
Immunotargeting of Cancer Stem Cells
 , , and
, , and
Abstract
:Simple Summary
Abstract
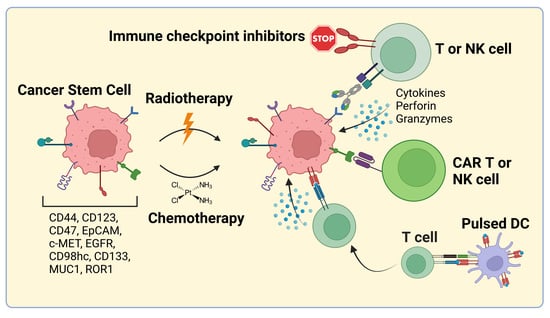
1. Introduction
1.1. CSC Definition and Clinical Significance
1.2. CSC-Directed Therapeutic Approaches
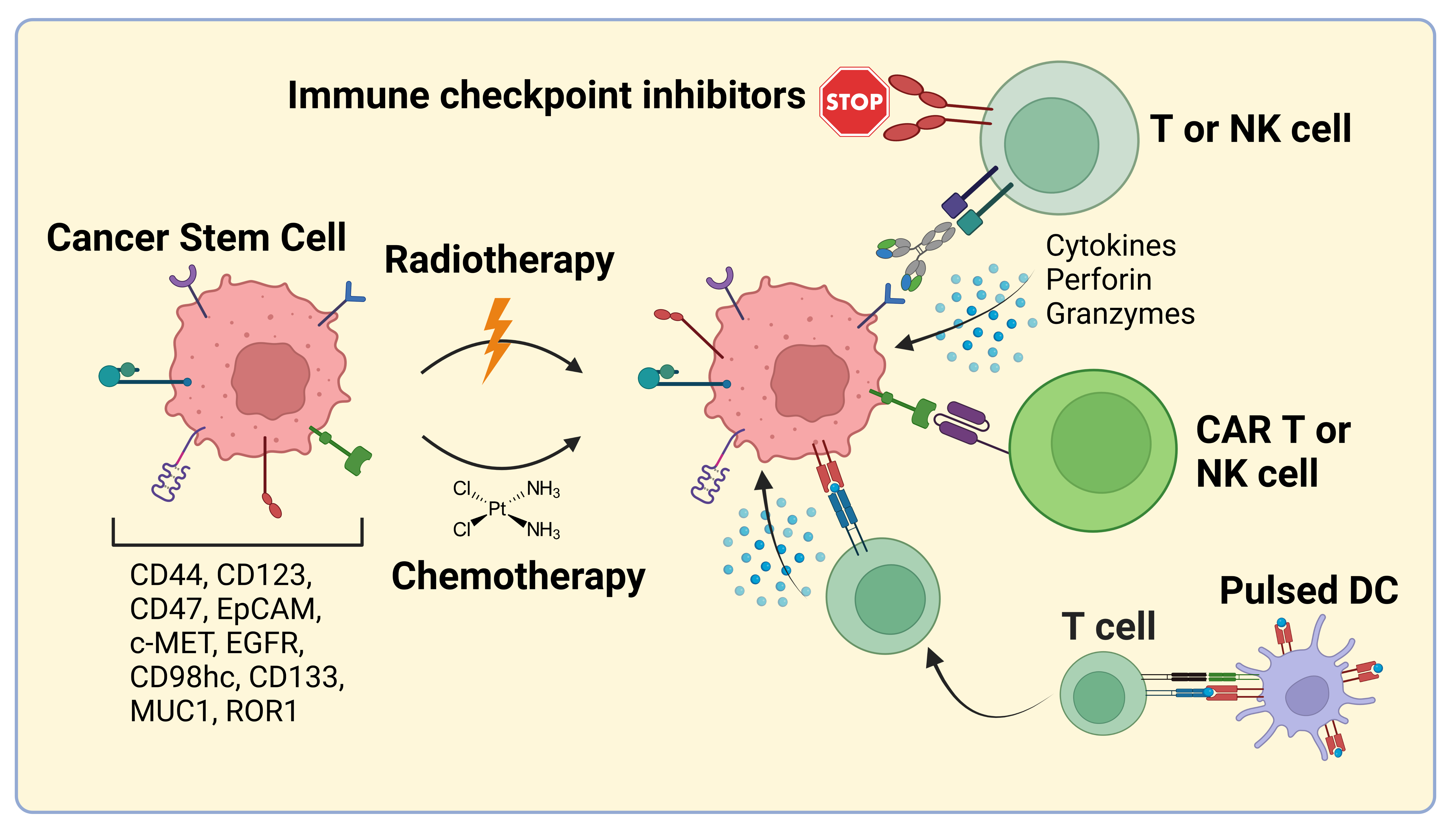
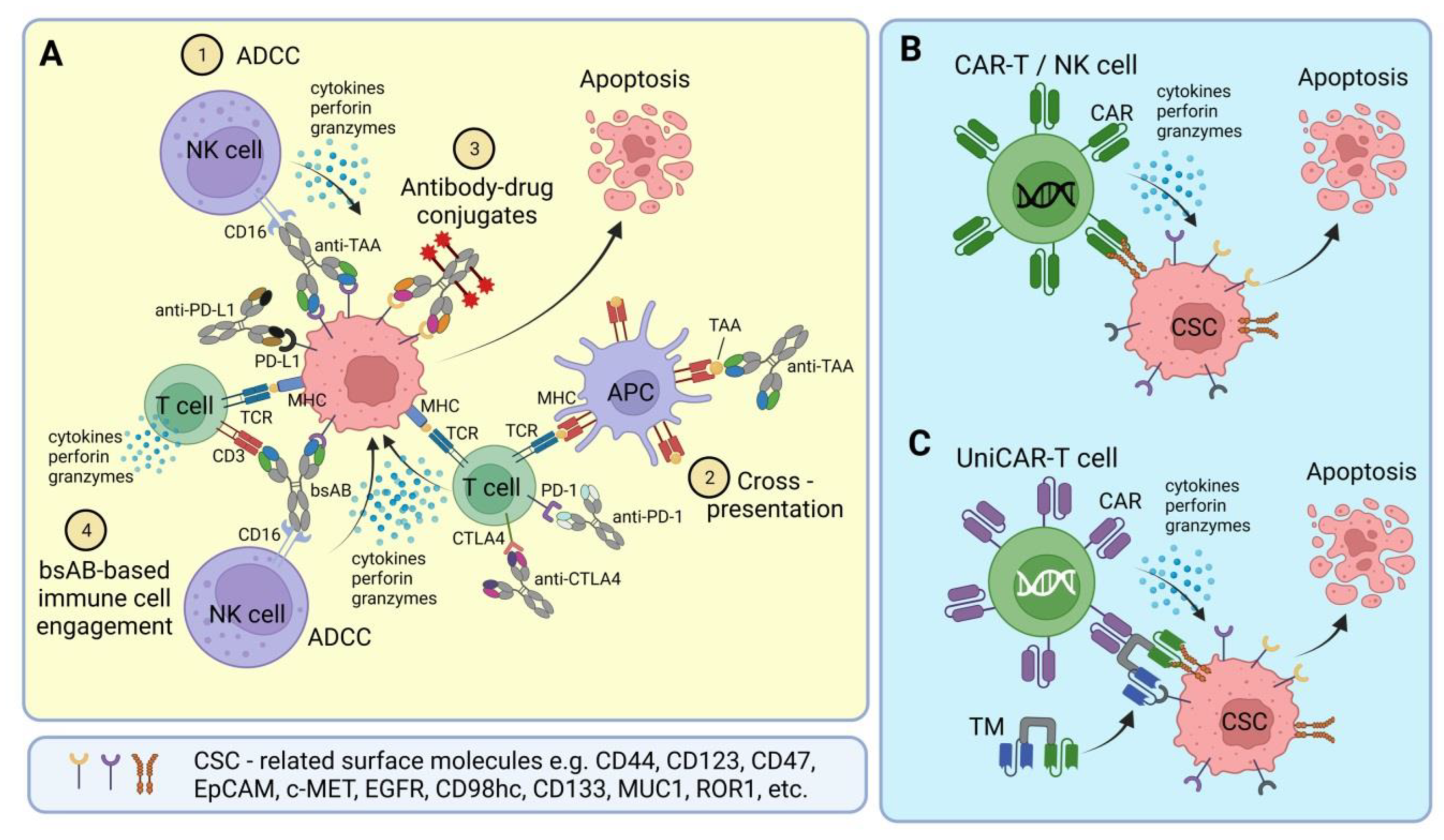
2. Bispecific Antibodies and Antibody-Drug Conjugates

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Specificity/Generic Name | Description | Tumor Entity Tested | Clinical Trials/Approvals | References |
|---|---|---|---|---|
| CD44v6/Bivatuzumab (BIWA 4) | mAb against CD44v6, (186)Re-labeled | Inoperable recurrent and/or metastatic HNSCC, NSCLC, breast cancer | Phase I: NCT02204059, NCT02204046, NCT02254018 Outcome: Antitumor effects and effective tumor targeting was observed. Administration is well tolerated. | [148,149] |
| CD44v6/Bivatuzumab—mertansine | mAb against CD44v6, conjugated mertansine | Incurable HNSCC or esophagus squamous cell carcinoma (ESCC), recurrent or metastatic breast cancer | Phase I: NCT02254044, NCT02254031, NCT02254005, NCT02254018 Outcome: one fatal drug-related adverse skin event had occurred. Further clinical development was discontinued. | [87,150,151,152] |
| CD44v6/RG7356 | mAb against CD44v6 | Advanced CD44-expressing solid malignancies. | Phase I study: NCT01358903 Outcome: acceptable safety profile, modest clinical efficacy was observed. The study was terminated due to the absence of a clinical and pharmacodynamic dose-response relationship | [88] |
| CD44v6/RG7356 | mAb against CD44v6 | AML | Phase I study; NCT01641250 Outcome: the treatment was generally safe and well tolerated. Out of 44 patients, two patients achieved complete or partial response and one patient had stable disease. | [89] |
| CD123/JNJ-56022473 /Talacotuzumab | 7G3 mAb against CD123 | Elderly high-risk MDS or AML failing hypomethylating agents | Phase II: NCT02992860 Talacotuzumab as a single agent; Outcome: limited clinical efficacy and significant toxicity | [153] |
| CD123/JNJ-56022473 /Talacotuzumab | 7G3 mAb against CD123 | CD123-positive AML | Phase II/III study: NCT02472145 Talacotuzumab in combination with decitabine versus decitabine alone; Outcome: no improvement in efficacy versus decitabine alone | [107] |
| CD123/IMGN632 | mAb G4723A against CD123 conjugated with DNA-alkylating payload of the IGN cytotoxic compounds | CD123-positive AML | Phase Ib/II study; NCT03386513 IMGN632 is given as monotherapy or in combination with AZA and/or VEN; Outcome: manageable toxicity profile; high ORR (of 75%) and CCR (of 40%) in high intensity cohort; ORR/CCR rates were even higher in the cohort of VEN-naïve patients (100%/60%, respectively) | [110,154] |
| CD47 IBI188/Letaplimab | mAb against CD47 | Newly diagnosed higher risk MDS | Phase I study: NCT04485065 The preliminary results suggest that IBI188 in combination with AZA showed a promising efficacy and a manageable toxicity profile | [99] |
| CD123 and CD3 Flotetuzumab/ MGD006 | bsAB (CD3ε × CD123) | Relapsed/refractory AML | Phase I/II study: NCT02152956 Outcome: acceptable safety profile, encouraging anti-leukemic activity (the complete remission rate (CRR)/CRR with partial hematological recovery was 26.7%; an overall response rate was 30.0% | [118] |
| CD47 and PD-1 HX009 | bsAB antibody binding CD47 and PD-1 | Relapsed or refractory lymphoma | Phase I/II study: NCT0409776, The preliminary results suggest that HX009 is well-tolerated and showed strong antitumor activity | [120,121] |
| EGFR and c-MET Amivantamab/Rybrevant/ JNJ-61186372 | bsAB antibody binding EGFR with one Fab and c-Met with the other Fab | Advanced or metastatic solid tumors including EGFR-mutated NSCLC | Amivantamab was approved by the US FDA for the treatment of patients with advanced or metastatic NSCLC with EGFR ex20ins mutations, whose disease has progressed on or after platinum-based chemotherapy. | [125,126,127] |
| EpCAM and CD3 Catumaxomab/Removab | EpCAM × CD3; trAb binding tumor cells, T cells, and accessory cells (e.g., macrophages, DC, and NK cells through its intact Fc region | Malignant ascites derived from epithelial tumors | Catumaxomab was approved in the European Union in April 2009 for the treatment of malignant ascites, but was withdrawn in 2017 for commercial reasons. | [112] |
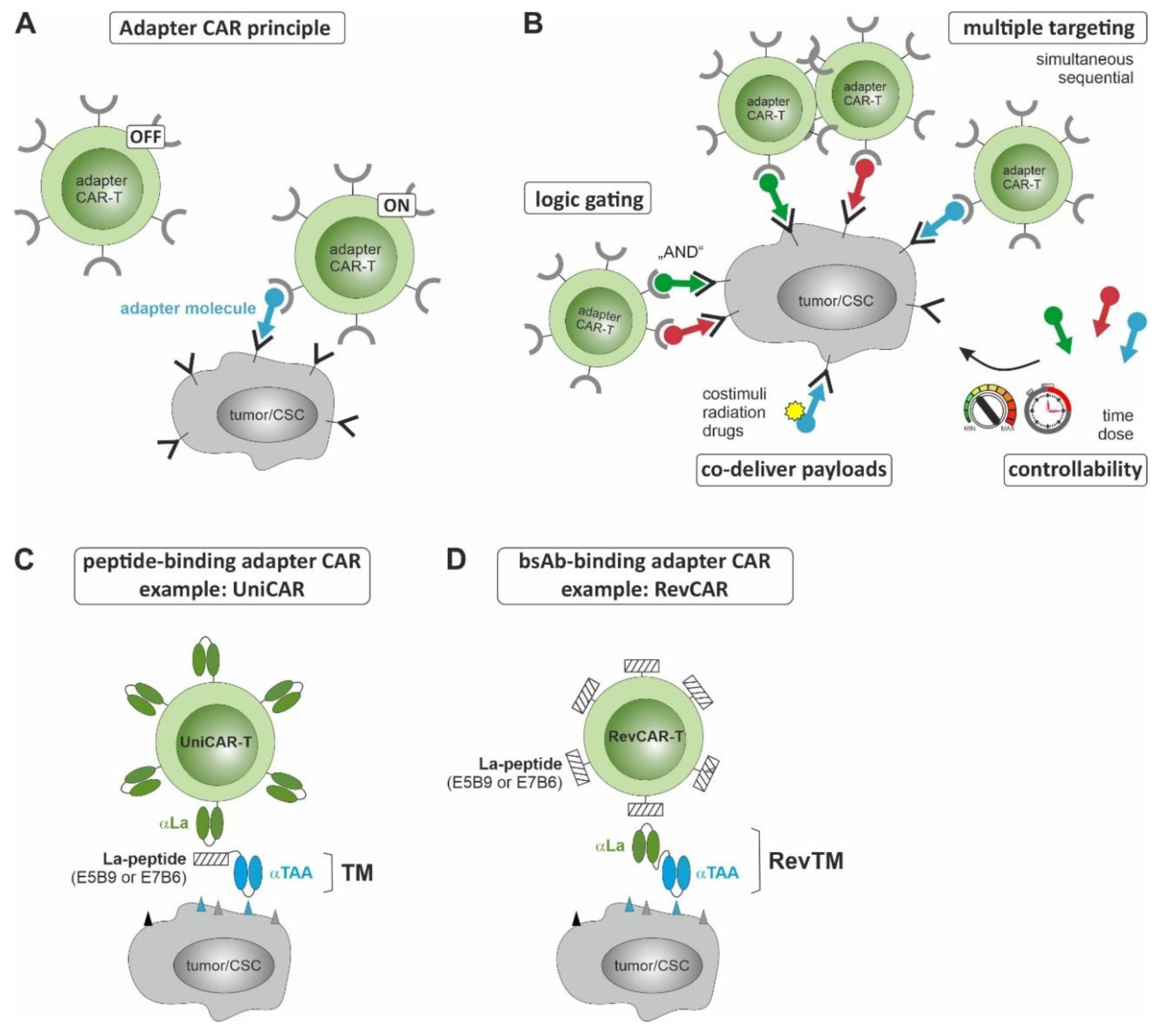
3. CSC-Targeted Immune Cells
| Specificity /Generic Name | Description | Tumor Entity Tested | Clinical Trials | References |
|---|---|---|---|---|
| CD44v6 | CAR-T cells | CD44v6 positive stomach cancer, lymphosarcoma | Phase I/II; NCT04427449 No results were posted. | |
| CD44v6 | MLM-CAR44.1 T cells; CD44v6 CAR-T cells were genetically modified to express herpes simplex virus (HSV)-TK Mut2 suicide gene to minimize toxicity | AML, Multiple Myeloma | Phase I/II; NCT04097301 Outcome: terminated due to the inability to close the study in a clinically relevant time frame. | [230] |
| CD133 | CAR-T cells | Relapsed and/or chemotherapy refractory advanced malignancies (liver cancer, pancreatic cancer, brain tumor, breast cancer, ovarian tumor, colorectal cancer, acute myeloid and lymphoid leukemia) | Phase I/II; NCT02541370 Outcome: out of 21 enrolled patients, 1 had a partial response, 14 had stable disease during 2–16.3 months, and 6 progressed after treatment start; hyperbilirubinemia was the most common high-grade adverse event | [231] |
| CD38-CART/ CD33-CART/ CD56-CART/ CD123-CART/ CD117-CART/ CD133-CART/ CD34-CART/ MUCl-CART | single CAR-T or double CAR-T cells with CD33,CD38, CD56,CD123, CD117,CD133, CD34 or MUCl | AML | Phase: n/a; NCT03473457 Outcome: terminated because the therapeutic effect was not as expected. No results were posted | |
| EpCAM | CAR-T cells | Nasopharyngeal carcinoma, breast cancer, gastric cancer and other EpCAM positive solid tumors | Phase I; NCT02915445 No results were posted | |
| EpCAM- and TM4SF1 | CAR-T cells | Refractory/recurrent advanced pancreatic cancer, colorectal cancer, gastric cancer or lung cancer | Phase: n/a; NCT04151186 No results were posted | |
| CD123 | CAR-NK cells | AML | Phase: I; NCT05574608 No results were posted | |
| MUC1 | CAR-NK cells | MUC1 positive relapsed or refractory solid tumor | Phase I/II; NCT02839954 No results were posted | |
| CD123 | Preconditioning (lymphodepletion) with cyclophosphamide and fludarabine followed by treatment with UniCAR-T and CD123 TM | Relapsed/refractory AML | Phase I; NCT04230265 Outcome: the initial results suggest that the treatment is well tolerated with mild adverse effects; out of three treated patients, one patient had a partial remission and two patients had complete remission with incomplete hematologic recovery | [220] |
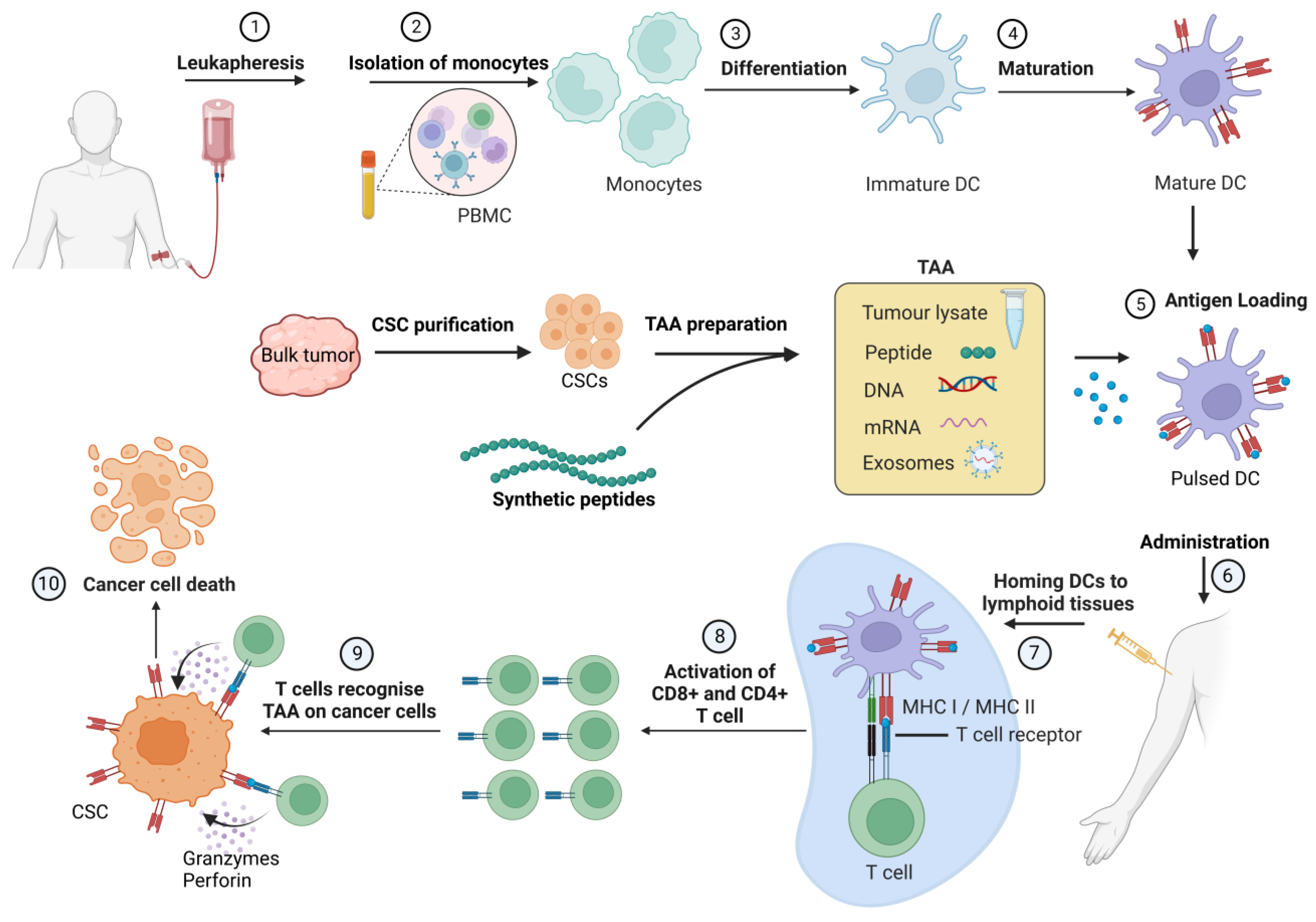
4. Cancer Stem Cell Vaccines
5. Preclinical and Clinical Trials of Combination Therapies with Immunotherapy and Conventional Therapies Targeting CSC Markers
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette |
| ACT | Adoptive cell therapy |
| ADCC | Antibody-dependent cellular cytotoxicity |
| ALDH | Aldehyde dehydrogenase |
| ALDHhigh | High ALDH activity |
| ALL | Acute lymphoblastic leukemia |
| AML | Acute myeloid leukemia |
| APC | Antigen-presenting cell |
| AZA | Azacitidine |
| BCMA | B-cell maturation antigen |
| BLL | B-lymphoid leukemia |
| BPDCN | Blastic plasmacytoid dendritic neoplasm |
| bsAB | Bispecific antibody |
| BTK | Bruton’s tyrosine kinase |
| CAF | Cancer-associated fibroblast |
| CAR | Chimeric antigen receptor |
| CCR | Composite complete remission rate |
| CD44v6 | CD44 isoform variant 6 |
| CEA | Carcinoembryonic antigen |
| CLL | Chronic lymphocytic leukemia |
| CRPC | Castration-resistant prostate cancer |
| CRR | Complete remission rate |
| CRS | Cytokine release syndrome |
| CSC | Cancer stem cell |
| CSC-DC | Dendritic CSC vaccination |
| CSC-TPDC | CSC-tumor pulsed DC |
| CTLA-4 | Cytotoxic T-lymphocyte-associated protein 4 |
| CTC | Circulating tumor cell |
| DC | Dendritic cell |
| EC | Endothelial cell |
| ECM | Extracellular matrix |
| EGFR | Epidermal growth factor receptor |
| EpCAM | Epithelial cell adhesion molecule |
| ESCC | Esophagus squamous cell carcinoma |
| ex20-ins | Insertions in exon 20 |
| FACS | Fluorescence-activated cell sorting |
| FAK | Focal adhesion kinase |
| FDA | Food and Drug Administration |
| FOLFOXIRI | Folinic acid, 5-fluorouracil, oxaliplatin and irinotecan |
| FTO | Fat mass and obesity-associated protein |
| GPC3 | Glypican-3 |
| GVHD | Graft versus host disease |
| H-TPDC | Heterogenous-tumor pulsed DC |
| HCL | Hairy cell leukemia |
| Hh | Hedgehog |
| HIF | Hypoxia-inducible transcriptional factor |
| HNSCC | Head and neck squamous cell carcinoma |
| ICANS | Immune effector cell-associated neurotoxicity syndrome |
| ICI | Immune checkpoint inhibitor |
| IGN | Indolinobenzodiazepine pseudodimer |
| IL-3R | Interleukin-3 receptor |
| iPSC | Induced pluripotent stem cell |
| LGR5 | Leucine-rich repeat-containing G protein-coupled receptor 5 |
| mAb | Monoclonal antibody |
| MDCS | Myeloid-derived suppressor cell |
| MDS | Myelodysplastic syndrome |
| MET | Mesenchymal-epithelial transition |
| MHC-I | Major histocompatibility complex class I |
| MMP-2 | Matrix metalloproteinase-2 |
| MsAb | Multi-specific antibody |
| MSC | Mesenchymal cells |
| MUC1 | Mucin 1 |
| ND | Nanodisk |
| NF-κB | Nuclear factor kappa B |
| NHL | Non-Hodgkin’s lymphoma |
| NK | Natural killer cell |
| NSCLC | Non-small cell lung cancer |
| ORR | Objective response rate |
| OS | Overall survival |
| PBMC | Peripheral blood mononuclear cell |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | Programmed death-ligand 1 |
| PFS | Progression-free survival |
| ROR1 | Receptor tyrosine kinase-like orphan receptor 1 |
| ROS | Reactive oxygen species |
| RT | Radiation therapy |
| RTK | Receptor tyrosine kinase |
