

DNA Repair Deficiency Regulates Immunity Response in Cancers: Molecular Mechanism and Approaches for Combining Immunotherapy

Abstract
:Simple Summary
Abstract
1. Introduction
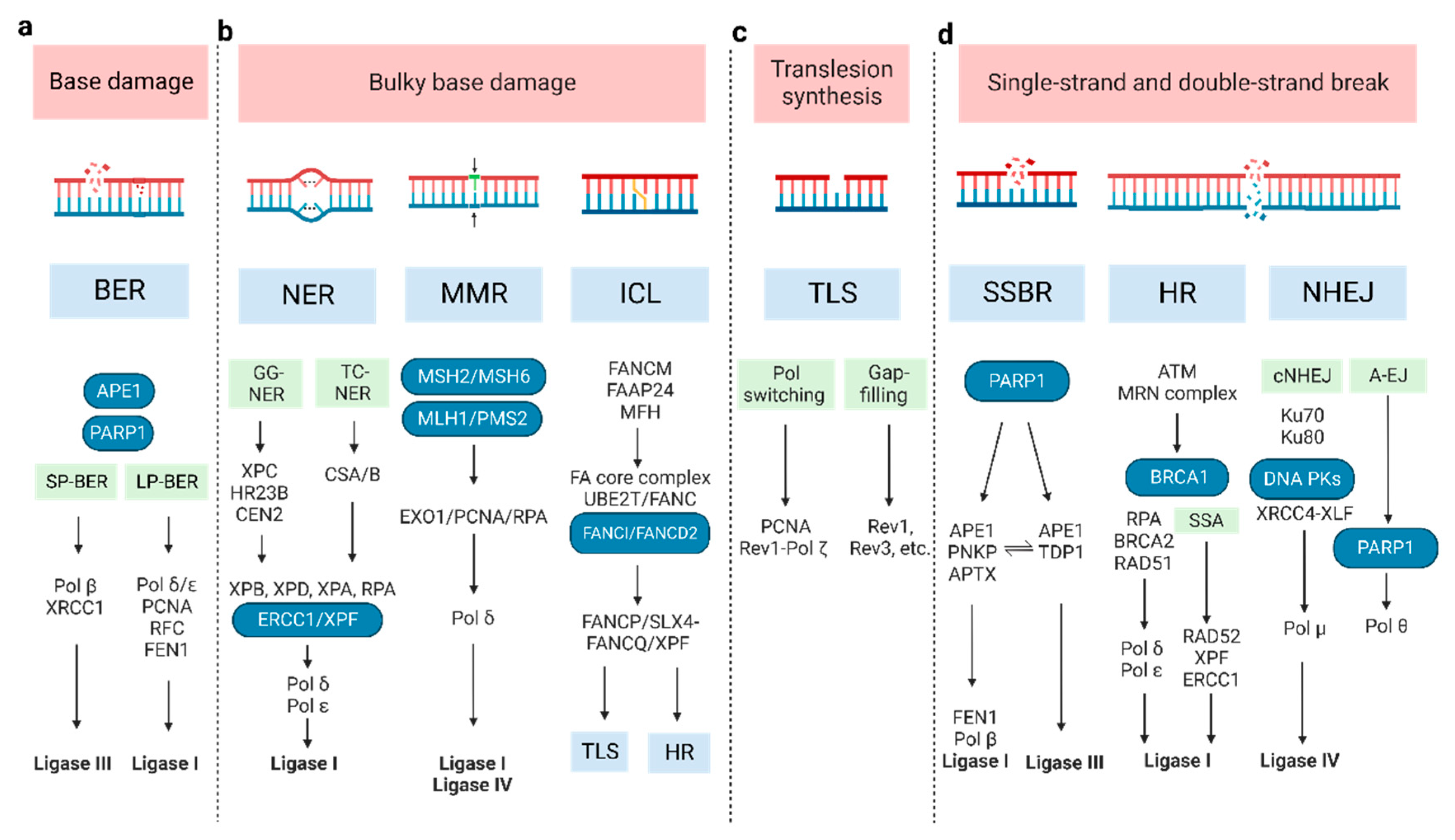
2. DNA Damage and Repair Pathway
2.1. Base Damage Repair
2.2. Bulky Base Damage Repair
2.3. Translesion Synthesis
2.4. SSB and DSB Break Repair
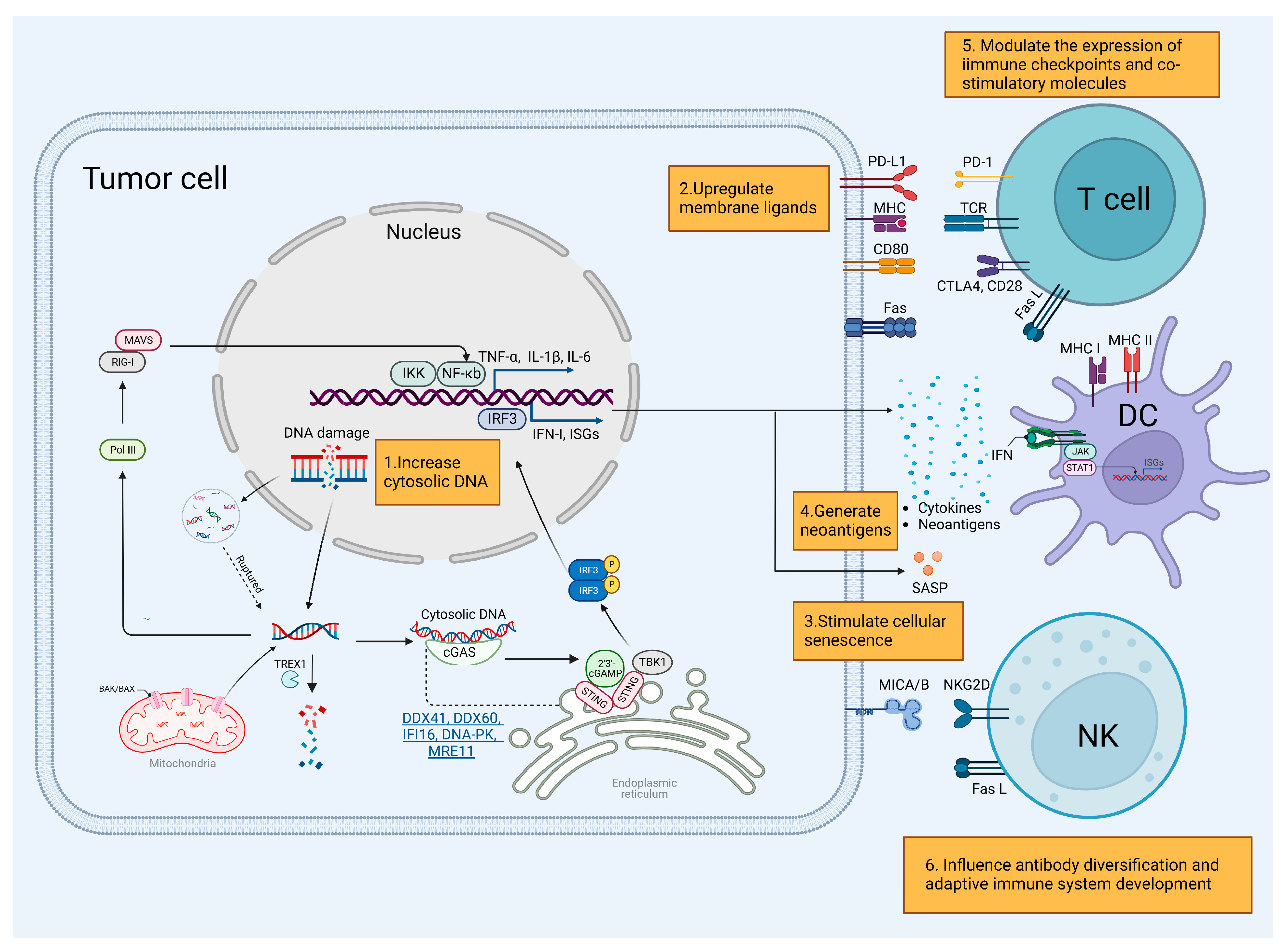
3. The Interplay between DDR Deficiency and Immune Response
3.1. Innate Immune Response
3.1.1. Cytosolic DNA Generation
3.1.2. Cytosolic Nucleic Acid Sensing Pathway
3.1.3. Other Mechanisms
3.2. Adaptive Immune Response
3.2.1. Influence Tumor Antigenicity
3.2.2. Immune Checkpoint Interaction
3.2.3. Induce Immunogenic Cell Death
3.2.4. Role in Immunogenic Diversity
4. Combining DDR Inhibition and Immunotherapy
4.1. Potential Mechanism and Clinical Implication
4.2. Treatment Strategies for Combining DDR Targets
4.3. DDR-Related Biomarkers for Predicting Immune Response
5. Conclusions and Future Prospects
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA. Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Pérez-Herrero, E.; Fernández-Medarde, A. Advanced Targeted Therapies in Cancer: Drug Nanocarriers, the Future of Chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hait, W.N. Targeted Cancer Therapeutics. Cancer Res. 2009, 69, 1263–1267. [Google Scholar] [CrossRef] [Green Version]
- Middleton, G.; Robbins, H.; Andre, F.; Swanton, C. A State-of-the-Art Review of Stratified Medicine in Cancer: Towards a Future Precision Medicine Strategy in Cancer. Ann. Oncol. 2022, 33, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zhang, T.; Zheng, L.; Liu, H.; Song, W.; Liu, D.; Li, Z.; Pan, C. Combination Strategies to Maximize the Benefits of Cancer Immunotherapy. J. Hematol. Oncol. 2021, 14, 156. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, L.; Livingstone, E.; Hassel, J.C.; Fluck, M.; Eigentler, T.; Loquai, C.; Haferkamp, S.; Gutzmer, R.; Meier, F.; Mohr, P.; et al. Adjuvant Nivolumab plus Ipilimumab or Nivolumab Monotherapy versus Placebo in Patients with Resected Stage IV Melanoma with No Evidence of Disease (IMMUNED): A Randomised, Double-Blind, Placebo-Controlled, Phase 2 Trial. Lancet 2020, 395, 1558–1568. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.-J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma (KEYNOTE-006): Post-Hoc 5-Year Results from an Open-Label, Multicentre, Randomised, Controlled, Phase 3 Study. Lancet Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef]
- Kim, T.K.; Vandsemb, E.N.; Herbst, R.S.; Chen, L. Adaptive Immune Resistance at the Tumour Site: Mechanisms and Therapeutic Opportunities. Nat. Rev. Drug Discov. 2022, 21, 529–540. [Google Scholar] [CrossRef]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Dobbelstein, M.; Sørensen, C.S. Exploiting Replicative Stress to Treat Cancer. Nat. Rev. Drug Discov. 2015, 14, 405–423. [Google Scholar] [CrossRef]
- Chatzinikolaou, G.; Karakasilioti, I.; Garinis, G.A. DNA Damage and Innate Immunity: Links and Trade-Offs. Trends Immunol. 2014, 35, 429–435. [Google Scholar] [CrossRef]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef] [Green Version]
- Nastasi, C.; Mannarino, L.; D’Incalci, M. DNA Damage Response and Immune Defense. Int. J. Mol. Sci. 2020, 21, 7504. [Google Scholar] [CrossRef]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, Density, and Location of Immune Cells within Human Colorectal Tumors Predict Clinical Outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [Green Version]
- Dexheimer, T.S. DNA Repair Pathways and Mechanisms. In DNA Repair of Cancer Stem Cells; Mathews, L.A., Cabarcas, S.M., Hurt, E.M., Eds.; Springer: Dordrecht, The Netherlands, 2013; pp. 19–32. [Google Scholar] [CrossRef]
- Hoeijmakers, J.H.J. DNA Damage, Aging, and Cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Sirbu, B.M.; Cortez, D. DNA Damage Response: Three Levels of DNA Repair Regulation. Cold Spring Harb. Perspect. Biol. 2013, 5, a012724. [Google Scholar] [CrossRef] [Green Version]
- Curtin, N.J. DNA Repair Dysregulation from Cancer Driver to Therapeutic Target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Christmann, M.; Tomicic, M.T.; Roos, W.P.; Kaina, B. Mechanisms of Human DNA Repair: An Update. Toxicology 2003, 193, 3–34. [Google Scholar] [CrossRef]
- Huang, R.; Zhou, P.-K. DNA Damage Repair: Historical Perspectives, Mechanistic Pathways and Clinical Translation for Targeted Cancer Therapy. Signal Transduct. Target. Ther. 2021, 6, 254. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA Damage, Repair, and Mutagenesis: DNA Damage and Repair. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [Green Version]
- Zharkov, D.O. Base Excision DNA Repair. Cell. Mol. Life Sci. 2008, 65, 1544–1565. [Google Scholar] [CrossRef]
- Fortini, P.; Dogliotti, E. Base Damage and Single-Strand Break Repair: Mechanisms and Functional Significance of Short- and Long-Patch Repair Subpathways. DNA Repair 2007, 6, 398–409. [Google Scholar] [CrossRef]
- Barakat, K.H.; Gajewski, M.M.; Tuszynski, J.A. DNA Polymerase Beta (Pol β) Inhibitors: A Comprehensive Overview. Drug Discov. Today 2012, 17, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.S.; Lasley, F.D.; Das, I.J.; Mendonca, M.S.; Dynlacht, J.R. Molecular Mechanisms of DNA Damage and Repair. In Basic Radiotherapy Physics and Biology; Springer International Publishing: Cham, Switzerland, 2014; pp. 201–209. [Google Scholar] [CrossRef]
- Shuck, S.C.; Short, E.A.; Turchi, J.J. Eukaryotic Nucleotide Excision Repair: From Understanding Mechanisms to Influencing Biology. Cell Res. 2008, 18, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Nouspikel, T. Nucleotide Excision Repair and Neurological Diseases. DNA Repair 2008, 7, 1155–1167. [Google Scholar] [CrossRef]
- Hanawalt, P.C.; Spivak, G. Transcription-Coupled DNA Repair: Two Decades of Progress and Surprises. Nat. Rev. Mol. Cell Biol. 2008, 9, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Sugasawa, K. Regulation of Damage Recognition in Mammalian Global Genomic Nucleotide Excision Repair. Mutat. Res. Mol. Mech. Mutagen. 2010, 685, 29–37. [Google Scholar] [CrossRef]
- Thoms, K.-M.; Kuschal, C.; Emmert, S. Lessons Learned from DNA Repair Defective Syndromes. Exp. Dermatol. 2007, 16, 532–544. [Google Scholar] [CrossRef]
- Poulogiannis, G.; Frayling, I.M.; Arends, M.J. DNA Mismatch Repair Deficiency in Sporadic Colorectal Cancer and Lynch Syndrome. Histopathology 2010, 56, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Ortega, J.; Lee, G.S.; Gu, L.; Yang, W.; Li, G.-M. Mispair-Bound Human MutS–MutL Complex Triggers DNA Incisions and Activates Mismatch Repair. Cell Res. 2021, 31, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.T.; Erdeniz, N.; Symington, L.S.; Liskay, R.M. EXO1-A Multi-Tasking Eukaryotic Nuclease. DNA Repair 2004, 3, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Lu, C.; Jin, Q.; Lu, H.; Chen, X.; Tian, L.; Zhang, Y.; Ortega, J.; Zhang, J.; Siteni, S.; et al. MLH1 Deficiency-Triggered DNA Hyperexcision by Exonuclease 1 Activates the CGAS-STING Pathway. Cancer Cell 2021, 39, 109–121.e5. [Google Scholar] [CrossRef]
- Niraj, J.; Färkkilä, A.; D’Andrea, A.D. The Fanconi Anemia Pathway in Cancer. Annu. Rev. Cancer Biol. 2019, 3, 457–478. [Google Scholar] [CrossRef]
