broad categories of ‘writers’, ‘readers’, and ‘erasers’. Methyl and acetyl groups are shown attached to DNA, and histones and are marked as ‘Me’ and ‘Ac’, respectively. Epigenetic inhibitors currently involved in clinical trials are listed in black, shown to be inhibiting their respective enzymes. Agents notated with an asterisk (*) are under investigation in clinical trials for non-urothelial cancers only.
Figure 1.
Schematic representation of the epigenome with enzymes grouped into three broad categories of ‘writers’, ‘readers’, and ‘erasers’. Methyl and acetyl groups are shown attached to DNA, and histones and are marked as ‘Me’ and ‘Ac’, respectively. Epigenetic inhibitors currently involved in clinical trials are listed in black, shown to be inhibiting their respective enzymes. Agents notated with an asterisk (*) are under investigation in clinical trials for non-urothelial cancers only.


{kind=link}
Table 1.
Summary of current human clinical trials investigating epigenetic inhibitors in bladder cancer.
Table 1.
Summary of current human clinical trials investigating epigenetic inhibitors in bladder cancer.
| Trial Identifier | Start Date | Expected End Date | Drug | Combination | Phase | Inclusion Cohort | Status | Results |
|---|---|---|---|---|---|---|---|---|
| DNMT inhibitors | ||||||||
| NCT04851834 | 25 August 2021 | November 2023 | NTX-301 | Platinum-based chemotherapy | 1/2 | Locally advanced or metastatic bladder cancer; refractory/intolerant to standard of care therapies | Recruiting | Pending |
| NCT03179943 | 27 November 2017 | July 2022 | Guadecitabine | Atezolizumab | 2 | Advanced or metastatic urothelial carcinoma; must have received/been ineligible for CTx; must have had received PD-L1 or PD-1 targeting agent | Active—not recruiting | Pending |
| ISRCTN16332228 | 1 March 2016 | 10 July 2018 | Guadecitabine | Cisplatin and gemcitabine | 1b/2a | Incurable metastatic bladder cancer | Completed | Guadecitabine 20 mg/m2 is the recommended dose [55] |
| NCT00978250 | 20 August 2009 | 11 April 2019 | 5-Fluoro-2′-Deoxycytidine | Tetrahydrouridine | 2 | Advanced or metastatic urothelial carcinoma; received at least one line of standard therapy | Completed | Well-tolerate; AUC increase 4-fold; progression-free survival above expected [58] |
| NCT02030067 | December 2013 | July 2019 | RX-3117 | N/A (monotherapy) | 1 | Advanced bladder cancer | Completed | Not reported |
| NCT00030615 | December 2001 | September 2008 | Decitabine | N/A (monotherapy) | 1 | Advanced or metastatic bladder cancer for which all other treatment has failed | Completed | Not reported |
| NCT02223052 | 27 October 2014 | 11 June 2018 | CC-486 (oral form of azacitidine) | N/A (monotherapy) | 1 | Metastatic or inoperable bladder cancer | Completed | Not reported |
| NCT01478685 | 29 November 2011 | 17 November 2015 | CC-486 (oral form of azacitidine) | Carboplatin or ABI-007 | 1 | Relapsed or refractory urothelial carcinoma of the bladder, renal pelvis, ureter, or urethra | Completed | CC-486 is tolerated as a priming agent with carboplatin and ABI-007 [56] |
| NCT00005639 | March 2000 | July 2005 | Azacitidine | Phenylbutyrate | 1 | Locally advanced or metastatic bladder cancer | Completed | Three doses were well-tolerated [57] |
| NCT02959437 | 27 February 2017 | 15 February 2019 | Azacitidine | Pembrolizumab and epacadostat | 1/2 | Advanced or metastatic solid tumour, which has failed prior standard therapy | Terminated (by sponsors) | Not reported |
| EZH2 inhibitors | ||||||||
| NCT03854474 | 17 May 2019 | 30 June 2023 | Tazemetostat (EPZ-6438) | Pembrolizumab | 1/2 | Locally advanced or metastatic urothelial carcinoma with progression during or following platinum-based CTx (or ineligible for CTx) | Recruiting | Pending |