| scFv | Single-chain variable region |
| SCID | Severe combined immunodeficient |
| SH2 | Src homology 2 |
| SHP-1 | Src homology 2 domain-containing phosphatase-1 |
| SHP-2 | Src homology 2 domain-containing phosphatase-2 |
| SIRPα | Signal regulatory protein α |
| STAT3 | Signal transducer and activator of transcription 3 |
| TAA | Tumor-associated antigen |
| TAM | Tumor-associated macrophage |
| TAP | The transporter associated with antigen processing |
| TAN | Tumor-associated neutrophil |
| TCR | T cell receptor |
| TM | Targeting module |
| TME | Tumor microenvironment |
| Treg | Regulatory T cell |
| trAb | Trifunctional antibody |
| TSP-1 | Thrombospondin 1 |
| UniCAR | Universal CAR |
| VEGF | Vascular endothelial growth factor |
| VEGFR-2 | Vascular endothelial growth factor receptor-2 |
| VEN | Venetoclax |
| VH | Heavy chain |
| VL | Light chain |
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Michael, A.; Caligiuri, J.; et al. A cell initiating human acute myeloid leukemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer Stem Cells: Current Status and Evolving Complexities. Cell Stem Cell 2012, 10, 717–728. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Peitzsch, C.; Tyutyunnykova, A.; Pantel, K.; Dubrovska, A. Cancer stem cells: The root of tumor recurrence and metastases. Semin. Cancer Biol. 2017, 44, 10–24. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.-B.S.; Zhang, H.; Damelin, M.; Geles, K.G.; Grindley, J.C.; Dirks, P.B. Tumour-initiating cells: Challenges and opportunities for anticancer drug discovery. Nat. Rev. Drug Discov. 2009, 8, 806–823. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Peng, M.; Li, N.; Song, Q.; Yao, Y.; Xu, B.; Liu, H.; Ruan, P. Combined use of EpCAM and FRalpha enables the high-efficiency capture of circulating tumor cells in non-small cell lung cancer. Sci. Rep. 2018, 8, 1188. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Wan, J.; Leng, D.; Zhang, Y.; Yang, S. Dual-targeting nanomicelles with CD133 and CD44 aptamers for enhanced delivery of gefitinib to two populations of lung cancer-initiating cells. Exp. Ther. Med. 2019, 19, 192–204. [Google Scholar] [CrossRef]
- Hadjimichael, C.; Chanoumidou, K.; Papadopoulou, N.; Arampatzi, P.; Papamatheakis, J.; Kretsovali, A. Common stemness regu-lators of embryonic and cancer stem cells. World J. Stem Cells 2015, 7, 1150–1184. [Google Scholar]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 Is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [Green Version]
- Ju, F.; Atyah, M.M.; Horstmann, N.; Gul, S.; Vago, R.; Bruns, C.J.; Zhao, Y.; Dong, Q.-Z.; Ren, N. Characteristics of the cancer stem cell niche and therapeutic strategies. Stem Cell Res. Ther. 2022, 13, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Han, M.-E.; Kim, H.-J.; Shin, D.H.; Hwang, S.-H.; Kang, C.-D.; Oh, S.-O. Overexpression of NRG1 promotes progression of gastric cancer by regulating the self-renewal of cancer stem cells. J. Gastroenterol. 2014, 50, 645–656. [Google Scholar] [CrossRef]
- Hu, Y.; Yan, C.; Mu, L.; Huang, K.; Li, X.; Tao, D.; Wu, Y.; Qin, J. Fibroblast-derived exosomes contribute to chemoresistance through priming cancer stem cells in colorectal cancer. PLoS ONE 2015, 10, e0125625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieto-Vila, M.; Takahashi, R.-U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef] [Green Version]
- Meurette, O.; Mehlen, P. Notch Signaling in the Tumor Microenvironment. Cancer Cell 2018, 34, 536–548. [Google Scholar] [CrossRef] [Green Version]
- Lau, E.Y.-T.; Ho, N.P.-Y.; Lee, T.K.-W. Cancer Stem Cells and Their Microenvironment: Biology and Therapeutic Implications. Stem Cells Int. 2017, 2017, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Grange, C.; Tapparo, M.; Collino, F.; Vitillo, L.; Damasco, C.; Deregibus, M.C.; Tetta, C.; Bussolati, B.; Camussi, G. Microvesicles Released from Human Renal Cancer Stem Cells Stimulate Angiogenesis and Formation of Lung Premetastatic Niche. Cancer Res. 2011, 71, 5346–5356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajani, J.A.; Song, S.; Hochster, H.S.; Steinberg, I.B. Cancer Stem Cells: The Promise and the Potential. Semin. Oncol. 2015, 42, S3–S17. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.; Dubrovska, A.; Linge, A.; Baumann, M. Cancer stem cells: Radioresistance, prediction of radiotherapy outcome and specific targets for combined treatments. Adv. Drug Deliv. Rev. 2016, 109, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Schulz, A.; Meyer, F.; Dubrovska, A.; Borgmann, K. Cancer Stem Cells and Radioresistance: DNA Repair and Beyond. Cancers 2019, 11, 862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cojoc, M.; Mäbert, K.; Muders, M.H.; Dubrovska, A. A role for cancer stem cells in therapy resistance: Cellular and molecular mechanisms. Semin. Cancer Biol. 2015, 31, 16–27. [Google Scholar] [CrossRef]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Pasello, M.; Giudice, A.M.; Scotlandi, K. The ABC subfamily A transporters: Multifaceted players with incipient potentialities in cancer. Semin. Cancer Biol. 2019, 60, 57–71. [Google Scholar] [CrossRef]
- Prager, B.C.; Xie, Q.; Bao, S.; Rich, J.N. Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell 2019, 24, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Liao, W.-T.; Ye, Y.-P.; Deng, Y.-J.; Bian, X.-W.; Ding, Y.-Q. Metastatic cancer stem cells: From the concept to therapeutics. Am. J. Stem Cells 2014, 3, 46–62. [Google Scholar]
- Garcia-Mayea, Y.; Mir, C.; Masson, F.; Paciucci, R.; Lleonart, M. Insights into new mechanisms and models of cancer stem cell multidrug resistance. Semin. Cancer Biol. 2020, 60, 166–180. [Google Scholar] [CrossRef]
- Ramos, E.K.; Hoffmann, A.D.; Gerson, S.L.; Liu, H. New Opportunities and Challenges to Defeat Cancer Stem Cells. Trends Cancer 2017, 3, 780–796. [Google Scholar] [CrossRef]
- Kolev, V.N.; Wright, Q.G.; Vidal, C.M.; Ring, J.E.; Shapiro, I.M.; Ricono, J.; Weaver, D.T.; Padval, M.V.; Pachter, J.A.; Xu, Q. PI3K/mTOR Dual Inhibitor VS-5584 Preferentially Targets Cancer Stem Cells. Cancer Res 2015, 75, 446–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mossahebi-Mohammadi, M.; Quan, M.; Zhang, J.-S.; Li, X. FGF Signaling Pathway: A Key Regulator of Stem Cell Pluripotency. Front. Cell Dev. Biol. 2020, 8, 79. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.N.; Ahn, D.H.; Kang, N.; Yeo, C.D.; Kim, Y.K.; Lee, K.Y.; Kim, T.J.; Lee, S.H.; Park, M.S.; Yim, H.W.; et al. TGF-beta induced EMT and stemness characteristics are associated with epigenetic reg-ulation in lung cancer. Sci. Rep. 2020, 10, 10597. [Google Scholar] [CrossRef]
- Kumar, V.; Vashishta, M.; Kong, L.; Wu, X.; Lu, J.J.; Guha, C.; Dwarakanath, B.S. The Role of Notch, Hedgehog, and Wnt Signaling Pathways in the Resistance of Tumors to Anticancer Therapies. Front. Cell Dev. Biol. 2021, 9, 650772. [Google Scholar] [CrossRef]
- Song, J.I.; Grandis, J.R. STAT signaling in head and neck cancer. Oncogene 2000, 19, 2489–2495. [Google Scholar] [CrossRef] [Green Version]
- Kaltschmidt, C.; Banz-Jansen, C.; Benhidjeb, T.; Beshay, M.; Förster, C.; Greiner, J.; Hamelmann, E.; Jorch, N.; Mertzlufft, F.; Pfitzenmaier, J.; et al. A Role for NF-kappaB in Organ Specific Cancer and Cancer Stem Cells. Cancers 2019, 11, 655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peitzsch, C.; Cojoc, M.; Hein, L.; Kurth, I.; Mäbert, K.; Trautmann, F.; Klink, B.; Schröck, E.; Wirth, M.P.; Krause, M.; et al. An Epigenetic Reprogramming Strategy to Resensitize Radioresistant Prostate Cancer Cells. Cancer Res. 2016, 76, 2637–2651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroon, P.; Berry, P.A.; Stower, M.J.; Rodrigues, G.; Mann, V.M.; Simms, M.; Bhasin, D.; Chettiar, S.; Li, C.; Li, P.-K.; et al. JAK-STAT Blockade Inhibits Tumor Initiation and Clonogenic Recovery of Prostate Cancer Stem-like Cells. Cancer Res. 2013, 73, 5288–5298. [Google Scholar] [CrossRef] [Green Version]
- Saygin, C.; Matei, D.; Majeti, R.; Reizes, O.; Lathia, J.D. Targeting Cancer Stemness in the Clinic: From Hype to Hope. Cell Stem. Cell 2018, 24, 25–40. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Dubrovska, A.; Salamone, R.J.; Walker, J.R.; Grandinetti, K.B.; Bonamy, G.M.C.; Orth, A.P.; Elliott, J.; Graus Porta, D.; Garcia-Echeverria, C.; et al. FGFR2 promotes breast tumorigenicity through maintenance of breast tumor-initiating cells. PLoS ONE 2013, 8, e51671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Z.; Goetz, J.A.; Singh, S.; Ogden, S.K.; Petty, W.J.; Black, C.C.; Memoli, V.A.; Dmitrovsky, E.; Robbins, D.J. Frequent requirement of hedgehog signaling in non-small cell lung carcinoma. Oncogene 2007, 26, 1046–1055. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Zhang, C.; Zhang, L.Y.; Dong, S.S.; Lu, L.H.; Chen, J.; Dai, Y.; Li, Y.; Kong, K.L.; Kwong, D.L.; et al. Wnt2 secreted by tumour fibroblasts promotes tumour progression in oesophageal cancer by activation of the Wnt/beta-catenin signalling pathway. Gut 2011, 60, 1635–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajakulendran, N.; Rowland, K.J.; Selvadurai, H.J.; Ahmadi, M.; Park, N.I.; Naumenko, S.; Dolma, S.; Ward, R.J.; So, M.; Lee, L.; et al. Wnt and Notch signaling govern self-renewal and differentiation in a subset of human glioblastoma stem cells. Genes Dev. 2019, 33, 498–510. [Google Scholar] [CrossRef] [PubMed]