- Zhang, J.; Dewar, J.M.; Budzowska, M.; Motnenko, A.; Cohn, M.A.; Walter, J.C. DNA Interstrand Cross-Link Repair Requires Replication-Fork Convergence. Nat. Struct. Mol. Biol. 2015, 22, 242–247. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, A.; D’Andrea, A. Fanconi Anemia Pathway. Curr. Biol. 2017, 27, R986–R988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shakeel, S.; Rajendra, E.; Alcón, P.; O’Reilly, F.; Chorev, D.S.; Maslen, S.; Degliesposti, G.; Russo, C.J.; He, S.; Hill, C.H.; et al. Structure of the Fanconi Anaemia Monoubiquitin Ligase Complex. Nature 2019, 575, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; van Twest, S.; Murphy, V.J.; Deans, A.J. ATR-Mediated FANCI Phosphorylation Regulates Both Ubiquitination and Deubiquitination of FANCD2. Front. Cell Dev. Biol. 2020, 8, 2. [Google Scholar] [CrossRef] [Green Version]
- Kolinjivadi, A.M.; Crismani, W.; Ngeow, J. Emerging Functions of Fanconi Anemia Genes in Replication Fork Protection Pathways. Hum. Mol. Genet. 2020, 29, R158–R164. [Google Scholar] [CrossRef]
- Peake, J.D.; Noguchi, E. Fanconi Anemia: Current Insights Regarding Epidemiology, Cancer, and DNA Repair. Hum. Genet. 2022, 141, 1811–1836. [Google Scholar] [CrossRef]
- Amunugama, R.; Walter, J.C. A New Varietal of DNA Interstrand Crosslink Repair. Cell Res. 2020, 30, 459–460. [Google Scholar] [CrossRef]
- Yang, W.; Gao, Y. Translesion and Repair DNA Polymerases: Diverse Structure and Mechanism. Annu. Rev. Biochem. 2018, 87, 239–261. [Google Scholar] [CrossRef] [PubMed]
- Bezalel-Buch, R.; Cheun, Y.K.; Roy, U.; Schärer, O.D.; Burgers, P.M. Bypass of DNA Interstrand Crosslinks by a Rev1–DNA Polymerase ζ Complex. Nucleic Acids Res. 2020, 48, 8461–8473. [Google Scholar] [CrossRef]
- Budzowska, M.; Graham, T.G.; Sobeck, A.; Waga, S.; Walter, J.C. Regulation of the Rev1–Pol ζ Complex during Bypass of a DNA Interstrand Cross-link. EMBO J. 2015, 34, 1971–1985. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.; Davidson, G.A.; Yang, K.; Zhuang, Z. Photo-Activatable Ub-PCNA Probes Reveal New Structural Features of the Saccharomyces Cerevisiae Polη/PCNA Complex. Nucleic Acids Res. 2021, 49, 9374–9388. [Google Scholar] [CrossRef] [PubMed]
- Weaver, T.M.; Click, T.H.; Khoang, T.H.; Todd Washington, M.; Agarwal, P.K.; Freudenthal, B.D. Mechanism of Nucleotide Discrimination by the Translesion Synthesis Polymerase Rev1. Nat. Commun. 2022, 13, 2876. [Google Scholar] [CrossRef]
- Masłowska, K.H.; Villafañez, F.; Laureti, L.; Iwai, S.; Pagès, V. Eukaryotic Stress–Induced Mutagenesis Is Limited by a Local Control of Translesion Synthesis. Nucleic Acids Res. 2022, 50, 2074–2080. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Fuchs, R.P. A Comprehensive View of Translesion Synthesis in Escherichia Coli. Microbiol. Mol. Biol. Rev. 2020, 84, e00002-20. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Wang, F.; Ma, X.; Yang, Y.; Wang, Z.; Liu, H.; Li, X.; Liu, Z.; Zhang, T.; Huang, M.; et al. Mismatch Repair Protein MSH2 Regulates Translesion DNA Synthesis Following Exposure of Cells to UV Radiation. Nucleic Acids Res. 2013, 41, 10312–10322. [Google Scholar] [CrossRef] [PubMed]
- Paniagua, I.; Tayeh, Z.; Falcone, M.; Hernández Pérez, S.; Cerutti, A.; Jacobs, J.J.L. MAD2L2 Promotes Replication Fork Protection and Recovery in a Shieldin-Independent and REV3L-Dependent Manner. Nat. Commun. 2022, 13, 5167. [Google Scholar] [CrossRef]
- Chen, D.; Gervai, J.Z.; Póti, Á.; Németh, E.; Szeltner, Z.; Szikriszt, B.; Gyüre, Z.; Zámborszky, J.; Ceccon, M.; d’Adda di Fagagna, F.; et al. BRCA1 Deficiency Specific Base Substitution Mutagenesis Is Dependent on Translesion Synthesis and Regulated by 53BP1. Nat. Commun. 2022, 13, 226. [Google Scholar] [CrossRef]
- Clairmont, C.S.; Sarangi, P.; Ponnienselvan, K.; Galli, L.D.; Csete, I.; Moreau, L.; Adelmant, G.; Chowdhury, D.; Marto, J.A.; D’Andrea, A.D. TRIP13 Regulates DNA Repair Pathway Choice through REV7 Conformational Change. Nat. Cell Biol. 2020, 22, 87–96. [Google Scholar] [CrossRef]
- Taglialatela, A.; Leuzzi, G.; Sannino, V.; Cuella-Martin, R.; Huang, J.-W.; Wu-Baer, F.; Baer, R.; Costanzo, V.; Ciccia, A. REV1-Polζ Maintains the Viability of Homologous Recombination-Deficient Cancer Cells through Mutagenic Repair of PRIMPOL-Dependent SsDNA Gaps. Mol. Cell 2021, 81, 4008–4025.e7. [Google Scholar] [CrossRef]
- Caldecott, K.W. Protein–Protein Interactions during Mammalian DNA Single-Strand Break Repair. Biochem. Soc. Trans. 2003, 31, 247–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldecott, K.W. DNA Single-Strand Break Repair and Human Genetic Disease. Trends Cell Biol. 2022, 32, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Blair, K.; Tehseen, M.; Raducanu, V.-S.; Shahid, T.; Lancey, C.; Rashid, F.; Crehuet, R.; Hamdan, S.M.; De Biasio, A. Mechanism of Human Lig1 Regulation by PCNA in Okazaki Fragment Sealing. Nat. Commun. 2022, 13, 7833. [Google Scholar] [CrossRef]
- Mengwasser, K.E.; Adeyemi, R.O.; Leng, Y.; Choi, M.Y.; Clairmont, C.; D’Andrea, A.D.; Elledge, S.J. Genetic Screens Reveal FEN1 and APEX2 as BRCA2 Synthetic Lethal Targets. Mol. Cell 2019, 73, 885–899.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, J.S.; Tumbale, P.P.; Arana, M.E.; Rana, J.A.; Williams, R.S.; Kunkel, T.A. High-Fidelity DNA Ligation Enforces Accurate Okazaki Fragment Maturation during DNA Replication. Nat. Commun. 2021, 12, 482. [Google Scholar] [CrossRef] [PubMed]
- Saha, L.K.; Wakasugi, M.; Akter, S.; Prasad, R.; Wilson, S.H.; Shimizu, N.; Sasanuma, H.; Huang, S.N.; Agama, K.; Pommier, Y.; et al. Topoisomerase I-Driven Repair of UV-Induced Damage in NER-Deficient Cells. Proc. Natl. Acad. Sci. USA 2020, 117, 14412–14420. [Google Scholar] [CrossRef]
- Lin, Y.; Raj, J.; Li, J.; Ha, A.; Hossain, M.A.; Richardson, C.; Mukherjee, P.; Yan, S. APE1 Senses DNA Single-Strand Breaks for Repair and Signaling. Nucleic Acids Res. 2020, 48, 1925–1940. [Google Scholar] [CrossRef] [Green Version]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA Double-Strand Break Repair-Pathway Choice in Somatic Mammalian Cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Cejka, P.; Symington, L.S. DNA End Resection: Mechanism and Control. Annu. Rev. Genet. 2021, 55, 285–307. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xu, D. Repair Pathway Choice for Double-Strand Breaks. Essays Biochem. 2020, 64, 765–777. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, J.; Bezak, E.; Kempson, I. Imaging DNA Double-Strand Breaks—Are We There Yet? Nat. Rev. Mol. Cell Biol. 2022, 23, 579–580. [Google Scholar] [CrossRef] [PubMed]
- Pardo, B.; Gómez-González, B.; Aguilera, A. DNA Repair in Mammalian Cells: DNA Double-Strand Break Repair: How to Fix a Broken Relationship. Cell. Mol. Life Sci. 2009, 66, 1039–1056. [Google Scholar] [CrossRef]
- Li, X.; Heyer, W.-D. Homologous Recombination in DNA Repair and DNA Damage Tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-H.; Paull, T.T. ATM Activation by DNA Double-Strand Breaks Through the Mre11-Rad50-Nbs1 Complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef]
- Abraham, R.T.; Tibbetts, R.S. Guiding ATM to Broken DNA. Science 2005, 308, 510–511. [Google Scholar] [CrossRef]
- Mailand, N.; Bekker-Jensen, S.; Faustrup, H.; Melander, F.; Bartek, J.; Lukas, C.; Lukas, J. RNF8 Ubiquitylates Histones at DNA Double-Strand Breaks and Promotes Assembly of Repair Proteins. Cell 2007, 131, 887–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorslund, T.; Ripplinger, A.; Hoffmann, S.; Wild, T.; Uckelmann, M.; Villumsen, B.; Narita, T.; Sixma, T.K.; Choudhary, C.; Bekker-Jensen, S.; et al. Histone H1 Couples Initiation and Amplification of Ubiquitin Signalling after DNA Damage. Nature 2015, 527, 389–393. [Google Scholar] [CrossRef] [Green Version]