| NCT03525795 | 14 December 2017 | 12 June 2019 | CPI-1205 | Ipilimumab | 1/2 | Unresectable or metastatic urothelial carcinoma (urethra, bladder, ureters, or renal pelvis) | Completed | Not reported |
| HDAC inhibitors | ||||||||
| NCT02619253 | 14 January 16 | 31 May 2023 | Vorinostat | Pembrolizumab | 1/2 | Urothelial cell carcinoma—previously treated and progressive disease, locally advanced or metastatic; must have received a prior platinum-based regimen in the metastatic setting | Active, not recruiting | Pending |
| NCT00045006 | July 2001 | July 2008 | Vorinostat | N/A (monotherapy) | 1 | Advanced or metastatic bladder cancer that is refractory to standard treatment | Completed | Not reported |
| NCT00565227 | April 2007 | November 2008 | Vorinostat | Docetaxel | 1 | Bladder/urothelial cancer that has progressed after chemotherapy | Terminated (toxicity) | Not reported |
| NCT00363883 | June 2006 | December 2010 | Vorinostat | N/A (monotherapy) | 2 | Bladder/urothelial TCC that has recurred or progressed on platinum-based CTx | Terminated (futility) | Limited efficacy and significant toxicity [59] |
| NCT05154994 | 14 January 2022 | 30 November 2023 | Belinostat | Tremelimumab and durvalumab | 1 | Urothelial carcinoma with metastatic disease or with unresectable, locally advanced disease | Recruiting | Pending |
| NCT00413075 | June 2006 | August 2011 | Belinostat | N/A (monotherapy) | 1 | Primary or metastatic solid tumour refractory to standard treatment | Completed | Not reported |
| NCT00413322 | September 2005 | March 2008 | Belinostat | 5-Fluorouracil | 1 | Advanced bladder cancer with progression on standard treatment | Completed | Not reported |
| NCT00421889 | August 2005 | February 2009 | Belinostat | Carboplatin or paclitaxel | 1/2 | Urothelial carcinoma, received up to three CTx regimens in advanced disease setting | Completed | No published results; partial response in 4/15 patients |
| NCT01638533 | 12 June 2012 | 29 November 2018 | Romidepsin | N/A (monotherapy) | 1 | Recurrent bladder cancer and concurrent hepatic impairment | Active, not recruiting | Similar toxicity to other HDAC inhibitors [60] |
| NCT00087295 | June 2004 | April 2006 | FR901228 (Romidepsin) | N/A (monotherapy) | 2 | Metastatic or poorly differentiated TCC; progression after one CTx regimen | Terminated (poor accrual) | Not reported |
| NCT01552434 | 16 March 2012 | 31 March 2022 | Valproic acid | Bevacizumab and temsirolimus | 1 | Metastatic urothelial cancer that is refractory to standard therapy | Active, not recruiting | Pending |
| NCT01738815 | December 2011 | May 2013 | Valproic acid | N/A (monotherapy) | 1 | Suspected or confirmed bladder tumour | Completed | Not reported |
| NCT03978624 | 23 September 2020 | 1 October 2023 | Entinostat | Pembrolizumab | 2 | MIBC ineligible for or refused neoadjuvant cisplatin-based CTx; pre-cystectomy | Recruiting | Pending |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Thompson, D.; Lawrentschuk, N.; Bolton, D. New Approaches to Targeting Epigenetic Regulation in Bladder Cancer. Cancers 2023, 15, 1856. https://doi.org/10.3390/cancers15061856
AMA Style
Thompson D, Lawrentschuk N, Bolton D. New Approaches to Targeting Epigenetic Regulation in Bladder Cancer. Cancers. 2023; 15(6):1856. https://doi.org/10.3390/cancers15061856
Chicago/Turabian StyleThompson, Daryl, Nathan Lawrentschuk, and Damien Bolton. 2023. "New Approaches to Targeting Epigenetic Regulation in Bladder Cancer" Cancers 15, no. 6: 1856. https://doi.org/10.3390/cancers15061856
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.