- van Groningen, T.; Akogul, N.; Westerhout, E.M.; Chan, A.; Hasselt, N.E.; Zwijnenburg, D.A.; Broekmans, M.; Stroeken, P.; Haneveld, F.; Hooijer, G.K.J.; et al. A NOTCH feed-forward loop drives reprogramming from adrenergic to mesenchymal state in neuroblastoma. Nat. Commun. 2019, 10, 1530. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Xing, H.; Lu, D.; Wang, J.; Li, B.; Tang, J.; Gu, F.; Hong, L. Role of Jagged1/STAT3 signalling in platinum-resistant ovarian cancer. J. Cell Mol. Med. 2019, 23, 4005–4018. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.-T.; Li, Y.-L.; Zhang, Y.-Q.; Xu, T.; Lu, B.; Fang, L.; Gao, J.-Q.; Yu, L.-S.; Zhu, D.-F.; Yang, B.; et al. KLF4 functions as an oncogene in promoting cancer stem cell-like characteristics in osteosarcoma cells. Acta Pharmacol. Sin. 2018, 40, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Rosner, M.H.; Vigano, M.A.; Ozato, K.; Timmons, P.M.; Poirie, F.; Rigby, P.W.J.; Staudt, L.M. A POU-domain transcription factor in early stem cells and germ cells of the mammalian embryo. Nature 1990, 345, 686–692. [Google Scholar] [CrossRef]
- Lundberg, I.V.; Edin, S.; Eklof, V.; Oberg, A.; Palmqvist, R.; Wikberg, M.L. SOX2 expression is associated with a cancer stem cell state and down-regulation of CDX2 in colorectal cancer. BMC Cancer 2016, 16, 471. [Google Scholar] [CrossRef] [Green Version]
- Nathansen, J.; Lukiyanchuk, V.; Hein, L.; Stolte, M.I.; Borgmann, K.; Löck, S.; Kurth, I.; Baumann, M.; Krause, M.; Linge, A.; et al. Oct4 confers stemness and radioresistance to head and neck squamous cell car-cinoma by regulating the homologous recombination factors PSMC3IP and RAD54L. Oncogene 2021, 40, 4214–4228. [Google Scholar] [CrossRef]
- Jeter, C.R.; Liu, B.; Liu, X.; Chen, X.; Liu, C.; Calhoun-Davis, T.; Repass, J.; Zaehres, H.; Shen, J.J.; Tang, D.G. NANOG promotes cancer stem cell characteristics and prostate cancer resistance to androgen deprivation. Oncogene 2011, 30, 3833–3845. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting Cancer Stem Cell Pathways for Cancer Therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Allen, F.; Maillard, I. Therapeutic Targeting of Notch Signaling: From Cancer to Inflammatory Disorders. Front. Cell Dev. Biol. 2021, 9, 649205. [Google Scholar] [CrossRef]
- Ruiu, R.; Tarone, L.; Rolih, V.; Barutello, G.; Bolli, E.; Riccardo, F.; Cavallo, F.; Conti, L. Cancer stem cell immunology and immunotherapy: Harnessing the immune system against cancer's source. Prog. Mol. Biol. Transl. Sci. 2019, 164, 119–188. [Google Scholar]
- Xiang, T.; Long, H.; He, L.; Han, X.; Lin, K.; Liang, Z.; Zhuo, W.; Xie, R.; Zhu, B. Interleukin-17 produced by tumor microenvironment promotes self-renewal of CD133+ cancer stem-like cells in ovarian cancer. Oncogene 2013, 34, 165–176. [Google Scholar] [CrossRef]
- Casbon, A.-J.; Reynaud, D.; Park, C.; Khuc, E.; Gan, D.D.; Schepers, K.; Passegué, E.; Werb, Z. Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc. Natl. Acad. Sci. USA 2015, 112, E566–E575. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, L.; Jungwirth, U.; Avgustinova, A.; Iravani, M.; Mills, A.P.; Haider, S.; Harper, J.; Isacke, C.M. Cancer-Associated Fibroblasts Suppress CD8+ T-cell Infiltration and Confer Resistance to Immune-Checkpoint Blockade. Cancer Res. 2022, 82, 2904–2917. [Google Scholar] [CrossRef]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The Tumor Microenvironment: A Milieu Hindering and Obstructing An-titumor Immune Responses. Front Immunol. 2020, 11, 940. [Google Scholar] [CrossRef]
- Vito, A.; El-Sayes, N.; Mossman, K. Hypoxia-Driven Immune Escape in the Tumor Microenvironment. Cells 2020, 9, 992. [Google Scholar] [CrossRef]
- Calcinotto, A.; Filipazzi, P.; Grioni, M.; Iero, M.; De Milito, A.; Ricupito, A.; Cova, A.; Canese, R.; Jachetti, E.; Rossetti, M.; et al. Modulation of Microenvironment Acidity Reverses Anergy in Human and Murine Tumor-Infiltrating T Lymphocytes. Cancer Res. 2012, 72, 2746–2756. [Google Scholar] [CrossRef] [Green Version]
- Schatton, T.; Schütte, U.; Frank, N.Y.; Zhan, Q.; Hoerning, A.; Robles, S.C.; Zhou, J.; Hodi, F.S.; Spagnoli, G.C.; Murphy, G.F.; et al. Modulation of T-Cell Activation by Malignant Melanoma Initiating Cells. Cancer Res. 2010, 70, 697–708. [Google Scholar] [CrossRef] [Green Version]
- Reim, F.; Dombrowski, Y.; Ritter, C.; Buttmann, M.; Haüsler, S.; Ossadnik, M.; Krockenberger, M.; Beier, D.; Beier, C.P.; Dietl, J.; et al. Immunoselection of Breast and Ovarian Cancer Cells with Trastuzumab and Natural Killer Cells: Selective Escape of CD44high/CD24low/HER2low Breast Cancer Stem Cells. Cancer Res. 2009, 69, 8058–8066. [Google Scholar] [CrossRef] [Green Version]
- Golan, H.; Shukrun, R.; Caspi, R.; Vax, E.; Pode-Shakked, N.; Goldberg, S.; Pleniceanu, O.; Bar-Lev, D.D.; Mark-Danieli, M.; Pri-Chen, S.; et al. In Vivo Expansion of Cancer Stemness Affords Novel Cancer Stem Cell Targets: Malignant Rhabdoid Tumor as an Example. Stem Cell Rep. 2018, 11, 795–810. [Google Scholar] [CrossRef] [Green Version]
- Bruttel, V.S.; Wischhusen, J. Cancer stem cell immunology: Key to understanding tumorigenesis and tumor immune escape? Front. Immunol. 2014, 5, 360. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Shin, J.H.; Longmire, M.; Wang, H.; Kohrt, H.E.; Chang, H.Y.; Sunwoo, J.B. CD44+ Cells in Head and Neck Squamous Cell Carcinoma Suppress T-Cell–Mediated Immunity by Selective Constitutive and Inducible Expression of PD-LClin. Cancer Res. 2016, 22, 3571–3581. [Google Scholar] [CrossRef] [Green Version]
- Miao, Y.; Yang, H.; Levorse, J.; Yuan, S.; Polak, L.; Sribour, M.; Singh, B.; Rosenblum, M.D.; Fuchs, E. Adaptive Immune Resistance Emerges from Tumor-Initiating Stem Cells. Cell 2019, 177, 1172–1186.e14. [Google Scholar] [CrossRef]
- Dong, P.; Xiong, Y.; Yue, J.; Hanley, S.J.B.; Watari, H. Tumor-Intrinsic PD-L1 Signaling in Cancer Initiation, Development and Treatment: Beyond Immune Evasion. Front. Oncol. 2018, 8, 386. [Google Scholar] [CrossRef] [Green Version]
- Shaim, H.; Shanley, M.; Basar, R.; Daher, M.; Gumin, J.; Zamler, D.B.; Uprety, N.; Wang, F.; Huang, Y.; Gabrusiewicz, K.; et al. Targeting the αv integrin/TGF-β axis improves natural killer cell function against glio-blastoma stem cells. J. Clin. Investig. 2021, 131, e142116. [Google Scholar] [CrossRef]
- Hsu, J.-M.; Xia, W.; Hsu, Y.-H.; Chan, L.-C.; Yu, W.-H.; Cha, J.-H.; Chen, C.-T.; Liao, H.-W.; Kuo, C.-W.; Khoo, K.-H.; et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Zavros, Y. Initiation and Maintenance of Gastric Cancer: A Focus on CD44 Variant Isoforms and Cancer Stem Cells. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Guo, Q.; Li, X.; Yang, X.; Ni, H.; Wang, T.; Zhao, Q.; Liu, H.; Xing, Y.; Xi, T.; et al. MiR-873/PD-L1 axis regulates the stemness of breast cancer cells. eBioMedicine 2019, 41, 395–407. [Google Scholar] [CrossRef] [Green Version]
- Sumransub, N.; Jirapongwattana, N.; Jamjuntra, P.; Thongchot, S.; Chieochansin, T.; Yenchitsomanus, P.; Thuwajit, P.; Warnnissorn, M.; O-Charoenrat, P.; Thuwajit, C. Breast cancer stem cell RNA-pulsed dendritic cells enhance tumor cell killing by effector T cells. Oncol. Lett. 2020, 19, 2422–2430. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Jiang, J.; Zhan, M.; Zhang, H.; Wang, Q.T.; Sun, S.N.; Guo, X.K.; Yin, H.; Wei, Y.; Liu, J.O.; et al. Targeting Neoantigens in Hepatocellular Carcinoma for Immunotherapy: A Futile Strategy? Hepatology 2021, 73, 414–421. [Google Scholar] [CrossRef] [Green Version]
- Herbrich, S.; Baran, N.; Cai, T.; Weng, C.; Aitken, M.J.L.; Post, S.M.; Henderson, J.; Shi, C.; Havranek, O.; Richard-Carpentier, G.; et al. Overexpression of CD200 is a stem cell-specific mechanism of immune evasion in AML. J. Immunother. Cancer 2021, 9, e002968. [Google Scholar] [CrossRef]
- Dianat-Moghadam, H.; Heidarifard, M.; Jahanban-Esfahlan, R.; Panahi, Y.; Hamishehkar, H.; Pouremamali, F.; Rahbarghazi, R.; Nouri, M. Cancer stem cells-emanated therapy resistance: Implications for liposomal drug delivery systems. J. Control. Release 2018, 288, 62–83. [Google Scholar] [CrossRef]
- Chernosky, N.M.; Tamagno, I. The Role of the Innate Immune System in Cancer Dormancy and Relapse. Cancers 2021, 13, 5621. [Google Scholar] [CrossRef]
- Cornel, A.M.; Mimpen, I.L.; Nierkens, S. MHC Class I Downregulation in Cancer: Underlying Mechanisms and Potential Targets for Cancer Immunotherapy. Cancers 2020, 12, 1760. [Google Scholar] [CrossRef]
- Akhter, Z.; Sharawat, S.K.; Kumar, V.; Kochat, V.; Equbal, Z.; Ramakrishnan, M.; Kumar, U.; Mathur, S.; Kumar, L.; Mukhopadhyay, A. Aggressive serous epithelial ovarian cancer is potentially propagated by EpCAM+CD45+ phenotype. Oncogene 2018, 37, 2089–2103. [Google Scholar] [CrossRef]
- Zhong, Y.; Guan, K.; Guo, S.; Zhou, C.; Wang, D.; Ma, W.; Zhang, Y.; Li, C.; Zhang, S. Spheres derived from the human SK-RC-42 renal cell carcinoma cell line are enriched in cancer stem cells. Cancer Lett. 2010, 299, 150–160. [Google Scholar] [CrossRef]
- Kushwah, R.; Guezguez, B.; Lee, J.B.; Hopkins, C.I.; Bhatia, M. Pleiotropic roles of Notch signaling in normal, malignant, and developmental hematopoiesis in the human. EMBO Rep. 2014, 15, 1128–1138. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Dontu, G.; Mantle, I.D.; Patel, S.; Ahn, N.-S.; Jackson, K.W.; Suri, P.; Wicha, M.S. Hedgehog Signaling and Bmi-1 Regulate Self-renewal of Normal and Malignant Human Mammary Stem Cells. Cancer Res 2006, 66, 6063–6071. [Google Scholar] [CrossRef] [Green Version]
- Kahn, M. Can we safely target the WNT pathway? Nat. Rev. Drug. Discov. 2014, 13, 513–532. [Google Scholar] [CrossRef] [Green Version]
- Tilley, L.P.; Liu, S.K.; Gilbertson, S.R.; Wagner, B.M.; Lord, P.F. Primary myocardial disease in the cat. A model for human car-dio-myopathy. Am. J. Pathol. 1977, 86, 493–522. [Google Scholar]
- Maloney, D.G.; Grillo-Lopez, A.J.; White, C.A.; Bodkin, D.; Schilder, R.J.; Neidhart, J.A.; Janakiraman, N.; Foon, K.A.; Liles, T.M.; Dallaire, B.K.; et al. IDEC-C2B8 (Rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin's lymphoma. Blood 1997, 90, 2188–2195. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Zhao, S.; Karnad, A.; Freeman, J.W. The biology and role of CD44 in cancer progression: Therapeutic implications. J. Hematol. Oncol. 2018, 11, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Morath, I.; Jung, C.; Lévêque, R.; Linfeng, C.; Toillon, R.-A.; Warth, A.; Orian-Rousseau, V. Differential recruitment of CD44 isoforms by ErbB ligands reveals an involvement of CD44 in breast cancer. Oncogene 2018, 37, 1472–1484. [Google Scholar] [CrossRef] [PubMed]