- Doil, C.; Mailand, N.; Bekker-Jensen, S.; Menard, P.; Larsen, D.H.; Pepperkok, R.; Ellenberg, J.; Panier, S.; Durocher, D.; Bartek, J.; et al. RNF168 Binds and Amplifies Ubiquitin Conjugates on Damaged Chromosomes to Allow Accumulation of Repair Proteins. Cell 2009, 136, 435–446. [Google Scholar] [CrossRef] [Green Version]
- Hustedt, N.; Durocher, D. The Control of DNA Repair by the Cell Cycle. Nat. Cell Biol. 2017, 19, 1–9. [Google Scholar] [CrossRef]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous Recombination and Human Health: The Roles of BRCA1, BRCA2, and Associated Proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef] [Green Version]
- Jensen, R.B.; Carreira, A.; Kowalczykowski, S.C. Purified Human BRCA2 Stimulates RAD51-Mediated Recombination. Nature 2010, 467, 678–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Li, Q.; Fan, J.; Holloman, W.K.; Pavletich, N.P. The BRCA2 Homologue Brh2 Nucleates RAD51 Filament Formation at a DsDNA–SsDNA Junction. Nature 2005, 433, 653–657. [Google Scholar] [CrossRef] [PubMed]
- Benitez, A.; Liu, W.; Palovcak, A.; Wang, G.; Moon, J.; An, K.; Kim, A.; Zheng, K.; Zhang, Y.; Bai, F.; et al. FANCA Promotes DNA Double-Strand Break Repair by Catalyzing Single-Strand Annealing and Strand Exchange. Mol. Cell 2018, 71, 621–628.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendez-Dorantes, C.; Bhargava, R.; Stark, J.M. Repeat-Mediated Deletions Can Be Induced by a Chromosomal Break Far from a Repeat, but Multiple Pathways Suppress Such Rearrangements. Genes Dev. 2018, 32, 524–536. [Google Scholar] [CrossRef] [Green Version]
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of Single-Strand Annealing and Its Role in Genome Maintenance. Trends Genet. 2016, 32, 566–575. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Rothenberg, E.; Ramsden, D.A.; Lieber, M.R. The Molecular Basis and Disease Relevance of Non-Homologous DNA End Joining. Nat. Rev. Mol. Cell Biol. 2020, 21, 765–781. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [Green Version]
- Gell, D.; Jackson, S.P. Mapping of Protein-Protein Interactions within the DNA-Dependent Protein Kinase Complex. Nucleic Acids Res. 1999, 27, 3494–3502. [Google Scholar] [CrossRef]
- Ahnesorg, P.; Smith, P.; Jackson, S.P. XLF Interacts with the XRCC4-DNA Ligase IV Complex to Promote DNA Nonhomologous End-Joining. Cell 2006, 124, 301–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Gelot, C.; Pantelidou, C.; Li, A.; Yücel, H.; Davis, R.E.; Färkkilä, A.; Kochupurakkal, B.; Syed, A.; Shapiro, G.I.; et al. A First-in-Class Polymerase Theta Inhibitor Selectively Targets Homologous-Recombination-Deficient Tumors. Nat. Cancer 2021, 2, 598–610. [Google Scholar] [CrossRef]
- Villanueva, M.T. A New Tool to Target DNA Repair. Nat. Rev. Cancer 2015, 15, 136. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T. Polθ Inhibitors Unchained. Nat. Cancer 2021, 2, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.R.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-Recombination-Deficient Tumours Are Dependent on Polθ-Mediated Repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Jackson, S.P.; Helleday, T. Drugging DNA Repair. Science 2016, 352, 1178–1179. [Google Scholar] [CrossRef]
- Kwon, J.; Bakhoum, S.F. The Cytosolic DNA-Sensing CGAS–STING Pathway in Cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic Progression Following DNA Damage Enables Pattern Recognition within Micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakhoum, S.; Ngo, B.; Bakhoum, A.; Cavallo-Fleming, J.A.; Murphy, C.W.; Powell, S.N.; Cantley, L. Chromosomal Instability Drives Metastasis Through a Cytosolic DNA Response. Int. J. Radiat. Oncol. 2018, 102, S118. [Google Scholar] [CrossRef]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA Stress Primes the Antiviral Innate Immune Response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zang, C.; Ren, M.; Shang, M.; Wang, Z.; Peng, X.; Zhang, Q.; Wen, X.; Xi, Z.; Zhou, C. Cellular Uptake of Extracellular Nucleosomes Induces Innate Immune Responses by Binding and Activating CGMP-AMP Synthase (CGAS). Sci. Rep. 2020, 10, 15385. [Google Scholar] [CrossRef]
- White, M.J.; McArthur, K.; Metcalf, D.; Lane, R.M.; Cambier, J.C.; Herold, M.J.; van Delft, M.F.; Bedoui, S.; Lessene, G.; Ritchie, M.E.; et al. Apoptotic Caspases Suppress MtDNA-Induced STING-Mediated Type I IFN Production. Cell 2014, 159, 1549–1562. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Guan, J.; Lu, S.; Jin, Q.; Rousseau, B.; Lu, T.; Stephens, D.; Zhang, H.; Zhu, J.; Yang, M.; et al. DNA Sensing in Mismatch Repair-Deficient Tumor Cells Is Essential for Anti-Tumor Immunity. Cancer Cell 2021, 39, 96–108.e6. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Srinivasan, K.; Abdisalaam, S.; Su, F.; Raj, P.; Dozmorov, I.; Mishra, R.; Wakeland, E.K.; Ghose, S.; Mukherjee, S.; et al. RAD51 Interconnects between DNA Replication, DNA Repair and Immunity. Nucleic Acids Res. 2017, 45, 4590–4605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costanzo, V. Brca2, Rad51 and Mre11: Performing Balancing Acts on Replication Forks. DNA Repair 2011, 10, 1060–1065. [Google Scholar] [CrossRef]
- Coquel, F.; Silva, M.-J.; Técher, H.; Zadorozhny, K.; Sharma, S.; Nieminuszczy, J.; Mettling, C.; Dardillac, E.; Barthe, A.; Schmitz, A.-L.; et al. SAMHD1 Acts at Stalled Replication Forks to Prevent Interferon Induction. Nature 2018, 557, 57–61. [Google Scholar] [CrossRef]
- Ciccia, A.; McDonald, N.; West, S.C. Structural and Functional Relationships of the XPF/MUS81 Family of Proteins. Annu. Rev. Biochem. 2008, 77, 259–287. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.S.W.; Zhang, W.Y.L.; Tan, N.Y.J.; Khatoo, M.; Suter, M.A.; Tripathi, S.; Cheung, F.S.G.; Lim, W.K.; Tan, P.H.; Ngeow, J.; et al. The DNA Structure-Specific Endonuclease MUS81 Mediates DNA Sensor STING-Dependent Host Rejection of Prostate Cancer Cells. Immunity 2016, 44, 1177–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cybulla, E.; Vindigni, A. Leveraging the Replication Stress Response to Optimize Cancer Therapy. Nat. Rev. Cancer 2023, 23, 6–24. [Google Scholar] [CrossRef]
- García-de-Teresa, B.; Rodríguez, A.; Frias, S. Chromosome Instability in Fanconi Anemia: From Breaks to Phenotypic Consequences. Genes 2020, 11, 1528. [Google Scholar] [CrossRef] [PubMed]
- Wardlaw, C.P.; Petrini, J.H.J. ISG15 Conjugation to Proteins on Nascent DNA Mitigates DNA Replication Stress. Nat. Commun. 2022, 13, 5971. [Google Scholar] [CrossRef]
- Sandy, Z.; da Costa, I.C.; Schmidt, C.K. More than Meets the ISG15: Emerging Roles in the DNA Damage Response and Beyond. Biomolecules 2020, 10, 1557. [Google Scholar] [CrossRef]
- Gratia, M.; Rodero, M.P.; Conrad, C.; Bou Samra, E.; Maurin, M.; Rice, G.I.; Duffy, D.; Revy, P.; Petit, F.; Dale, R.C.; et al. Bloom Syndrome Protein Restrains Innate Immune Sensing of Micronuclei by CGAS. J. Exp. Med. 2019, 216, 1199–1213. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.; Schubert, M.; Tijhuis, A.E.; Requesens, M.; Roorda, M.; van den Brink, A.; Ruiz, L.A.; Bakker, P.L.; van der Sluis, T.; Pieters, W.; et al. CGAS–STING Drives the IL-6-Dependent Survival of Chromosomally Instable Cancers. Nature 2022, 607, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.-A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal Instability Drives Metastasis through a Cytosolic DNA Response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef] [Green Version]