- Tijink, B.M.; Buter, J.; de Bree, R.; Giaccone, G.; Lang, M.S.; Staab, A.; Leemans, C.R.; van Dongen, G.A. A Phase I Dose Escalation Study with Anti-CD44v6 Bivatuzumab Mertansine in Patients with Incurable Squamous Cell Carcinoma of the Head and Neck or Esophagus. Clin. Cancer Res. 2006, 12, 6064–6072. [Google Scholar] [CrossRef] [Green Version]
- Menke-van der Houven van Oordt, C.W.; Gomez-Roca, C.; van Herpen, C.; Coveler, A.L.; Mahalingam, D.; Verheul, H.M.W.; van der Graaf, W.T.A.; Christen, R.; Rüttinger, D.; Weigand, S.; et al. First-in-human phase I clinical trial of RG7356, an anti-CD44 humanized antibody, in patients with advanced, CD44-expressing solid tumors. Oncotarget. 2016, 7, 80046–80058. [Google Scholar] [CrossRef] [Green Version]
- Vey, N.; Delaunay, J.; Martinelli, G.; Fiedler, W.; Raffoux, E.; Prebet, T.; Gomez-Roca, C.; Papayannidis, C.; Kebenko, M.; Paschka, P.; et al. Phase I clinical study of RG7356, an anti-CD44 humanized antibody, in patients with acute myeloid leukemia. Oncotarget 2016, 7, 32532–32542. [Google Scholar] [CrossRef] [Green Version]
- Matlung, H.L.; Szilagyi, K.; Barclay, N.A.; van den Berg, T.K. The CD47-SIRPalpha signaling axis as an innate immune checkpoint in cancer. Immunol. Rev. 2017, 276, 145–164. [Google Scholar] [CrossRef]
- Barclay, A.N.; Van den Berg, T.K. The interaction between signal regulatory protein alpha (SIRPalpha) and CD47: Structure, function, and therapeutic target. Annu. Rev. Immunol. 2014, 32, 25–50. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.G.; Lindberg, F.P.; Dimitry, J.M.; Brown, E.J.; Frazier, W.A. Thrombospondin modulates alpha v beta 3 function through integrin-associated protein. J. Cell Biol. 1996, 135, 533–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayat, S.M.G.; Bianconi, V.; Pirro, M.; Jaafari, M.R.; Hatamipour, M.; Sahebkar, A. CD47: Role in the immune system and application to cancer therapy. Cell. Oncol. 2019, 43, 19–30. [Google Scholar] [CrossRef]
- Gao, L.; Chen, K.; Gao, Q.; Wang, X.; Sun, J.; Yang, Y.-G. CD47 deficiency in tumor stroma promotes tumor progression by enhancing angiogenesis. Oncotarget 2016, 8, 22406–22413. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Lu, H.; Xiang, L.; Bullen, J.W.; Zhang, C.; Samanta, D.; Gilkes, D.M.; He, J.; Semenza, G.L. HIF-1 regulates CD47 expression in breast cancer cells to promote evasion of phagocytosis and maintenance of cancer stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, E6215–E6223. [Google Scholar] [CrossRef] [Green Version]
- Cioffi, M.; Trabulo, S.; Hidalgo, M.; Costello, E.; Greenhalf, W.; Erkan, M.; Kleeff, J.; Sainz, B.; Heeschen, C. Inhibition of CD47 Effectively Targets Pancreatic Cancer Stem Cells via Dual Mechanisms. Clin. Cancer Res. 2015, 21, 2325–2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.L.; Wu, C.C.; Hsu, Y.T.; Hsu, Y.C. Immune vulnerability of ovarian cancer stem-like cells due to low CD47 expression is protected by surrounding bulk tumor cells. Oncoimmunology 2020, 9, 1803530. [Google Scholar] [CrossRef]
- Candas-Green, D.; Xie, B.; Huang, J.; Fan, M.; Wang, A.; Menaa, C.; Zhang, Y.; Zhang, L.; Jing, D.; Azghadi, S.; et al. Dual blockade of CD47 and HER2 eliminates radioresistant breast cancer cells. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Miao, M.; Wu, D.; Xie, S.; Hong, M.; Chang, C.; Yu, L.; Gao, S.; Li, Y.; Li, Y.; Zhu, Z.; et al. A Phase 1b Study to Evaluate Safety and Efficacy of IBI188 in Combination with Azacitidine (AZA) As a First-Line Treatment in Subjects with Newly Diagnosed Higher Risk Myelodysplastic Syndrome. Blood 2022, 140, 4045–4046. [Google Scholar] [CrossRef]
- Testa, U.; Pelosi, E.; Castelli, G. CD123 as a Therapeutic Target in the Treatment of Hematological Malignancies. Cancers 2019, 11, 1358. [Google Scholar] [CrossRef] [Green Version]
- El Achi, H.; Dupont, E.; Paul, S.; Khoury, J.D. CD123 as a Biomarker in Hematolymphoid Malignancies: Principles of Detection and Targeted Therapies. Cancers 2020, 12, 3087. [Google Scholar] [CrossRef] [PubMed]
- Pemmaraju, N.; Lane, A.A.; Sweet, K.L.; Stein, A.S.; Vasu, S.; Blum, W.; Rizzieri, D.A.; Wang, E.S.; Duvic, M.; Sloan, J.M.; et al. Tagraxofusp in Blastic Plasmacytoid Dendritic-Cell Neoplasm. N. Engl. J. Med. 2019, 380, 1628–1637. [Google Scholar] [CrossRef] [PubMed]
- Alkharabsheh, O.; Frankel, A.E. Clinical Activity and Tolerability of SL-401 (Tagraxofusp): Recombinant Diphtheria Toxin and Interleukin-3 in Hematologic Malignancies. Biomedicines 2019, 7, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.H.; Biondo, M.; Busfield, S.J.; Arruda, A.; Yang, X.; Vairo, G.; Minden, M.D. CD123 target validation and preclinical evaluation of ADCC activity of anti-CD123 antibody CSL362 in combination with NKs from AML patients in remission. Blood Cancer J. 2017, 7, e567. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.M.; Yee, D.; Busfield, S.J.; McManus, J.F.; Cummings, N.; Vairo, G.; Wei, A.; Ramshaw, H.; Powell, J.; Lopez, A.F.; et al. Efficacy of an Fc-modified anti-CD123 antibody (CSL362) combined with chemotherapy in xenograft models of acute myelogenous leukemia in immunodeficient mice. Haematologica 2015, 100, 914–926. [Google Scholar] [CrossRef]
- Nievergall, E.; Ramshaw, H.S.; Yong, A.S.M.; Biondo, M.; Busfield, S.J.; Vairo, G.; Lopez, A.F.; Hughes, T.P.; White, D.L.; Hiwase, D.K. Monoclonal antibody targeting of IL-3 receptor α with CSL362 effectively depletes CML progenitor and stem cells. Blood 2014, 123, 1218–1228. [Google Scholar] [CrossRef] [Green Version]
- Montesinos, P.; Roboz, G.J.; Bulabois, C.-E.; Subklewe, M.; Platzbecker, U.; Ofran, Y.; Papayannidis, C.; Wierzbowska, A.; Shin, H.J.; Doronin, V.; et al. Safety and efficacy of talacotuzumab plus decitabine or decitabine alone in patients with acute myeloid leukemia not eligible for chemotherapy: Results from a multicenter, randomized, phase 2/3 study. Leukemia 2020, 35, 62–74. [Google Scholar] [CrossRef] [Green Version]
- Kovtun, Y.; Jones, G.E.; Adams, S.; Harvey, L.; Audette, C.A.; Wilhelm, A.; Bai, C.; Rui, L.; Laleau, R.; Liu, F.; et al. A CD123-targeting antibody-drug conjugate, IMGN632, designed to eradicate AML while sparing normal bone marrow cells. Blood Adv. 2018, 2, 848–858. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.C.T.; Han, L.; Mary Kuruvilla, V.; Adams, S.; Callum, S.M.; Harutyunyan, K.; Lane, A.A.; Kovtun, Y.; Daver, N.G.; Pemmaraju, N.; et al. Pre-Clinical Efficacy of CD123-Targeting Antibody-Drug Conjugate IMGN632 in Blastic Plasmacytoid Dentritic Cell Neoplasm (BPDCN) Models. Blood 2018, 132 (Suppl. S1), 3956. [Google Scholar] [CrossRef]
- Daver, N.; Aribi, A.; Montesinos, P.; Roboz, G.J.; Wang, E.S.; Walter, R.B.; Jeyakumar, D.; DeAngelo, D.J.; Erba, H.P.; Advani, A.; et al. Safety and Efficacy from a Phase 1b/2 Study of IMGN632 in Combination with Aza-citidine and Venetoclax for Patients with CD123-Positive Acute Myeloid Leukemia. Blood 2021, 138 (Suppl. S1), 372. [Google Scholar] [CrossRef]
- Brinkmann, U.; Kontermann, R.E. Bispecific antibodies. Science 2021, 372, 916–917. [Google Scholar] [CrossRef] [PubMed]
- Linke, R.; Klein, A.; Seimetz, D. Catumaxomab: Clinical development and future directions. MABS 2010, 2, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Chen, K.; Lei, Q.; Ma, P.; Yuan, A.Q.; Zhao, Y.; Jiang, Y.; Fang, H.; Xing, S.; Fang, Y.; et al. The state of the art of bispecific antibodies for treating human malignancies. EMBO Mol. Med. 2021, 13, e14291. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, Y.; Fan, D.; Xiong, D. The development of bispecific antibodies and their applications in tumor immune escape. Exp. Hematol. Oncol. 2017, 6, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. mAbs 2016, 9, 182–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Mo, Y.; Tang, M.; Shen, J.; Qi, Y.; Zhao, W.; Huang, Y.; Xu, Y.; Qian, C. Bispecific Antibodies: From Research to Clinical Application. Front. Immunol. 2021, 12, 626616. [Google Scholar] [CrossRef]
- Sweet-Jones, J.; Ahmad, M.; Martin, A.C. Antibody markup language (AbML)—A notation language for antibody-based drug formats and software for creating and rendering AbML (abYdraw). mAbs 2022, 14, 2101183. [Google Scholar] [CrossRef]
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S.; Godwin, J.E.; Arellano, M.L.; Sweet, K.L.; Emadi, A.; et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood 2021, 137, 751–762. [Google Scholar] [CrossRef]
- Simon, S.; Labarriere, N. PD-1 expression on tumor-specific T cells: Friend or foe for immunotherapy? Oncoimmunology 2017, 7, e1364828. [Google Scholar] [CrossRef]
- Roohullah, A.; Ganju, V.; Zhang, F.; Zhang, L.; Yu, T.; Wilkinson, K.; Cooper, A.; de Souza, P. First-in-human phase 1 dose escalation study of HX009, a novel recombinant humanized anti-PD-1 and CD47 bispecific antibody, in patients with advanced malignancies. J. Clin. Oncol. 2021, 39, 2517. [Google Scholar] [CrossRef]
- An, X.; Li, H.; Ke, H.; Xiong, L.; Zhang, F.; Zang, M.; Tu, X.; Wang, J.; Liu, D.; Chen, C.; et al. 491 Preclinical pharmacology modeling of HX009, a clinical stage first-in-class PD-1xCD47 BsAb, for anti-lymphoma applications. J. Immuno. Ther. Cancer 2022, 10, 491. [Google Scholar]
- Wang, Y.; Ni, H.; Zhou, S.; He, K.; Gao, Y.; Wu, W.; Wu, M.; Wu, Z.; Qiu, X.; Zhou, Y.; et al. Tumor-selective blockade of CD47 signaling with a CD47/PD-L1 bispecific antibody for enhanced anti-tumor activity and limited toxicity. Cancer Immunol. Immunother. 2021, 70, 365–376. [Google Scholar] [CrossRef]
- Arcila, M.E.; Nafa, K.; Chaft, J.E.; Rekhtman, N.; Lau, C.; Reva, B.A.; Zakowski, M.F.; Kris, M.G.; Ladanyi, M. EGFR exon 20 insertion mutations in lung adenocarcinomas: Prevalence, molecular het-erogeneity, and clinicopathologic characteristics. Mol Cancer Ther. 2013, 12, 220–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, N.; Salgia, R. Synergism of EGFR and c-Met pathways, cross-talk and inhibition, in non-small cell lung cancer. J. Carcinog. 2008, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Moores, S.L.; Chiu, M.L.; Bushey, B.S.; Chevalier, K.; Luistro, L.; Dorn, K.; Brezski, R.J.; Haytko, P.; Kelly, T.; Wu, S.-J.; et al. A Novel Bispecific Antibody Targeting EGFR and cMet Is Effective against EGFR Inhibitor–Resistant Lung Tumors. Cancer Res. 2016, 76, 3942–3953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syed, Y.Y. Amivantamab: First Approval. Drugs 2021, 81, 1349–1353. [Google Scholar] [CrossRef]
- Parums, D.V. Editorial: Current Status of Two Adjuvanted Malaria Vaccines and the World Health Organization (WHO) Strategy to Eradicate Malaria by 2030. Med. Sci. Monit. 2023, 29, e939357. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wu, J.; Hynes, M.; Dosch, J.; Sarkar, B.; Welling, T.H.; di Magliano, M.P.; Simeone, D.M. c-Met Is a Marker of Pancreatic Cancer Stem Cells and Therapeutic Target. Gastroenterology 2011, 141, 2218–2227.e5. [Google Scholar] [CrossRef]
- Yan, B.; Jiang, Z.; Cheng, L.; Chen, K.; Zhou, C.; Sun, L.; Qian, W.; Li, J.; Cao, J.; Xu, Q.; et al. Paracrine HGF/c-MET enhances the stem cell-like potential and glycolysis of pancreatic cancer cells via activation of YAP/HIF-1α. Exp. Cell Res. 2018, 371, 63–71. [Google Scholar] [CrossRef]
- van Leenders, G.J.; Sookhlall, R.; Teubel, W.J.; Reneman, S.; Sacchetti, A.; Vissers, K.J.; van Weerden, W.; Jenster, G. Activation of c-MET induces a stem-like phenotype in human prostate cancer. PLoS ONE 2011, 6, e26753. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Lu, C.; Hsiang, Y.; Pi, S.; Chen, C.; Cheng, K.; Pan, H.; Chien, P.; Chen, Y. c-Met inhibition is required for the celecoxib-attenuated stemness property of human colorectal cancer cells. J. Cell. Physiol. 2018, 234, 10336–10344. [Google Scholar] [CrossRef]
- Imrich, S.; Hachmeister, M.; Gires, O. EpCAM and its potential role in tumor-initiating cells. Cell Adhes. Migr. 2012, 6, 30–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seimetz, D.; Lindhofer, H.; Bokemeyer, C. Development and approval of the trifunctional antibody catumaxomab (an-ti-EpCAM x anti-CD3) as a targeted cancer immunotherapy. Cancer Treat Rev. 2010, 36, 458–467. [Google Scholar] [CrossRef]
- Jager, M.; Schoberth, A.; Ruf, P.; Hess, J.; Hennig, M.; Schmalfeldt, B.; Wimberger, P.; Strohlein, M.; Theissen, B.; Heiss, M.M.; et al. Immunomonitoring results of a phase II/III study of malignant ascites patients treated with the trifunctional antibody catumaxomab (anti-EpCAM x anti-CD3). Cancer Res. 2012, 72, 24–32. [Google Scholar] [CrossRef] [Green Version]
- Morris, E.C.; Neelapu, S.S.; Giavridis, T.; Sadelain, M. Cytokine release syndrome and associated neurotoxicity in cancer im-mu-notherapy. Nat. Rev. Immunol. 2022, 22, 85–96. [Google Scholar] [CrossRef]
- Esfandiari, A.; Cassidy, S.; Webster, R.M. Bispecific antibodies in oncology. Nat. Rev. Drug Discov. 2022, 21, 411–412. [Google Scholar] [CrossRef]
- Gera, N. The evolution of bispecific antibodies. Expert Opin. Biol. Ther. 2022, 22, 945–949. [Google Scholar] [CrossRef]
- Krief, P.; Boucheix, C.; Billard, C.; Mishal, Z.; van Agthoven, A.; Fiers, W.; Azzarone, B. Modulation of expression of class II histocompatibility antigens by secretion of a cellular inhibitor in K562 leukemic cells. Eur. J. Immunol. 1987, 17, 1021–1025. [Google Scholar] [CrossRef]
- Peitzsch, C.; Perrin, R.; Hill, R.P.; Dubrovska, A.; Kurth, I. Hypoxia as a biomarker for radioresistant cancer stem cells. Int. J. Radiat. Biol. 2014, 90, 636–652. [Google Scholar] [CrossRef] [PubMed]
- Cruz, E.; Kayser, V. Monoclonal antibody therapy of solid tumors: Clinical limitations and novel strategies to enhance treatment efficacy. Biol. Targets Ther. 2019, 13, 33–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef]
- Prasetyanti, P.R.; Medema, J.P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 2017, 16, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, F.M.; Schniewind, I.; Besso, M.J.; Lange, S.; Linge, A.; Patil, S.G.; Loeck, S.; Klusa, D.; Dietrich, A.; Voss-Boehme, A.; et al. Plasticity within Aldehyde Dehydrogenase–Positive Cells Determines Prostate Cancer Radiosensitivity. Mol. Cancer Res. 2022, 20, 794–809. [Google Scholar] [CrossRef]
- Schniewind, I.; Hadiwikarta, W.W.; Grajek, J.; Poleszczuk, J.; Richter, S.; Peitzsch, M.; Muller, J.; Klusa, D.; Beyreuther, E.; Lock, S.; et al. Cellular plasticity upon proton irradiation determines tumor cell radio-sen-sitivity. Cell Rep. 2022, 38, 110422. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.-T.; Ryu, C.J. Cancer stem cell surface markers on normal stem cells. BMB Rep. 2017, 50, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Betts, A.; van der Graaf, P.H. Mechanistic Quantitative Pharmacology Strategies for the Early Clinical Development of Bispecific Antibodies in Oncology. Clin. Pharm. Ther. 2020, 108, 528–541. [Google Scholar] [CrossRef] [PubMed]
- Elshiaty, M.; Schindler, H.; Christopoulos, P. Principles and Current Clinical Landscape of Multispecific Antibodies against Cancer. Int. J. Mol. Sci. 2021, 22, 5632. [Google Scholar] [CrossRef]
- Börjesson, P.K.E.; Postema, E.J.; Roos, J.C.; Colnot, D.R.; Marres, H.A.M.; Van Schie, M.H.; Stehle, G.; De Bree, R.; Snow, G.B.; Oyen, W.J.G.; et al. Phase I therapy study with (186)Re-labeled humanized monoclonal antibody BIWA 4 (bivatuzumab) in patients with head and neck squamous cell carcinoma. Clin. Cancer Res. 2003, 9, 3961. [Google Scholar]
- Colnot, D.R.; Quak, J.J.; Roos, J.C.; Van Lingen, A.; Wilhelm, A.J.; Van Kamp, G.J.; Huijgens, P.C.; Snow, G.B.; Van Dongen, G.A. Phase I therapy study of 186Re-labeled chimeric monoclonal antibody U36 in patients with squamous cell carcinoma of the head and neck. J. Nucl. Med. 2000, 41, 1999–2010. [Google Scholar] [PubMed]
- Riechelmann, H.; Sauter, A.; Golze, W.; Hanft, G.; Schroen, C.; Hoermann, K.; Erhardt, T.; Gronau, S. Phase I trial with the CD44v6-targeting immunoconjugate bivatuzumab mertansine in head and neck squamous cell carcinoma. Oral. Oncol. 2008, 44, 823–829. [Google Scholar] [CrossRef]
- Rupp, U.; Schoendorf-Holland, E.; Eichbaum, M.; Schuetz, F.; Lauschner, I.; Schmidt, P.; Staab, A.; Hanft, G.; Huober, J.; Sinn, H.-P.; et al. Safety and pharmacokinetics of bivatuzumab mertansine in patients with CD44v6-positive metastatic breast cancer: Final results of a phase I study. Anti-Cancer Drugs 2007, 18, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Sauter, A.; Kloft, C.; Gronau, S.; Bogeschdorfer, F.; Erhardt, T.; Golze, W.; Schroen, C.; Staab, A.; Riechelmann, H.; Hoermann, K. Pharmacokinetics, immunogenicity and safety of bivatuzumab mertansine, a novel CD44v6-targeting immunoconjugate, in patients with squamous cell carcinoma of the head and neck. Int. J. Oncol. 2007, 30, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Kubasch, A.S.; Schulze, F.; Giagounidis, A.; Götze, K.S.; Krönke, J.; Sockel, K.; Middeke, J.M.; Chermat, F.; Gloaguen, S.; Puttrich, M.; et al. Single agent talacotuzumab demonstrates limited efficacy but considerable toxicity in elderly high-risk MDS or AML patients failing hypomethylating agents. Leukemia 2019, 34, 1182–1186. [Google Scholar] [CrossRef] [Green Version]
- Daver, N.G.; Montesinos, P.; DeAngelo, D.J.; Roboz, G.J.; Wang, E.S.; Walter, R.B.; Jeyakumar, D.; DeAngelo, D.J.; Erba, H.P.; Advani, A.; et al. A phase I/II study of IMGN632, a novel CD123-targeting antibody-drug conjugate, in patients with relapsed/refractory acute myeloid leukemia, blastic plasmacytoid dendritic cell neoplasm, and other CD123-positive hematologic malignancies. J. Clin. Oncol. 2020, 38 (Suppl. S15), TPS7563. [Google Scholar] [CrossRef]
- Voskoboinik, I.; Whisstock, J.C.; Trapani, J.A. Perforin and granzymes: Function, dysfunction and human pathology. Nat. Rev. Immunol. 2015, 15, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Wolf, N.K.; Kissiov, D.U.; Raulet, D.H. Roles of natural killer cells in immunity to cancer, and applications to immunotherapy. Nat. Rev. Immunol. 2022, 23, 90–105. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immu-no-therapy of patients with metastatic melanoma. A preliminary report. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [Green Version]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A Phase I Study on Adoptive Immunotherapy Using Gene-Modified T Cells for Ovarian Cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Riviere, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Krause, A.; Guo, H.-F.; Latouche, J.-B.; Tan, C.; Cheung, N.-K.V.; Sadelain, M. Antigen-dependent CD28 Signaling Selectively Enhances Survival and Proliferation in Genetically Modified Activated Human Primary T Lymphocytes. J. Exp. Med. 1998, 188, 619–626. [Google Scholar] [CrossRef] [Green Version]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X.-S.; Matsushita, M.; Plotkin, J.; Riviere, I.; Sadelain, M. Chimeric Antigen Receptors Combining 4-1BB and CD28 Signaling Domains Augment PI3kinase/AKT/Bcl-XL Activation and CD8+ T Cell–mediated Tumor Eradication. Mol. Ther. 2010, 18, 413–420. [Google Scholar] [CrossRef]
- Jayaraman, J.; Mellody, M.P.; Hou, A.J.; Desai, R.P.; Fung, A.W.; Pham, A.H.T.; Chen, Y.Y.; Zhao, W. CAR-T design: Elements and their synergistic function. eBiomedicine 2020, 58, 102931. [Google Scholar] [CrossRef] [PubMed]
- Billadeau, D.D.; Upshaw, J.L.; Schoon, R.A.; Dick, C.J.; Leibson, P.J. NKG2D-DAP10 triggers human NK cell–mediated killing via a Syk-independent regulatory pathway. Nat. Immunol. 2003, 4, 557–564. [Google Scholar] [CrossRef]