- McArthur, K.; Whitehead, L.W.; Heddleston, J.M.; Li, L.; Padman, B.S.; Oorschot, V.; Geoghegan, N.D.; Chappaz, S.; Davidson, S.; San Chin, H.; et al. BAK/BAX Macropores Facilitate Mitochondrial Herniation and MtDNA Efflux during Apoptosis. Science 2018, 359, eaao6047. [Google Scholar] [CrossRef] [Green Version]
- Kanneganti, T.-D.; Kundu, M.; Green, D.R. Innate Immune Recognition of MtDNA—An Undercover Signal? Cell Metab. 2015, 21, 793–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, M.P.; Onji, M.; Fukui, R.; Kawane, K.; Shibata, T.; Saitoh, S.; Ohto, U.; Shimizu, T.; Barber, G.N.; Miyake, K. DNase II-Dependent DNA Digestion Is Required for DNA Sensing by TLR9. Nat. Commun. 2015, 6, 5853. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.-G.; Lindahl, T.; Barnes, D.E. Trex1 Exonuclease Degrades SsDNA to Prevent Chronic Checkpoint Activation and Autoimmune Disease. Cell 2007, 131, 873–886. [Google Scholar] [CrossRef] [Green Version]
- Ablasser, A.; Chen, Z.J. CGAS in Action: Expanding Roles in Immunity and Inflammation. Science 2019, 363, eaat8657. [Google Scholar] [CrossRef]
- Du, H.; Xu, T.; Cui, M. CGAS-STING Signaling in Cancer Immunity and Immunotherapy. Biomed. Pharmacother. 2021, 133, 110972. [Google Scholar] [CrossRef]
- Hopfner, K.-P.; Hornung, V. Molecular Mechanisms and Cellular Functions of CGAS–STING Signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef] [PubMed]
- Wan, D.; Jiang, W.; Hao, J. Research Advances in How the CGAS-STING Pathway Controls the Cellular Inflammatory Response. Front. Immunol. 2020, 11, 615. [Google Scholar] [CrossRef] [PubMed]
- Burdette, D.L.; Monroe, K.M.; Sotelo-Troha, K.; Iwig, J.S.; Eckert, B.; Hyodo, M.; Hayakawa, Y.; Vance, R.E. STING Is a Direct Innate Immune Sensor of Cyclic Di-GMP. Nature 2011, 478, 515–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Bai, X.; Chen, Z.J. Structures and Mechanisms in the CGAS-STING Innate Immunity Pathway. Immunity 2020, 53, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro-Silva, C.; Aydin, Ö.Z.; Mesquita-Ribeiro, R.; Slyskova, J.; Helfricht, A.; Marteijn, J.A.; Hoeijmakers, J.H.J.; Lans, H.; Vermeulen, W. DNA Damage Sensitivity of SWI/SNF-Deficient Cells Depends on TFIIH Subunit P62/GTF2H1. Nat. Commun. 2018, 9, 4067. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Yang, L.; Wang, C.; Zhao, W.; Ju, Z.; Zhang, W.; Shen, J.; Peng, Y.; An, C.; Luu, Y.T.; et al. Inhibition of the ATM/Chk2 Axis Promotes CGAS/STING Signaling in ARID1A-Deficient Tumors. J. Clin. Investig. 2020, 130, 5951–5966. [Google Scholar] [CrossRef] [PubMed]
- Chabanon, R.M.; Morel, D.; Eychenne, T.; Colmet-Daage, L.; Bajrami, I.; Dorvault, N.; Garrido, M.; Meisenberg, C.; Lamb, A.; Ngo, C.; et al. PBRM1 Deficiency Confers Synthetic Lethality to DNA Repair Inhibitors in Cancer. Cancer Res. 2021, 81, 2888–2902. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic Chromatin Triggers Inflammation in Senescence and Cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Shang, G.; Gui, X.; Zhang, X.; Bai, X.; Chen, Z.J. Structural Basis of STING Binding with and Phosphorylation by TBK1. Nature 2019, 567, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-D.; Wu, J.; Gao, D.; Wang, H.; Sun, L.; Chen, Z.J. Pivotal Roles of CGAS-CGAMP Signaling in Antiviral Defense and Immune Adjuvant Effects. Science 2013, 341, 1390–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, H.; Barber, G.N. STING Is an Endoplasmic Reticulum Adaptor That Facilitates Innate Immune Signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yum, S.; Li, M.; Fang, Y.; Chen, Z.J. TBK1 Recruitment to STING Activates Both IRF3 and NF-ΚB That Mediate Immune Defense against Tumors and Viral Infections. Proc. Natl. Acad. Sci. USA 2021, 118, e2100225118. [Google Scholar] [CrossRef]
- Brzostek-Racine, S.; Gordon, C.; Van Scoy, S.; Reich, N.C. The DNA Damage Response Induces IFN. J. Immunol. 2011, 187, 5336–5345. [Google Scholar] [CrossRef] [Green Version]
- Parker, B.S.; Rautela, J.; Hertzog, P.J. Antitumour Actions of Interferons: Implications for Cancer Therapy. Nat. Rev. Cancer 2016, 16, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, H.; Wu, X.; Ma, D.; Wu, J.; Wang, L.; Jiang, Y.; Fei, Y.; Zhu, C.; Tan, R.; et al. Nuclear CGAS Suppresses DNA Repair and Promotes Tumorigenesis. Nature 2018, 563, 131–136. [Google Scholar] [CrossRef]
- Bai, J.; Liu, F. Nuclear CGAS: Sequestration and Beyond. Protein Cell 2022, 13, 90–101. [Google Scholar] [CrossRef]
- Chen, H.; Chen, H.; Zhang, J.; Wang, Y.; Simoneau, A.; Yang, H.; Levine, A.S.; Zou, L.; Chen, Z.; Lan, L. CGAS Suppresses Genomic Instability as a Decelerator of Replication Forks. Sci. Adv. 2020, 6, eabb8941. [Google Scholar] [CrossRef]
- Zhang, Z.; Yuan, B.; Bao, M.; Lu, N.; Kim, T.; Liu, Y.-J. The Helicase DDX41 Senses Intracellular DNA Mediated by the Adaptor STING in Dendritic Cells. Nat. Immunol. 2011, 12, 959–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, G.N. STING-Dependent Signaling. Nat. Immunol. 2011, 12, 929–930. [Google Scholar] [CrossRef]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 Is an Innate Immune Sensor for Intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [Green Version]
- Cao, X. Self-Regulation and Cross-Regulation of Pattern-Recognition Receptor Signalling in Health and Disease. Nat. Rev. Immunol. 2016, 16, 35–50. [Google Scholar] [CrossRef]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) Is a Cytosolic DNA Sensor and an Activator of Innate Immune Response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey Daniel, R.; Latz, E.; Fitzgerald, K.A. AIM2 Recognizes Cytosolic DsDNA and Forms a Caspase-1-Activating Inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [Green Version]
- Chiu, Y.-H.; MacMillan, J.B.; Chen, Z.J. RNA Polymerase III Detects Cytosolic DNA and Induces Type I Interferons through the RIG-I Pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mankan, A.K.; Schmidt, T.; Chauhan, D.; Goldeck, M.; Höning, K.; Gaidt, M.; Kubarenko, A.V.; Andreeva, L.; Hopfner, K.; Hornung, V. Cytosolic RNA:DNA Hybrids Activate the cGAS –STING Axis. EMBO J. 2014, 33, 2937–2946. [Google Scholar] [CrossRef] [Green Version]
- Gasser, S.; Orsulic, S.; Brown, E.J.; Raulet, D.H. The DNA Damage Pathway Regulates Innate Immune System Ligands of the NKG2D Receptor. Nature 2005, 436, 1186–1190. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.; Gowen, B.G.; Zhang, L.; Wang, L.; Lau, S.; Iannello, A.; Xu, J.; Rovis, T.L.; Xiong, N.; Raulet, D.H. A Shed NKG2D Ligand That Promotes Natural Killer Cell Activation and Tumor Rejection. Science 2015, 348, 136–139. [Google Scholar] [CrossRef] [Green Version]
- Tang, K.-F.; Ren, H.; Cao, J.; Zeng, G.-L.; Xie, J.; Chen, M.; Wang, L.; He, C.-X. Decreased Dicer Expression Elicits DNA Damage and Up-Regulation of MICA and MICB. J. Cell Biol. 2008, 182, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Davalos, A.R.; Kawahara, M.; Malhotra, G.K.; Schaum, N.; Huang, J.; Ved, U.; Beausejour, C.M.; Coppe, J.-P.; Rodier, F.; Campisi, J. P53-Dependent Release of Alarmin HMGB1 Is a Central Mediator of Senescent Phenotypes. J. Cell Biol. 2013, 201, 613–629. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA Damage Response Induces Inflammation and Senescence by Inhibiting Autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef] [Green Version]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular Senescence and the Senescent Secretory Phenotype: Therapeutic Opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J. Aging, Cellular Senescence, and Cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Setton, J.; Lee, N.Y.; Riaz, N.; Powell, S.N. The Therapeutic Significance of Mutational Signatures from DNA Repair Deficiency in Cancer. Nat. Commun. 2018, 9, 3292. [Google Scholar] [CrossRef]