- Töpfer, K.; Cartellieri, M.; Michen, S.; Wiedemuth, R.; Müller, N.; Lindemann, D.; Bachmann, M.; Füssel, M.; Schackert, G.; Temme, A. DAP12-Based Activating Chimeric Antigen Receptor for NK Cell Tumor Immunotherapy. J. Immunol. 2015, 194, 3201–3212. [Google Scholar] [CrossRef] [Green Version]
- Curio, S.; Jonsson, G.; Marinović, S. A summary of current NKG2D-based CAR clinical trials. Immunother. Adv. 2021, 1, ltab018. [Google Scholar] [CrossRef]
- Wang, X.; Rivière, I. Clinical manufacturing of CAR T cells: Foundation of a promising therapy. Mol. Ther. Oncolytics 2016, 3, 16015. [Google Scholar] [CrossRef] [Green Version]
- Barros, L.R.C.; Couto, S.C.F.; Santurio, D.D.S.; Paixão, E.A.; Cardoso, F.; da Silva, V.J.; Klinger, P.; Ribeiro, P.D.A.C.; Rós, F.A.; Oliveira, T.G.M.; et al. Systematic Review of Available CAR-T Cell Trials around the World. Cancers 2022, 14, 2667. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Lee, S.; Sharma, P.; George, B.; Scott, J. Summary of US Food and Drug Administration Chimeric Antigen Receptor (CAR) T-Cell Biologics License Application Approvals from a Statistical Perspective. J. Clin. Oncol. 2022, 40, 3501–3509. [Google Scholar] [CrossRef]
- Braendstrup, P.; Levine, B.L.; Ruella, M. The long road to the first FDA-approved gene therapy: Chimeric antigen receptor T cells targeting CD. Cytotherapy 2020, 22, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Yi, D.; Gergis, M.; Hsu, J.; Yang, Y.; Bi, X.; Aljurf, M.; Gergis, U. Next-Generation Chimeric Antigen Receptor T Cells. Hematol. Stem Cell Ther. 2022, 15, 11. [Google Scholar] [CrossRef]
- Sengsayadeth, S.; Savani, B.N.; Oluwole, O.; Dholaria, B. Overview of approved CAR-T therapies, ongoing clinical trials, and its impact on clinical practice. eJHaem 2022, 3 (Suppl. S1), 6–10. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.G.; Mortensson, E.; Williams, A.C. Targeting LGR5 in Colorectal Cancer: Therapeutic gold or too plastic? Br. J. Cancer 2018, 118, 1410–1418. [Google Scholar] [CrossRef]
- Li, W.; Zhang, N.; Jin, C.; Long, M.D.; Rajabi, H.; Yasumizu, Y.; Fushimi, A.; Yamashita, N.; Hagiwara, M.; Zheng, R.; et al. MUC1-C drives stemness in progression of colitis to colorectal cancer. J. Clin. Investig. 2020, 5, e137112. [Google Scholar] [CrossRef]
- Kufe, D.W. Chronic activation of MUC1-C in wound repair promotes progression to cancer stem cells. J. Cancer Metastasis Treat. 2022, 8, 12. [Google Scholar] [CrossRef]
- Shah, P.D.; Huang, A.C.C.; Xu, X.; Zhang, P.Z.; Orlowski, R.; Matlawski, T.; Shea, J.; Cervini, A.; Amaravadi, R.K.; Tchou, J.C.; et al. Phase I trial of autologous cMET-directed CAR-T cells administered intravenously in patients with melanoma & breast carcinoma. J. Clin. Oncol. 2020, 38 (Suppl. S15), 10035. [Google Scholar]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, A.; Lynn, R.C.; Poussin, M.; Eiva, M.A.; Shaw, L.C.; O’Connor, R.S.; Minutolo, N.G.; Casado-Medrano, V.; Lopez, G.; Matsuyama, T.; et al. CAR-T cell-mediated depletion of immunosuppressive tumor-associated macrophages promotes endogenous antitumor immunity and augments adoptive immunotherapy. Nat. Commun. 2021, 12, 1–17. [Google Scholar]
- Baur, K.; Heim, D.; Beerlage, A.; Poerings, A.S.; Kopp, B.; Medinger, M.; Dirks, J.C.; Passweg, J.R.; Holbro, A. Dasatinib for treatment of CAR T-cell therapy-related complications. J. Immunother. Cancer 2022, 10, e005956. [Google Scholar] [CrossRef]
- Ran, T.; Eichmüller, S.B.; Schmidt, P.; Schlander, M. Cost of decentralized CAR T-cell production in an academic nonprofit setting. Int. J. Cancer 2020, 147, 3438–3445. [Google Scholar] [CrossRef] [PubMed]
- Zah, E.; Nam, E.; Bhuvan, V.; Tran, U.; Ji, B.Y.; Gosliner, S.B.; Wang, X.; Brown, C.E.; Chen, Y.Y. Systematically optimized BCMA/CS1 bispecific CAR-T cells robustly control heterogeneous multiple myeloma. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Kloss, C.C.; Condomines, M.; Cartellieri, M.; Bachmann, M.; Sadelain, M. Combinatorial antigen recognition with balanced sig-naling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol. 2013, 31, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Bielamowicz, K.; Fousek, K.; Byrd, T.T.; Samaha, H.; Mukherjee, M.; Aware, N.; Wu, M.-F.; Orange, J.S.; Sumazin, P.; Man, T.-K.; et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro-Oncol. 2018, 20, 506–518. [Google Scholar] [CrossRef] [Green Version]
- Philip, B.; Kokalaki, E.; Mekkaoui, L.; Thomas, S.; Straathof, K.; Flutter, B.; Marin, V.; Marafioti, T.; Chakraverty, R.; Linch, D.; et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood 2014, 124, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.S.; Lamb, L.S.; Egoldman, F.; Stasi, A.E. Improving the safety of cell therapy products by suicide gene transfer. Front. Pharmacol. 2014, 5, 254. [Google Scholar] [CrossRef]
- Raes, L.; Stremersch, S.; Fraire, J.C.; Brans, T.; Goetgeluk, G.; De Munter, S.; Van Hoecke, L.; Verbeke, R.; Van Hoeck, J.; Xiong, R.; et al. Intracellular Delivery of mRNA in Adherent and Suspension Cells by Vapor Nanobubble Photoporation. Nano-Micro Lett. 2020, 12, 1–17. [Google Scholar] [CrossRef]
- Foster, J.B.; Barrett, D.M.; Karikó, K. The Emerging Role of In Vitro-Transcribed mRNA in Adoptive T Cell Immunotherapy. Mol. Ther. 2019, 27, 747–756. [Google Scholar] [CrossRef] [Green Version]
- Arndt, C.; Fasslrinner, F.; Loureiro, L.R.; Koristka, S.; Feldmann, A.; Bachmann, M. Adaptor CAR Platforms—Next Generation of T Cell-Based Cancer Immunotherapy. Cancers 2020, 12, 1302. [Google Scholar] [CrossRef] [PubMed]
- Koristka, M.S.; Cartellieri, M.; Feldmann, A.; Arndt, M.C.; Loff, M.S.; Michalk, M.I.; Aliperta, M.R.; Von Bonin, M.; Bornhäuser, M.; Ehninger, A.; et al. Flexible Antigen-Specific Redirection of Human Regulatory T Cells Via a Novel Universal Chimeric Antigen Receptor System. Blood 2014, 124, 3494. [Google Scholar] [CrossRef]
- Cartellieri, M.; Loff, S.; von Bonin, M.; Arndt, C.; Loff, S.; Ehninger, A.; von Bonin, M.; Bejestani, E.P.; Ehninger, G.; Bachmann, M.P. Unicar: A Novel Modular Retargeting Platform Technology for CAR T Cells. Blood 2015, 125, 5549. [Google Scholar] [CrossRef]
- Cao, Y.; Rodgers, D.T.; Du, J.; Ahmad, I.; Hampton, E.N.; Ma, J.S.Y.; Mazagova, M.; Choi, S.-H.; Yun, H.Y.; Xiao, H.; et al. Design of Switchable Chimeric Antigen Receptor T Cells Targeting Breast Cancer. Angew. Chem. Int. Ed. 2016, 55, 7520–7524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamada, K.; Geng, D.; Sakoda, Y.; Bansal, N.; Srivastava, R.; Li, Z.; Davila, E. Redirecting Gene-Modified T Cells toward Various Cancer Types Using Tagged Antibodies. Clin. Cancer Res. 2012, 18, 6436–6445. [Google Scholar] [CrossRef] [Green Version]
- Urbanska, K.; Lanitis, E.; Poussin, M.; Lynn, R.C.; Gavin, B.P.; Kelderman, S.; Yu, J.; Scholler, N.; Powell, D.J., Jr. A Universal Strategy for Adoptive Immunotherapy of Cancer through Use of a Novel T-cell Antigen Receptor. Cancer Res. 2012, 72, 1844–1852. [Google Scholar] [CrossRef] [Green Version]
- Rong, L.; Lim, R.M.; Yin, X.; Tan, L.; Yang, J.H.; Xie, J. Site-Specific Dinitrophenylation of Single-Chain Antibody Fragments for Redirecting a Universal CAR-T Cell against Cancer Antigens. J. Mol. Biol. 2022, 434, 167513. [Google Scholar] [CrossRef]
- Urbanska, K.; Lynn, R.C.; Stashwick, C.; Thakur, A.; Lum, L.G.; Powell, D.J. Targeted cancer immunotherapy via combination of designer bispecific antibody and novel gene-engineered T cells. J. Transl. Med. 2014, 12, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Klesmith, J.R.; Su, L.; Wu, L.; Klesmith, J.R.; Su, L.; Wu, L.; Schrack, I.A.; Dufort, F.J.; Birt, A.; Ambrose, C.; et al. Retargeting CD19 Chimeric Antigen Receptor T Cells via Engineered CD19-Fusion Proteins. Mol. Pharm. 2019, 16, 3544–3558. [Google Scholar] [CrossRef] [PubMed]
- Karches, C.H.; Benmebarek, M.-R.; Schmidbauer, M.L.; Kurzay, M.; Klaus, R.; Geiger, M.; Rataj, F.; Cadilha, B.L.; Lesch, S.; Heise, C.; et al. Bispecific Antibodies Enable Synthetic Agonistic Receptor-Transduced T Cells for Tumor Immunotherapy. Clin. Cancer Res. 2019, 25, 5890–5900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldmann, A.; Hoffmann, A.; Bergmann, R.; Koristka, S.; Berndt, N.; Arndt, C.; Loureiro, L.R.; Kittel-Boselli, E.; Mitwasi, N.; Kegler, A.; et al. Versatile chimeric antigen receptor platform for controllable and combinatorial T cell therapy. Oncoimmunology 2020, 9, 1785608. [Google Scholar] [CrossRef]
- Borrok, M.J.; Li, Y.; Harvilla, P.B.; Maruthachalam, B.V.; Tamot, N.; Prokopowitz, C.; Chen, J.; Venkataramani, S.; Grewal, I.S.; Ganesan, R.; et al. Conduit CAR: Redirecting CAR T-Cell Specificity with A Universal and Adaptable Bispecific Antibody Platform. Cancer Res. Commun. 2022, 2, 146–157. [Google Scholar] [CrossRef]
- Carmo-Fonseca, M.; Pfeifer, K.; Schröder, H.C.; Vaz, M.; Fonseca, J.; Müller, W.E.; Bachmann, M. Identification of La ribonucleoproteins as a component of interchromatin granules. Exp. Cell Res. 1989, 185, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, T.; Arndt, C.; Loureiro, L.R.; Kegler, A.; Puentes-Cala, E.; Soto, J.A.; Kurien, B.T.; Feldmann, A.; Berndt, N.; Bachmann, M.P. A Small Step, a Giant Leap: Somatic Hypermutation of a Single Amino Acid Leads to Anti-La Autoreactivity. Int. J. Mol. Sci. 2021, 22, 12046. [Google Scholar] [CrossRef]
- Bachmann, M.; Bartsch, T.; Bippes, C.; Bachmann, D.; Puentes-Cala, E.; Bachmann, J.; Bartsch, H.; Arndt, C.; Koristka, S.; Loureiro, L.; et al. T Cell Mediated Conversion of a Non-Anti-La Reactive B Cell to an Autoreactive Anti-La B Cell by Somatic Hypermutation. Int. J. Mol. Sci. 2021, 22, 1198. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M. The UniCAR system: A modular CAR T cell approach to improve the safety of CAR T cells. Immunol. Lett. 2019, 211, 13–22. [Google Scholar] [CrossRef]