- Permata, T.B.M.; Hagiwara, Y.; Sato, H.; Yasuhara, T.; Oike, T.; Gondhowiardjo, S.; Held, K.D.; Nakano, T.; Shibata, A. Base Excision Repair Regulates PD-L1 Expression in Cancer Cells. Oncogene 2019, 38, 4452–4466. [Google Scholar] [CrossRef]
- Golan, T.; O’Kane, G.M.; Denroche, R.E.; Raitses-Gurevich, M.; Grant, R.C.; Holter, S.; Wang, Y.; Zhang, A.; Jang, G.H.; Stossel, C.; et al. Genomic Features and Classification of Homologous Recombination Deficient Pancreatic Ductal Adenocarcinoma. Gastroenterology 2021, 160, 2119–2132.e9. [Google Scholar] [CrossRef]
- Jiricny, J. Postreplicative Mismatch Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012633. [Google Scholar] [CrossRef]
- Ma, X.; Dong, L.; Liu, X.; Ou, K.; Yang, L. POLE/POLD1 Mutation and Tumor Immunotherapy. J. Exp. Clin. Cancer Res. 2022, 41, 216. [Google Scholar] [CrossRef]
- Amodio, V.; Lamba, S.; Chilà, R.; Cattaneo, C.M.; Mussolin, B.; Corti, G.; Rospo, G.; Berrino, E.; Tripodo, C.; Pisati, F.; et al. Genetic and Pharmacological Modulation of DNA Mismatch Repair Heterogeneous Tumors Promotes Immune Surveillance. Cancer Cell 2023, 41, 196–209.e5. [Google Scholar] [CrossRef] [PubMed]
- De Mattos-Arruda, L.; Vazquez, M.; Finotello, F.; Lepore, R.; Porta, E.; Hundal, J.; Amengual-Rigo, P.; Ng, C.K.Y.; Valencia, A.; Carrillo, J.; et al. Neoantigen Prediction and Computational Perspectives towards Clinical Benefit: Recommendations from the ESMO Precision Medicine Working Group. Ann. Oncol. 2020, 31, 978–990. [Google Scholar] [CrossRef]
- Kiyotani, K.; Chan, H.T.; Nakamura, Y. Immunopharmacogenomics towards Personalized Cancer Immunotherapy Targeting Neoantigens. Cancer Sci. 2018, 109, 542–549. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Zou, Z.; Du, J.; Su, S.; Shao, J.; Meng, F.; Yang, J.; Xu, Q.; Ding, N.; Yang, Y.; et al. Neoantigen Identification Strategies Enable Personalized Immunotherapy in Refractory Solid Tumors. J. Clin. Investig. 2019, 129, 2056–2070. [Google Scholar] [CrossRef] [PubMed]
- Roudko, V.; Greenbaum, B.; Bhardwaj, N. Computational Prediction and Validation of Tumor-Associated Neoantigens. Front. Immunol. 2020, 11, 27. [Google Scholar] [CrossRef] [PubMed]
- Richters, M.M.; Xia, H.; Campbell, K.M.; Gillanders, W.E.; Griffith, O.L.; Griffith, M. Best Practices for Bioinformatic Characterization of Neoantigens for Clinical Utility. Genome Med. 2019, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-C.; Robbins, P.F. Targeting Neoantigens for Cancer Immunotherapy: Table 1. Int. Immunol. 2016, 28, 365–370. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Jiang, M.; He, K.; Wang, H.; Chen, P.; Guo, H.; Zhao, W.; Lu, H.; He, Y.; Zhou, C. DNA Damage Response and Repair Gene Alterations Increase Tumor Mutational Burden and Promote Poor Prognosis of Advanced Lung Cancer. Front. Oncol. 2021, 11, 708294. [Google Scholar] [CrossRef]
- Georgoulias, G.; Zaravinos, A. Genomic Landscape of the Immunogenicity Regulation in Skin Melanomas with Diverse Tumor Mutation Burden. Front. Immunol. 2022, 13, 1006665. [Google Scholar] [CrossRef]
- Chen, M.; Huang, B.; Zhu, L.; Wang, Q.; Pang, Y.; Cheng, M.; Lian, H.; Liu, M.; Zhao, K.; Xu, S.; et al. DNA Damage Response Evaluation Provides Novel Insights for Personalized Immunotherapy in Glioma. Front. Immunol. 2022, 13, 875648. [Google Scholar] [CrossRef] [PubMed]
- Lou, S.; Wang, Y.; Zhang, J.; Yin, X.; Zhang, Y.; Wang, Y.; Xue, Y. Patient-Level DNA Damage Repair Pathway Profiles and Anti-Tumor Immunity for Gastric Cancer. Front. Immunol. 2022, 12, 806324. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zheng, S.; Liu, Y.; Li, X.; Wu, J.; Sun, Y.; Liu, G. DNA Damage Response and PD-1/PD-L1 Pathway in Ovarian Cancer. DNA Repair 2021, 102, 103112. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, L. Aneuploidy and Immune Evasion—A Biomarker of Response. Nat. Rev. Clin. Oncol. 2017, 14, 140. [Google Scholar] [CrossRef]
- Spurr, L.F.; Weichselbaum, R.R.; Pitroda, S.P. Tumor Aneuploidy Predicts Survival Following Immunotherapy across Multiple Cancers. Nat. Genet. 2022, 54, 1782–1785. [Google Scholar] [CrossRef]
- Spurr, L.F.; Martinez, C.A.; Kang, W.; Chen, M.; Zha, Y.; Hseu, R.; Gutiontov, S.I.; Turchan, W.T.; Lynch, C.M.; Pointer, K.B.; et al. Highly Aneuploid Non-Small Cell Lung Cancer Shows Enhanced Responsiveness to Concurrent Radiation and Immune Checkpoint Blockade. Nat. Cancer 2022, 3, 1498–1512. [Google Scholar] [CrossRef]
- Zanetti, M. Chromosomal Chaos Silences Immune Surveillance. Science 2017, 355, 249–250. [Google Scholar] [CrossRef]
- Davoli, T.; Uno, H.; Wooten, E.C.; Elledge, S.J. Tumor Aneuploidy Correlates with Markers of Immune Evasion and with Reduced Response to Immunotherapy. Science 2017, 355, eaaf8399. [Google Scholar] [CrossRef] [Green Version]
- Sloan, E.A.; Ring, K.L.; Willis, B.C.; Modesitt, S.C.; Mills, A.M. PD-L1 Expression in Mismatch Repair-Deficient Endometrial Carcinomas, Including Lynch Syndrome-Associated and MLH1 Promoter Hypermethylated Tumors. Am. J. Surg. Pathol. 2017, 41, 326–333. [Google Scholar] [CrossRef]
- Favier, A.; Varinot, J.; Uzan, C.; Duval, A.; Brocheriou, I.; Canlorbe, G. The Role of Immunohistochemistry Markers in Endometrial Cancer with Mismatch Repair Deficiency: A Systematic Review. Cancers 2022, 14, 3783. [Google Scholar] [CrossRef]
- Ramchander, N.C.; Ryan, N.A.J.; Walker, T.D.J.; Harries, L.; Bolton, J.; Bosse, T.; Evans, D.G.; Crosbie, E.J. Distinct Immunological Landscapes Characterize Inherited and Sporadic Mismatch Repair Deficient Endometrial Cancer. Front. Immunol. 2020, 10, 3023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, A.M.; Dill, E.A.; Moskaluk, C.A.; Dziegielewski, J.; Bullock, T.N.; Dillon, P.M. The Relationship Between Mismatch Repair Deficiency and PD-L1 Expression in Breast Carcinoma. Am. J. Surg. Pathol. 2018, 42, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-Associated B7-H1 Promotes T-Cell Apoptosis: A Potential Mechanism of Immune Evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [Green Version]
- Chabanon, R.M.; Muirhead, G.; Krastev, D.B.; Adam, J.; Morel, D.; Garrido, M.; Lamb, A.; Hénon, C.; Dorvault, N.; Rouanne, M.; et al. PARP Inhibition Enhances Tumor Cell–Intrinsic Immunity in ERCC1-Deficient Non–Small Cell Lung Cancer. J. Clin. Investig. 2019, 129, 1211–1228. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zheng, K.; Xiong, H.; Huang, Y.; Chen, X.; Zhou, Y.; Qin, W.; Su, J.; Chen, R.; Qiu, H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Provides a New Combination Therapy in Pancreatic Cancer. Front. Immunol. 2021, 12, 762989. [Google Scholar] [CrossRef]
- Sato, H.; Jeggo, P.A.; Shibata, A. Regulation of Programmed Death-ligand 1 Expression in Response to DNA Damage in Cancer Cells: Implications for Precision Medicine. Cancer Sci. 2019, 110, 3415–3423. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA Double-Strand Break Repair Pathway Regulates PD-L1 Expression in Cancer Cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouw, K.W.; Konstantinopoulos, P.A. From Checkpoint to Checkpoint: DNA Damage ATR/Chk1 Checkpoint Signalling Elicits PD-L1 Immune Checkpoint Activation. Br. J. Cancer 2018, 118, 933–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.E.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. J. Natl. Cancer Inst. 2017, 109, djw199. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.; Qin, K.; Lin, A.; Jiang, A.; Cheng, Q.; Liu, Z.; Zhang, J.; Luo, P. The Role of DNA Damage Repair (DDR) System in Response to Immune Checkpoint Inhibitor (ICI) Therapy. J. Exp. Clin. Cancer Res. 2022, 41, 268. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hamrouni, A.; Wolowiec, D.; Coiteux, V.; Kuliczkowski, K.; Hetuin, D.; Saudemont, A.; Quesnel, B. Plasma Cells from Multiple Myeloma Patients Express B7-H1 (PD-L1) and Increase Expression after Stimulation with IFN-γ and TLR Ligands via a MyD88-, TRAF6-, and MEK-Dependent Pathway. Blood 2007, 110, 296–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like Receptor 4–Dependent Contribution of the Immune System to Anticancer Chemotherapy and Radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef]
- Blank, C.; Gajewski, T.F.; Mackensen, A. Interaction of PD-L1 on Tumor Cells with PD-1 on Tumor-Specific T Cells as a Mechanism of Immune Evasion: Implications for Tumor Immunotherapy. Cancer Immunol. Immunother. 2005, 54, 307–314. [Google Scholar] [CrossRef]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on Tumor Cells in the Escape from Host Immune System and Tumor Immunotherapy by PD-L1 Blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajiwara, Y.; Tazawa, H.; Yamada, M.; Kanaya, N.; Fushimi, T.; Kikuchi, S.; Kuroda, S.; Ohara, T.; Noma, K.; Yoshida, R.; et al. Oncolytic Virus-Mediated Reducing of Myeloid-Derived Suppressor Cells Enhances the Efficacy of PD-L1 Blockade in Gemcitabine-Resistant Pancreatic Cancer. Cancer Immunol. Immunother. 2022. In press. [Google Scholar] [CrossRef]
- Al Nabhani, S.; Al Harthy, A.; Al Riyami, M.; Al Sinawi, S.; Al Rashdi, A.; Al Husseni, S.; Kumar, S. Programmed Death-Ligand 1 (PD-L1) Expression in Bladder Cancer and Its Correlation with Tumor Grade, Stage, and Outcome. Oman Med. J. 2022, 37, e441. [Google Scholar] [CrossRef]
- Tu, X.; Qin, B.; Zhang, Y.; Zhang, C.; Kahila, M.; Nowsheen, S.; Yin, P.; Yuan, J.; Pei, H.; Li, H.; et al. PD-L1 (B7-H1) Competes with the RNA Exosome to Regulate the DNA Damage Response and Can Be Targeted to Sensitize to Radiation or Chemotherapy. Mol. Cell 2019, 74, 1215–1226.e4. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the Pd-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Brunner, S.; Herndler-Brandstetter, D.; Arnold, C.R.; Wiegers, G.J.; Villunger, A.; Hackl, M.; Grillari, J.; Moreno-Villanueva, M.; Bürkle, A.; Grubeck-Loebenstein, B. Upregulation of MiR-24 Is Associated with a Decreased DNA Damage Response upon Etoposide Treatment in Highly Differentiated CD8 + T Cells Sensitizing Them to Apoptotic Cell Death. Aging Cell 2012, 11, 579–587. [Google Scholar] [CrossRef] [Green Version]
- Yan, Q.; Zhang, B.; Ling, X.; Zhu, B.; Mei, S.; Yang, H.; Zhang, D.; Huo, J.; Zhao, Z. CTLA-4 Facilitates DNA Damage–Induced Apoptosis by Interacting with PP2A. Front. Cell Dev. Biol. 2022, 10, 728771. [Google Scholar] [CrossRef]
- Ryan, A.E.; Shanahan, F.; O’Connell, J.; Houston, A.M. Addressing the “Fas Counterattack” Controversy: Blocking Fas Ligand Expression Suppresses Tumor Immune Evasion of Colon Cancer In Vivo. Cancer Res. 2005, 65, 9817–9823. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Powis de Tenbossche, C.G.; Cané, S.; Colau, D.; van Baren, N.; Lurquin, C.; Schmitt-Verhulst, A.-M.; Liljeström, P.; Uyttenhove, C.; Van den Eynde, B.J. Resistance to Cancer Immunotherapy Mediated by Apoptosis of Tumor-Infiltrating Lymphocytes. Nat. Commun. 2017, 8, 1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upadhyay, R.; Boiarsky, J.A.; Pantsulaia, G.; Svensson-Arvelund, J.; Lin, M.J.; Wroblewska, A.; Bhalla, S.; Scholler, N.; Bot, A.; Rossi, J.M.; et al. A Critical Role for Fas-Mediated Off-Target Tumor Killing in T-Cell Immunotherapy. Cancer Discov. 2021, 11, 599–613. [Google Scholar] [CrossRef]
- Raats, D.A.; Frenkel, N.; van Schelven, S.J.; Rinkes, I.H.; Laoukili, J.; Kranenburg, O. CD95 Ligand Induces Senescence in Mismatch Repair-Deficient Human Colon Cancer via Chronic Caspase-Mediated Induction of DNA Damage. Cell Death Dis. 2017, 8, e2669. [Google Scholar] [CrossRef] [PubMed]
- Gullickson, P.; Xu, Y.W.; Niedernhofer, L.J.; Thompson, E.L.; Yousefzadeh, M.J. The Role of DNA Repair in Immunological Diversity: From Molecular Mechanisms to Clinical Ramifications. Front. Immunol. 2022, 13, 834889. [Google Scholar] [CrossRef]
- Bednarski, J.J.; Sleckman, B.P. At the Intersection of DNA Damage and Immune Responses. Nat. Rev. Immunol. 2019, 19, 231–242. [Google Scholar] [CrossRef]
- Hoolehan, W.; Harris, J.C.; Byrum, J.N.; Simpson, D.A.; Rodgers, K.K. An Updated Definition of V(D)J Recombination Signal Sequences Revealed by High-Throughput Recombination Assays. Nucleic Acids Res. 2022, 50, 11696–11711. [Google Scholar] [CrossRef]
- Alt, F.W.; Zhang, Y.; Meng, F.-L.; Guo, C.; Schwer, B. Mechanisms of Programmed DNA Lesions and Genomic Instability in the Immune System. Cell 2013, 152, 417–429. [Google Scholar] [CrossRef] [Green Version]
- Johnston, R.; Mathias, B.; Crowley, S.J.; Schmidt, H.A.; White, L.S.; Mosammaparast, N.; Green, A.M.; Bednarski, J.J. Nuclease-independent Functions of RAG1 Direct Distinct DNA Damage Responses in B Cells. EMBO Rep. 2023, 24, e55429. [Google Scholar] [CrossRef]
- Picard, C.; Bobby Gaspar, H.; Al-Herz, W.; Bousfiha, A.; Casanova, J.-L.; Chatila, T.; Crow, Y.J.; Cunningham-Rundles, C.; Etzioni, A.; Franco, J.L.; et al. International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee Report on Inborn Errors of Immunity. J. Clin. Immunol. 2018, 38, 96–128. [Google Scholar] [CrossRef] [Green Version]
- Niewolik, D.; Schwarz, K. Physical ARTEMIS:DNA-PKcs Interaction Is Necessary for V(D)J Recombination. Nucleic Acids Res. 2022, 50, 2096–2110. [Google Scholar] [CrossRef]
- Thientosapol, E.S.; Sharbeen, G.; Lau, K.K.E.; Bosnjak, D.; Durack, T.; Stevanovski, I.; Weninger, W.; Jolly, C.J. Proximity to AGCT Sequences Dictates MMR-Independent versus MMR-Dependent Mechanisms for AID-Induced Mutation via UNG2. Nucleic Acids Res. 2016, 45, 3146–3157. [Google Scholar] [CrossRef] [Green Version]
- Safavi, S.; Larouche, A.; Zahn, A.; Patenaude, A.-M.; Domanska, D.; Dionne, K.; Rognes, T.; Dingler, F.; Kang, S.-K.; Liu, Y.; et al. The Uracil-DNA Glycosylase UNG Protects the Fitness of Normal and Cancer B Cells Expressing AID. NAR Cancer 2021, 2, zcaa019. [Google Scholar] [CrossRef]
- Roco, J.A.; Mesin, L.; Binder, S.C.; Nefzger, C.; Gonzalez-Figueroa, P.; Canete, P.F.; Ellyard, J.; Shen, Q.; Robert, P.A.; Cappello, J.; et al. Class-Switch Recombination Occurs Infrequently in Germinal Centers. Immunity 2019, 51, 337–350.e7. [Google Scholar] [CrossRef]
- Weitering, T.J.; Takada, S.; Weemaes, C.M.R.; van Schouwenburg, P.A.; van der Burg, M. ATM: Translating the DNA Damage Response to Adaptive Immunity. Trends Immunol. 2021, 42, 350–365. [Google Scholar] [CrossRef]
- Bahjat, M.; Guikema, J. The Complex Interplay between DNA Injury and Repair in Enzymatically Induced Mutagenesis and DNA Damage in B Lymphocytes. Int. J. Mol. Sci. 2017, 18, 1876. [Google Scholar] [CrossRef] [Green Version]