- Arndt, C.; Feldmann, A.; Koristka, S.; Schäfer, M.; Bergmann, R.; Mitwasi, N.; Berndt, N.; Bachmann, D.; Kegler, A.; Schmitz, M.; et al. A theranostic PSMA ligand for PET imaging and retargeting of T cells expressing the universal chimeric antigen receptor UniCAR. Oncoimmunology 2019, 8, 1659095. [Google Scholar] [CrossRef] [Green Version]
- Albert, S.; Arndt, C.; Koristka, S.; Berndt, N.; Bergmann, R.; Feldmann, A.; Schmitz, M.; Pietzsch, J.; Steinbach, J.; Bachmann, M. From mono- to bivalent: Improving theranostic properties of target modules for redi-rection of UniCAR T cells against EGFR-expressing tumor cells in vitro and in vivo. Oncotarget 2018, 9, 25597–25616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldmann, A.; Arndt, C.; Bergmann, R.; Loff, S.; Cartellieri, M.; Bachmann, D.; Aliperta, R.; Hetzenecker, M.; Ludwig, F.; Albert, S.; et al. Retargeting of T lymphocytes to PSCA- or PSMA positive prostate cancer cells using the novel modular chimeric antigen receptor platform technology “UniCAR”. Oncotarget 2017, 8, 31368–31385. [Google Scholar] [CrossRef] [Green Version]
- Koristka, S.; Ziller-Walter, P.; Bergmann, R.; Arndt, C.; Feldmann, A.; Kegler, A.; Cartellieri, M.; Ehninger, A.; Ehninger, G.; Bornhäuser, M.; et al. Anti-CAR-engineered T cells for epitope-based elimination of autologous CAR T cells. Cancer Immunol. Immunother. 2019, 68, 1401–1415. [Google Scholar] [CrossRef] [Green Version]
- Loureiro, L.R.; Feldmann, A.; Bergmann, R.; Koristka, S.; Berndt, N.; Máthé, D.; Hegedüs, N.; Szigeti, K.; Videira, P.A.; Bachmann, M.; et al. Extended half-life target module for sustainable UniCAR T-cell treatment of STn-expressing cancers. J. Exp. Clin. Cancer Res. 2020, 39, 77. [Google Scholar] [CrossRef]
- Mitwasi, N.; Feldmann, A.; Bergmann, R.; Berndt, N.; Arndt, C.; Koristka, S.; Kegler, A.; Jureczek, J.; Hoffmann, A.; Ehninger, A.; et al. Development of novel target modules for retargeting of UniCAR T cells to GD2 positive tumor cells. Oncotarget 2017, 8, 108584–108603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kittel-Boselli, E.; Soto, K.E.G.; Loureiro, L.R.; Hoffmann, A.; Bergmann, R.; Arndt, C.; Koristka, S.; Mitwasi, N.; Kegler, A.; Bartsch, T.; et al. Targeting Acute Myeloid Leukemia Using the RevCAR Platform: A Pro-grammable, Switchable and Combinatorial Strategy. Cancers 2021, 13, 4785. [Google Scholar] [CrossRef]
- Stock, S.; Benmebarek, M.-R.; Kluever, A.-K.; Darowski, D.; Jost, C.; Stubenrauch, K.-G.; Benz, J.; Freimoser-Grundschober, A.; Moessner, E.; Umana, P.; et al. Chimeric antigen receptor T cells engineered to recognize the P329G-mutated Fc part of effector-silenced tumor antigen-targeting human IgG1 antibodies enable modular targeting of solid tumors. J. Immunother. Cancer 2022, 10, e005054. [Google Scholar] [CrossRef]
- Kuo, Y.-C.; Kuo, C.-F.; Jenkins, K.; Hung, A.F.-H.; Chang, W.-C.; Park, M.; Aguilar, B.; Starr, R.; Hibbard, J.; Brown, C.; et al. Antibody-based redirection of universal Fabrack-CAR T cells selectively kill antigen bearing tumor cells. J. Immunother. Cancer 2022, 10, e003752. [Google Scholar] [CrossRef]
- Cartellieri, M.; Feldmann, A.; Koristka, S.; Arndt, C.; Loff, S.; Ehninger, A.V.; Von Bonin, M.; Bejestani, E.P.; Ehninger, G.; Bachmann, M.P. Switching CAR T cells on and off: A novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J. 2016, 6, e458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loff, S.; Dietrich, J.; Meyer, J.-E.; Riewaldt, J.; Spehr, J.; von Bonin, M.; Gründer, C.; Swayampakula, M.; Franke, K.; Feldmann, A.; et al. Rapidly Switchable Universal CAR-T Cells for Treatment of CD123-Positive Leukemia. Mol. Ther. Oncolytics 2020, 17, 408–420. [Google Scholar] [CrossRef] [PubMed]
- Minutolo, N.G.; Sharma, P.; Poussin, M.; Shaw, L.C.; Brown, D.P.; Hollander, E.E.; Smole, A.; Rodriguez-Garcia, A.; Hui, J.Z.; Zappala, F.; et al. Quantitative Control of Gene-Engineered T-Cell Activity through the Covalent Attachment of Targeting Ligands to a Universal Immune Receptor. J. Am. Chem. Soc. 2020, 142, 6554–6568. [Google Scholar] [CrossRef]
- Arndt, C.; Loureiro, L.R.; Feldmann, A.; Jureczek, J.; Bergmann, R.; Máthé, D.; Hegedüs, N.; Berndt, N.; Koristka, S.; Mitwasi, N.; et al. UniCAR T cell immunotherapy enables efficient elimination of radioresistant cancer cells. Oncoimmunology 2020, 9, 1743036. [Google Scholar] [CrossRef] [Green Version]
- Wermke, M.; Kraus, S.; Ehninger, A.; Bargou, R.C.; Goebeler, M.E.; Middeke, J.M.; Kreissig, C.; von Bonin, M.; Koedam, J.; Pehl, M.; et al. Proof of concept for a rapidly switchable universal CAR-T platform with UniCAR-T-CD123 in relapsed/refractory AML. Blood 2021, 137, 3145–3148. [Google Scholar] [CrossRef]
- Ehninger, G.; Kraus, S.; Sala, E.; Metzelder, S.K.; Vucinic, V.; Fiedler, W.; Goebeler, M.E.; Middeke, J.M.; von Bonin, M.; Kreissig, C.; et al. Phase 1 Dose Escalation Study of the Rapidly Switchable Universal CAR-T Therapy Unicar-T-CD123 in Relapsed/Refractory AML. Blood 2022, 140, 2367–2368. [Google Scholar] [CrossRef]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. eBiomedicine 2020, 59, 102975. [Google Scholar] [CrossRef]
- Simonetta, F.; Alvarez, M.; Negrin, R.S. Natural Killer Cells in Graft-versus-Host-Disease after Allogeneic Hematopoietic Cell Transplantation. Front. Immunol. 2017, 8, 465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; Defor, T.E.; Burns, L.J.; et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjorklund, A.T.; Carlsten, M.; Sohlberg, E.; Liu, L.L.; Clancy, T.; Karimi, M.; Cooley, S.; Miller, J.S.; Klimkowska, M.; Schaffer, M.; et al. Complete Remission with Reduction of High-Risk Clones following Hap-loidentical NK-Cell Therapy against MDS and AML. Clin. Cancer Res. 2018, 24, 1834–1844. [Google Scholar] [CrossRef] [Green Version]
- Du, W.; Cui, L.; Zhang, J.; Zhang, H.; Liu, R.; Yang, W.; Zhang, Y. Generation of universal natural killer cells from a cryopreserved cord blood mononuclear cell-derived induced pluripotent stem cell library. FEBS Open Bio 2022, 12, 1771–1781. [Google Scholar] [CrossRef] [PubMed]
- Kerbauy, L.N.; Marin, N.D.; Kaplan, M.; Banerjee, P.P.; Berrien-Elliott, M.M.; Becker-Hapak, M.; Basar, R.; Foster, M.; Garcia Melo, L.; Neal, C.C.; et al. Combining AFM13, a Bispecific CD30/CD16 Antibody, with Cytokine-Activated Blood and Cord Blood-Derived NK Cells Facilitates CAR-like Responses Against CD30(+) Malignancies. Clin. Cancer Res. 2021, 27, 3744–3756. [Google Scholar] [CrossRef]
- Heipertz, E.L.; Zynda, E.R.; Stav-Noraas, T.E.; Hungler, A.D.; Boucher, S.E.; Kaur, N.; Vemuri, M.C. Current Perspectives on “Off-The-Shelf” Allogeneic NK and CAR-NK Cell Therapies. Front. Immunol. 2021, 12, 732135. [Google Scholar] [CrossRef] [PubMed]
- Valeri, A.; García-Ortiz, A.; Castellano, E.; Córdoba, L.; Maroto-Martín, E.; Encinas, J.; Leivas, A.; Río, P.; Martínez-López, J. Overcoming tumor resistance mechanisms in CAR-NK cell therapy. Front. Immunol. 2022, 13, 953849. [Google Scholar] [CrossRef]
- Carrabba, M.G.C.; Hudecek, M.; Quintarelli, C.; Briones, J.; Hajek, R.; Sierra, J.; Locatelli, F.; Einsele, H.; Bordignon, C.; Ciceri, F.; et al. Phase I-IIa Clinical Trial to Assess Safety and Efficacy of MLM-CAR44.1, a CD44v6 Directed CAR-T in Relapsed/Refractory Acute Myeloid Leukemia (AML) and Multiple Myeloma (MM). Blood 2018, 132 (Suppl. S1), 5790. [Google Scholar] [CrossRef]
- Dai, H.; Tong, C.; Shi, D.; Chen, M.; Guo, Y.; Chen, D.; Han, X.; Wang, H.; Wang, Y.; Shen, P. Efficacy and biomarker analysis of CD133-directed CAR T cells in advanced hepatocellular car-cinoma: A single-arm, open-label, phase II trial. Oncoimmunology 2020, 9, 1846926. [Google Scholar] [CrossRef]
- Rowley, D.A.; Fitch, F.W. The road to the discovery of dendritic cells, a tribute to Ralph Steinman. Cell. Immunol. 2012, 273, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Laureano, R.S.; Sprooten, J.; Vanmeerbeerk, I.; Borras, D.M.; Govaerts, J.; Naulaerts, S.; Berneman, Z.N.; Beuselinck, B.; Bol, K.F.; Borst, J.; et al. Trial watch: Dendritic cell (DC)-based immunotherapy for cancer. Oncoimmunology 2022, 11, 2096363. [Google Scholar] [CrossRef] [PubMed]
- van Willigen, W.W.; Bloemendal, M.; Gerritsen, W.R.; Schreibelt, G.; de Vries, I.J.M.; Bol, K.F. Dendritic Cell Cancer Therapy: Vac-cinating the Right Patient at the Right Time. Front Immunol. 2018, 9, 2265. [Google Scholar] [CrossRef] [PubMed]
- Teramoto, K.; Ozaki, Y.; Hanaoka, J.; Sawai, S.; Tezuka, N.; Fujino, S.; Daigo, Y.; Kontani, K. Predictive biomarkers and effectiveness of MUC1-targeted dendritic-cell-based vaccine in patients with refractory non-small cell lung cancer. Ther. Adv. Med Oncol. 2016, 9, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Ogasawara, M.; Miyashita, M.; Yamagishi, Y.; Ota, S. Dendritic cell vaccination combined with a conventional chemotherapy for patients with relapsed or advanced pancreatic ductal adenocarcinoma: A single-center phase I/II trial. Ther. Apher. Dial. 2021, 25, 415–424. [Google Scholar] [CrossRef]
- Nagai, K.; Adachi, T.; Harada, H.; Eguchi, S.; Sugiyama, H.; Miyazaki, Y. Dendritic Cell-based Immunotherapy Pulsed with Wilms Tumor 1 Peptide and Mucin 1 as an Adjuvant Therapy for Pancreatic Ductal Adenocarcinoma After Curative Resection: A Phase I/IIa Clinical Trial. Anticancer. Res. 2020, 40, 5765–5776. [Google Scholar] [CrossRef]
- Lepisto, A.J.; Moser, A.J.; Zeh, H.; Lee, K.; Bartlett, D.; McKolanis, J.R.; Geller, B.A.; Schmotzer, A.; Potter, D.P.; Whiteside, T.; et al. A phase I/II study of a MUC1 peptide pulsed autologous dendritic cell vaccine as adjuvant therapy in patients with resected pancreatic and biliary tumors. Cancer Ther. 2008, 6, 955–964. [Google Scholar]
- Westdorp, H.; Creemers, J.H.A.; van Oort, I.M.; Schreibelt, G.; Gorris, M.A.J.; Mehra, N.; Simons, M.; de Goede, A.L.; van Rossum, M.M.; Croockewit, A.J.; et al. Blood-derived dendritic cell vaccinations induce immune responses that correlate with clinical outcome in patients with chemo-naive castration-resistant prostate cancer. J. Immunother. Cancer 2019, 7, 302. [Google Scholar] [CrossRef]