- Stratigopoulou, M.; van Dam, T.P.; Guikema, J.E.J. Base Excision Repair in the Immune System: Small DNA Lesions with Big Consequences. Front. Immunol. 2020, 11, 1084. [Google Scholar] [CrossRef]
- Rotte, A.; Jin, J.Y.; Lemaire, V. Mechanistic Overview of Immune Checkpoints to Support the Rational Design of Their Combinations in Cancer Immunotherapy. Ann. Oncol. 2018, 29, 71–83. [Google Scholar] [CrossRef] [Green Version]
- André, T.; Tougeron, D.; Piessen, G.; de la Fouchardière, C.; Louvet, C.; Adenis, A.; Jary, M.; Tournigand, C.; Aparicio, T.; Desrame, J.; et al. Neoadjuvant Nivolumab Plus Ipilimumab and Adjuvant Nivolumab in Localized Deficient Mismatch Repair/Microsatellite Instability–High Gastric or Esophagogastric Junction Adenocarcinoma: The GERCOR NEONIPIGA Phase II Study. J. Clin. Oncol. 2023, 41, 255–265. [Google Scholar] [CrossRef]
- André, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability–High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.-J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in Patients with Metastatic DNA Mismatch Repair-Deficient or Microsatellite Instability-High Colorectal Cancer (CheckMate 142): An Open-Label, Multicentre, Phase 2 Study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Sahin, I.H.; Akce, M.; Alese, O.; Shaib, W.; Lesinski, G.B.; El-Rayes, B.; Wu, C. Immune Checkpoint Inhibitors for the Treatment of MSI-H/MMR-D Colorectal Cancer and a Perspective on Resistance Mechanisms. Br. J. Cancer 2019, 121, 809–818. [Google Scholar] [CrossRef]
- Jiang, M.; Jia, K.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; He, Y.; Zhou, C. Alterations of DNA Damage Response Pathway: Biomarker and Therapeutic Strategy for Cancer Immunotherapy. Acta Pharm. Sin. B 2021, 11, 2983–2994. [Google Scholar] [CrossRef]
- Wang, D.-R.; Wu, X.-L.; Sun, Y.-L. Therapeutic Targets and Biomarkers of Tumor Immunotherapy: Response versus Non-Response. Signal Transduct. Target. Ther. 2022, 7, 331. [Google Scholar] [CrossRef]
- Grimaldi, A.; Cammarata, I.; Martire, C.; Focaccetti, C.; Piconese, S.; Buccilli, M.; Mancone, C.; Buzzacchino, F.; Berrios, J.R.G.; D’Alessandris, N.; et al. Combination of Chemotherapy and PD-1 Blockade Induces T Cell Responses to Tumor Non-Mutated Neoantigens. Commun. Biol. 2020, 3, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donlon, N.E.; Power, R.; Hayes, C.; Reynolds, J.V.; Lysaght, J. Radiotherapy, Immunotherapy, and the Tumour Microenvironment: Turning an Immunosuppressive Milieu into a Therapeutic Opportunity. Cancer Lett. 2021, 502, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Shi, Y.; Lees-Miller, S.P.; Tainer, J.A. Function and Molecular Mechanism of the DNA Damage Response in Immunity and Cancer Immunotherapy. Front. Immunol. 2021, 12, 797880. [Google Scholar] [CrossRef]
- Brown, J.S.; O’Carrigan, B.; Jackson, S.P.; Yap, T.A. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov. 2017, 7, 20–37. [Google Scholar] [CrossRef] [Green Version]
- Cetin, B.; Wabl, C.A.; Gumusay, O. The DNA Damaging Revolution. Crit. Rev. Oncol. Hematol. 2020, 156, 103117. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA Repair Defect in BRCA Mutant Cells as a Therapeutic Strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Ledermann, J.A.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Overall Survival in Patients with Platinum-Sensitive Recurrent Serous Ovarian Cancer Receiving Olaparib Maintenance Monotherapy: An Updated Analysis from a Randomised, Placebo-Controlled, Double-Blind, Phase 2 Trial. Lancet Oncol. 2016, 17, 1579–1589. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib Maintenance Therapy in Patients with Platinum-Sensitive Relapsed Serous Ovarian Cancer: A Preplanned Retrospective Analysis of Outcomes by BRCA Status in a Randomised Phase 2 Trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib Maintenance Therapy in Platinum-Sensitive Relapsed Ovarian Cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higuchi, T.; Flies, D.B.; Marjon, N.A.; Mantia-Smaldone, G.; Ronner, L.; Gimotty, P.A.; Adams, S.F. CTLA-4 Blockade Synergizes Therapeutically with PARP Inhibition in BRCA1-Deficient Ovarian Cancer. Cancer Immunol. Res. 2015, 3, 1257–1268. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Kim, H.-J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.K.; Konstantinopoulos, P.A. Combined PARP and Immune Checkpoint Inhibition in Ovarian Cancer. Trends Cancer 2019, 5, 524–528. [Google Scholar] [CrossRef]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Ramalingam, S.S.; Thara, E.; Awad, M.M.; Dowlati, A.; Haque, B.; Stinchcombe, T.E.; Dy, G.K.; Spigel, D.R.; Lu, S.; Iyer Singh, N.; et al. JASPER: Phase 2 Trial of First-line Niraparib plus Pembrolizumab in Patients with Advanced Non–Small Cell Lung Cancer. Cancer 2022, 128, 65–74. [Google Scholar] [CrossRef]
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.; Mita, M.; McCann, G.; Tan, A.R.; Wahner-Hendrickson, A.E.; Forero, A.; Anders, C.; Wulf, G.M.; et al. Open-Label Clinical Trial of Niraparib Combined with Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA Oncol. 2019, 5, 1132. [Google Scholar] [CrossRef] [Green Version]
- Houl, J.H.; Ye, Z.; Brosey, C.A.; Balapiti-Modarage, L.P.F.; Namjoshi, S.; Bacolla, A.; Laverty, D.; Walker, B.L.; Pourfarjam, Y.; Warden, L.S.; et al. Selective Small Molecule PARG Inhibitor Causes Replication Fork Stalling and Cancer Cell Death. Nat. Commun. 2019, 10, 5654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fathers, C.; Drayton, R.M.; Solovieva, S.; Bryant, H.E. Inhibition of Poly(ADP-Ribose) Glycohydrolase (PARG) Specifically Kills BRCA2-Deficient Tumor Cells. Cell Cycle 2012, 11, 990–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, T.A.; Krebs, M.G.; Postel-Vinay, S.; Bang, Y.J.; El-Khoueiry, A.; Abida, W.; Harrington, K.; Sundar, R.; Carter, L.; Castanon-Alvarez, E.; et al. Phase I Modular Study of AZD6738, a Novel Oral, Potent and Selective Ataxia Telangiectasia Rad3-Related (ATR) Inhibitor in Combination (Combo) with Carboplatin, Olaparib or Durvalumab in Patients (Pts) with Advanced Cancers. Eur. J. Cancer 2016, 69, S2. [Google Scholar] [CrossRef]
- Morales-Juarez, D.A.; Jackson, S.P. Clinical Prospects of WRN Inhibition as a Treatment for MSI Tumours. Npj Precis. Oncol. 2022, 6, 85. [Google Scholar] [CrossRef]
- Kelderman, S.; Schumacher, T.N.; Kvistborg, P. Mismatch Repair-Deficient Cancers Are Targets for Anti-PD-1 Therapy. Cancer Cell 2015, 28, 11–13. [Google Scholar] [CrossRef] [Green Version]
- Diaz, L.A.; Le, D.T. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 373, 1979. [Google Scholar] [CrossRef] [Green Version]
- Diaz, L.A.; Shiu, K.-K.; Kim, T.-W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab versus Chemotherapy for Microsatellite Instability-High or Mismatch Repair-Deficient Metastatic Colorectal Cancer (KEYNOTE-177): Final Analysis of a Randomised, Open-Label, Phase 3 Study. Lancet Oncol. 2022, 23, 659–670. [Google Scholar] [CrossRef]
- Romero, D. New First-Line Therapy for DMMR/MSI-H CRC. Nat. Rev. Clin. Oncol. 2021, 18, 63. [Google Scholar] [CrossRef]
- Zhao, P.; Li, L.; Jiang, X.; Li, Q. Mismatch Repair Deficiency/Microsatellite Instability-High as a Predictor for Anti-PD-1/PD-L1 Immunotherapy Efficacy. J. Hematol. Oncol. 2019, 12, 54. [Google Scholar] [CrossRef] [PubMed]
- Durbin, R.P. Letter: Acid Secretion by Gastric Mucous Membrane. Am. J. Physiol. 1975, 229, 1726. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Hellmann, M.D.; Ciuleanu, T.-E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef]
- High TMB Predicts Immunotherapy Benefit. Cancer Discov. 2018, 8, 668. [CrossRef] [Green Version]
- Killock, D. Nivolumab–Ipilimumab—Exploiting the Mutation Burden of NSCLCs. Nat. Rev. Clin. Oncol. 2018, 15, 403. [Google Scholar] [CrossRef]
- Ready, N.; Hellmann, M.D.; Awad, M.M.; Otterson, G.A.; Gutierrez, M.; Gainor, J.F.; Borghaei, H.; Jolivet, J.; Horn, L.; Mates, M.; et al. First-Line Nivolumab Plus Ipilimumab in Advanced Non–Small-Cell Lung Cancer (CheckMate 568): Outcomes by Programmed Death Ligand 1 and Tumor Mutational Burden as Biomarkers. J. Clin. Oncol. 2019, 37, 992–1000. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 Human Cancer Genomes Reveals the Landscape of Tumor Mutational Burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [Green Version]
- Ding, K.; He, Y.; Wei, J.; Fu, S.; Wang, J.; Chen, Z.; Zhang, H.; Qu, Y.; Liang, K.; Gong, X.; et al. A Score of DNA Damage Repair Pathway with the Predictive Ability for Chemotherapy and Immunotherapy Is Strongly Associated with Immune Signaling Pathway in Pan-Cancer. Front. Immunol. 2022, 13, 943090. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X.; Deng, X.; Zhang, Y.; Liao, R.; Li, Y.; Yang, H.; Chen, K. DNA Damage Repair Status Predicts Opposite Clinical Prognosis Immunotherapy and Non-Immunotherapy in Hepatocellular Carcinoma. Front. Immunol. 2021, 12, 676922. [Google Scholar] [CrossRef] [PubMed]
- Bruand, M.; Barras, D.; Mina, M.; Ghisoni, E.; Morotti, M.; Lanitis, E.; Fahr, N.; Desbuisson, M.; Grimm, A.; Zhang, H.; et al. Cell-Autonomous Inflammation of BRCA1-Deficient Ovarian Cancers Drives Both Tumor-Intrinsic Immunoreactivity and Immune Resistance via STING. Cell Rep. 2021, 36, 109412. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Target | Drug | Tumor Type |
|---|---|---|
| PARP | Olaparib, Niraparib, Veliparib, Rucaparib | TNBC, SCLC, NSCLC, ovarian cancer, bladder cancer, prostate, colorectal cancers, pancreatic cancer, advanced solid tumors |
| ATM | AZD-1390, M-4076, XRD-0394, AZD0156, M3541 | Solid tumors |
| ATR | Berzosertib, Ceralasertib, RP-3500, ART-0380, ATRN-119, M-4344, M-1774, Elimusertib | Ovarian, Advanced NSCLC, SCLC, Gynae or neuroendocrine, HNSCC, relapsed CLL, PLL, B-cell lymphomas |
| DNA-PK | M3814, AZD-7648, CC-115, BR2002, BR101801 | GBM, HNSCC, prostate, ES, CLL |
| MEK1/2 | Selumetinib, Binimetinib, Cobinetinib, Trametinib | Melanoma, colorectal cancer, NF1-associated neurofibroma |
| WEE1 | Adavosertib, ZN-c3, IMP7068, SY-4835, Debio0123 | Endometrial serous carcinoma, osteosarcoma, solid tumor, NSCLC, gastric carcinoma, AML, other myeloid malignancies |
| CHK1/2 | MK8776, LY2603618, CCT245737, LY2606368 | NSCLC, refractory SCLC, relapsed AML, relapsed lymphoma, pancreatic carcinoma, ovarian, breast, prostate, pediatric solid tumors |
| BER | TRC102 | GBM, lymphoma, hematologic malignancies, NSCLC |
| ICBs | DDRi | Study Identifier (Status) | Tumor Type | Phase | Primary Endpoint |
|---|---|---|---|---|---|
| Pembrolizumab | Niraparib | NCT02657889 (completed) | Ovarian, advanced TNBC | I/II | DLTs, ORR |
| NCT04475939 (recruiting) | Advanced or metastatic NSCLC | III | 3-year PFS, 5-year OS | ||
| NCT03308942 (completed) | NSCLC | II | ORR | ||
| Olaparib | NCT04191135 (not recruiting) | TNBC | II/III | PFS | |
| NCT04548752 (recruiting) | Inherited BRCA-mutation pancreatic cancer | II | PFS | ||
| NCT02861573 (recruiting) | Metastatic castration-resistant prostate cancer | Ib/II | ORR | ||
| Nivolumab | Rucaparib | NCT03338790 (not recruiting) | Metastatic castration-resistant prostate cancer | I | ORR |
| NCT03522246 (not recruiting) | Ovarian cancer | III | 7-year PFS | ||
| NCT03572478 (terminated) | Prostate or endometrial cancer | Ib/II | DLTs | ||
| Veliparib | NCT02944396 (completed) | Metastatic or advanced NSCLC | II | PFS | |
| Camrelizumab (SHR-1210) | Apatinib | NCT03394287 (completed) | Advanced TNBC | II | ORR |
| Tislelizumab (BGB-A317) | Pamiparib (BGB-290) | NCT02660034 (completed) | Advanced solid tumors | I | AEs |
| Dostarlimab (TSR-042) | Niraparib | NCT03307785 (not recruiting) | Solid tumor | I/II | DLTs |
| NCT03602859 (not recruiting) | III or IV nonmucinous epithelial ovarian cancer | III | 5-year PFS | ||
| Durvalumab | Olaparib | NCT03167619 (completed) | Advanced TNBC | II | 1-year PFS |
| NCT02734004 (not recruiting) | Ovarian, breast, SCLC, gastric cancers | I/II | Disease control rate | ||
| NCT02546661 (not recruiting) | Muscle-invasive bladder cancer | I | AEs | ||
| NCT03459846 (not recruiting) | Urinary bladder neoplasms | II | PFS | ||
| NCT02484404 (recruiting) | Recurrent ovarian cancer | I/II | ORR | ||
| NCT03334617 (recruiting) | Advanced NSCLC | II | ORR | ||
| Durvalumab | Olaparib | NCT03534492 (completed) | Resectable urothelial bladder cancer | II | Pathological complete response rate |
| Durvalumab+ Tremelimumab | Olaparib | NCT02953457 (not recruiting) | Recurrent or refractory ovarian, fallopian tube, or primary peritoneal cancer with BRCA1 or BRCA2 mutation | II | DLTs |
| Durvalumab | Olaparib | NCT03851614 (not recruiting) | Mismatch repair proficient colorectal cancer, pancreatic adenocarcinoma, leiomyosarcoma | II | Changes in genomic and immune biomarkers |
| Ceralasertib | NCT02264678 (recruiting) | Advanced Solid Tumors | I/II | AEs | |
| Olaparib | NCT02484404 (recruiting) | Advanced Solid Tumors and Advanced or Recurrent Ovarian, TN BC, Lung, Prostate, and Colorectal Cancers | I/II | ORR | |
| Adavosertib (AZD1775) | NCT02617277 (not recruiting) | Advanced solid tumors | I | DLTs | |
| Ipilimumab or Nivolumab | Niraparib | NCT03404960 (not recruiting) | Pancreatic adenocarcinoma | I/II | PFS |
| Avelumab | Talazoparib | NCT03330405 (not recruiting) | Locally advanced (primary or recurrent) or metastatic solid tumors | Ib/II | DLTs |
| NCT03565991. (not recruiting) | BRCA1/2 or ATM alterations tumor | II | Confirmed Objective Response | ||
| NCT03637491 (terminated) | Locally advanced or metastatic RAS-mutant solid tumors | Ib/II | DLTs | ||
| NCT03642132 (completed) | Ovarian cancer | III | PFS | ||
| NCT03964532 (not recruiting) | Advanced breast cancer | I/II | AEs | ||
| Atezolizumab | Olaparib | NCT02849496 (not recruiting) | Mutant Non-HER2-positive breast cancer | II | PFS |
| Rucaparib | NCT03101280 (completed) | Advanced gynecologic cancers, TNBC | IB | AEs | |
| Niraparib | NCT03598270 (not recruiting) | Recurrent ovarian cancer | III | PFS | |
| Tremelimumab | Olaparib | NCT02571725 (not recruiting) | BRCA-deficient ovarian cancer | I/II | DLT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Nowsheen, S.; Deng, M. DNA Repair Deficiency Regulates Immunity Response in Cancers: Molecular Mechanism and Approaches for Combining Immunotherapy. Cancers 2023, 15, 1619. https://doi.org/10.3390/cancers15051619
Xu Y, Nowsheen S, Deng M. DNA Repair Deficiency Regulates Immunity Response in Cancers: Molecular Mechanism and Approaches for Combining Immunotherapy. Cancers. 2023; 15(5):1619. https://doi.org/10.3390/cancers15051619
Chicago/Turabian StyleXu, Yi, Somaira Nowsheen, and Min Deng. 2023. "DNA Repair Deficiency Regulates Immunity Response in Cancers: Molecular Mechanism and Approaches for Combining Immunotherapy" Cancers 15, no. 5: 1619. https://doi.org/10.3390/cancers15051619