- Liao, F.; Zhang, J.; Hu, Y.; Najafabadi, A.H.; Moon, J.J.; Wicha, M.S.; Kaspo, B.; Whitfield, J.; Chang, A.E.; Li, Q. Efficacy of an ALDH peptide-based dendritic cell vaccine targeting cancer stem cells. Cancer Immunol. Immunother. 2022, 71, 1959–1973. [Google Scholar] [CrossRef]
- Visus, C.; Wang, Y.; Lozano-Leon, A.; Ferris, R.L.; Silver, S.; Szczepanski, M.J.; Brand, R.E.; Ferrone, C.R.; Whiteside, T.L.; Ferrone, S.; et al. Targeting ALDH (bright) human carcinoma-initiating cells with ALDH1A1-specific CD8(+) T cells. Clin Cancer Res. 2011, 17, 6174–6184. [Google Scholar] [CrossRef] [Green Version]
- Püschel, J.; Dubrovska, A.; Gorodetska, I. The Multifaceted Role of Aldehyde Dehydrogenases in Prostate Cancer Stem Cells. Cancers 2021, 13, 4703. [Google Scholar] [CrossRef] [PubMed]
- Cojoc, M.; Peitzsch, C.; Kurth, I.; Trautmann, F.; Kunz-Schughart, L.A.; Telegeev, G.D.; Stakhovsky, E.A.; Walker, J.R.; Simin, K.; Lyle, S.; et al. Aldehyde Dehydrogenase Is Regulated by beta-Catenin/TCF and Promotes Radioresistance in Prostate Cancer Progenitor Cells. Cancer Res. 2015, 75, 1482–1494. [Google Scholar] [CrossRef] [Green Version]
- Mukha, A.; Kahya, U.; Linge, A.; Chen, O.; Lock, S.; Lukiyanchuk, V.; Richter, S.; Alves, T.C.; Peitzsch, M.; Telychko, V.; et al. GLS-driven glutamine catabolism contributes to prostate cancer radiosensitivity by reg-ulating the redox state, stemness and ATG5-mediated autophagy. Theranostics 2021, 11, 7844–7868. [Google Scholar] [CrossRef]
- Ning, N.; Pan, Q.; Zheng, F.; Teitz-Tennenbaum, S.; Egenti, M.; Yet, J.; Li, M.; Ginestier, C.; Wicha, M.S.; Moyer, J.S.; et al. Cancer Stem Cell Vaccination Confers Significant Antitumor Immunity. Cancer Res 2012, 72, 1853–1864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köseer, A.S.; Loureiro, L.R.; Jureczek, J.; Mitwasi, N.; Soto, K.E.G.; Aepler, J.; Bartsch, T.; Feldmann, A.; Kunz-Schughart, L.A.; Linge, A.; et al. Validation of CD98hc as a Therapeutic Target for a Combination of Radiation and Immunotherapies in Head and Neck Squamous Cell Carcinoma. Cancers 2022, 14, 1677. [Google Scholar] [CrossRef] [PubMed]
- Hassani Najafabadi, A.; Zhang, J.; Aikins, M.E.; Najaf Abadi, Z.I.; Liao, F.; Qin, Y.; Okeke, E.B.; Scheetz, L.M.; Nam, J.; Xu, Y.; et al. Cancer Immunotherapy via Targeting Cancer Stem Cells Using Vaccine Nanodiscs. Nano Lett. 2020, 20, 7783–7792. [Google Scholar] [CrossRef]
- Zheng, F.; Dang, J.; Zhang, H.; Xu, F.; Ba, D.; Zhang, B.; Cheng, F.; Chang, A.E.; Wicha, M.S.; Li, Q. Cancer Stem Cell Vaccination with PD-L1 and CTLA-4 Blockades Enhances the Eradication of Melanoma Stem Cells in a Mouse Tumor Model. J. Immunother. 2018, 41, 361–368. [Google Scholar] [CrossRef]
- Hu, Y.; Lu, L.; Xia, Y.; Chen, X.; Chang, A.E.; Hollingsworth, R.E.; Hurt, E.; Owen, J.; Moyer, J.S.; Prince, M.E.; et al. Therapeutic Efficacy of Cancer Stem Cell Vaccines in the Adjuvant Setting. Cancer Res. 2016, 76, 4661–4672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Ashmawy, N.E.; Salem, M.L.; Khedr, E.G.; El-Zamarany, E.A.; Ibrahim, A.O. Dual-targeted therapeutic strategy combining CSC–DC-based vaccine and cisplatin overcomes chemo-resistance in experimental mice model. Clin. Transl. Oncol. 2019, 22, 1155–1165. [Google Scholar] [CrossRef]
- Feng, D.; Gip, P.; McKenna, B.K.M.; Zhao, F.; Mata, O.; Choi, B.T.S.; Duan, M.J.; Sompalli, M.K.; Majeti, R.; Weissman, I.L.; et al. Combination Treatment with 5F9 and Azacitidine Enhances Phagocytic Elimination of Acute Myeloid Leukemia. Blood 2018, 132, 2729. [Google Scholar] [CrossRef]
- Sallman, D.A.; Asch, A.S.; Al Malki, M.M.; Lee, D.J.; Donnellan, W.B.; Marcucci, G.; Kambhampati, S.; Daver, N.G.; Garcia-Manero, G.; Komrokji, R.S.; et al. The First-in-Class Anti-CD47 Antibody Magrolimab (5F9) in Combination with Azacitidine Is Effective in MDS and AML Patients: Ongoing Phase 1b Results. Blood 2019, 134, 569. [Google Scholar] [CrossRef]
- Yu, J.; Chen, L.; Cui, B.; Wu, C.; Choi, M.Y.; Chen, Y.; Zhang, L.; Rassenti, L.Z.; Ii, G.F.W.; Kipps, T.J. Cirmtuzumab inhibits Wnt5a-induced Rac1 activation in chronic lymphocytic leukemia treated with ibrutinib. Leukemia 2016, 31, 1333–1339. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Choi, M.Y.; Siddiqi, T.; Barrientos, J.C.; Wierda, W.G.; Isufi, I.; Tuscano, J.M.; Lamanna, N.; Subbiah, S.; Koff, J.L.; et al. Phase 1/2 study of cirmtuzumab and ibrutinib in mantle cell lymphoma (MCL) or chronic lymphocytic leukemia (CLL). J. Clin. Oncol. 2021, 39 (Suppl. S15), 7556. [Google Scholar] [CrossRef]
- Su, R.; Dong, L.; Li, Y.; Gao, M.; Han, L.; Wunderlich, M.; Deng, X.; Li, H.; Huang, Y.; Gao, L.; et al. Targeting FTO Suppresses Cancer Stem Cell Maintenance and Immune Evasion. Cancer Cell 2020, 38, 79–96.e11. [Google Scholar] [CrossRef]
- Wu, X.; Huang, S. HER2-specific chimeric antigen receptor-engineered natural killer cells combined with apatinib for the treatment of gastric cancer. Bull. Du Cancer 2019, 106, 946–958. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Xu, J.; Liu, B.; He, X.; Zhou, L.; Hu, X.; Qiao, F.; Zhang, A.; Xu, X.; Zhang, H.; et al. IL6 blockade potentiates the anti-tumor effects of γ-secretase inhibitors in Notch3-expressing breast cancer. Cell Death Differ. 2017, 25, 330–339. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, H.; Ding, J.; Liu, H.; Li, H.; Li, H.; Lu, M.; Miao, Y.; Li, L.; Zheng, J. Combination Therapy with EpCAM-CAR-NK-92 Cells and Regorafenib against Human Colorectal Cancer Models. J. Immunol. Res. 2018, 2018, 4263520. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Sun, B.; Cai, H.; Xuan, Y. Simultaneously target of normal and stem cells-like gastric cancer cells via cisplatin and an-ti-CD133 CAR-T combination therapy. Cancer Immunol. Immunother. 2021, 70, 2795–2803. [Google Scholar] [CrossRef]
- Klapdor, R.; Wang, S.; Hacker, U.; Büning, H.; Morgan, M.; Dörk, T.; Hillemanns, P.; Schambach, A. Improved Killing of Ovarian Cancer Stem Cells by Combining a Novel Chimeric Antigen Receptor–Based Immunotherapy and Chemotherapy. Hum. Gene Ther. 2017, 28, 886–896. [Google Scholar] [CrossRef]
- Fleurence, J.; Bahri, M.; Fougeray, S.; Faraj, S.; Vermeulen, S.; Pinault, E.; Geraldo, F.; Oliver, L.; Véziers, J.; Marquet, P.; et al. Impairing temozolomide resistance driven by glioma stem-like cells with adjuvant immunotherapy targeting O-acetyl GD2 ganglioside. Int. J. Cancer 2019, 146, 424–438. [Google Scholar] [CrossRef]
- Madhav, A.; Andres, A.; Duong, F.; Mishra, R.; Haldar, S.; Liu, Z.; Angara, B.; Gottlieb, R.; Zumsteg, Z.S.; Bhowmick, N.A. Antagonizing CD105 enhances radiation sensitivity in prostate cancer. Oncogene 2018, 37, 4385–4397. [Google Scholar] [CrossRef]
- Nishiga, Y.; Drainas, A.P.; Baron, M.; Bhattacharya, D.; Barkal, A.A.; Ahrari, Y.; Mancusi, R.; Ross, J.B.; Takahashi, N.; Thomas, A.; et al. Radiotherapy in combination with CD47 blockade elicits a macrophage-mediated abscopal effect. Nat. Cancer 2022, 3, 1351–1366. [Google Scholar] [CrossRef]
- Darragh, L.B.; Gadwa, J.; Pham, T.T.; Van Court, B.; Neupert, B.; Olimpo, N.A.; Nguyen, K.; Nguyen, D.; Knitz, M.W.; Hoen, M.; et al. Elective nodal irradiation mitigates local and systemic immunity generated by com-bination radiation and immunotherapy in head and neck tumors. Nat. Commun. 2022, 13, 7015. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.C.; Eisenberg, P.D.; Manikhas, G.; Chugh, R.; Gubens, M.A.; Stagg, R.J.; Kapoun, A.M.; Xu, L.; Dupont, J.; Sikic, B. A Phase I Dose Escalation and Expansion Study of the Anticancer Stem Cell Agent Demcizumab (Anti-DLL4) in Patients with Previously Treated Solid Tumors. Clin. Cancer Res. 2014, 20, 6295–6303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Andre, F.; Tesniere, A.; Kroemer, G. The anticancer immune response: Indispensable for therapeutic success? J. Clin. Investig. 2008, 118, 1991–2001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ménard, C.; Martin, F.; Apetoh, L.; Bouyer, F.; Ghiringhelli, F. Cancer chemotherapy: Not only a direct cytotoxic effect, but also an adjuvant for antitumor immunity. Cancer Immunol. Immunother 2008, 57, 1579–1587. [Google Scholar] [CrossRef]
- Formenti, S.C.; Demaria, S. Systemic effects of local radiotherapy. Lancet Oncol. 2009, 10, 718–726. [Google Scholar] [CrossRef] [Green Version]
- Opzoomer, J.W.; Sosnowska, D.; Anstee, J.E.; Spicer, J.F.; Arnold, J.N. Cytotoxic Chemotherapy as an Immune Stimulus: A Molecular Perspective on Turning Up the Immunological Heat on Cancer. Front. Immunol. 2019, 10, 1654. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Liu, X.; Chen, D.; Yu, J. Radiotherapy combined with immunotherapy: The dawn of cancer treatment. Signal Transduct. Target. Ther. 2022, 7, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.; Brehm, M.; Bridges, S.; Ferguson, S.; Kumar, P.; Mirochnitchenko, O.; Palucka, K.; Pelanda, R.; Sanders-Beer, B.; Shultz, L.D.; et al. Humanized immune system mouse models: Progress, challenges and opportunities. Nat. Immunol. 2019, 20, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Graham, A.L. Naturalizing mouse models for immunology. Nat. Immunol. 2021, 22, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.A.; Hughes, A.M.; Walton, J.; Coenen-Stass, A.M.L.; Magiera, L.; Mooney, L.; Bell, S.; Staniszewska, A.; Sandin, L.C.; Barry, S.T.; et al. Longitudinal immune characterization of syngeneic tumor models to enable model selection for immune oncology drug discovery. J. Immunother. Cancer 2019, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- DuPage, M.; Jacks, T. Genetically engineered mouse models of cancer reveal new insights about the antitumor immune response. Curr. Opin. Immunol. 2013, 25, 192–199. [Google Scholar] [CrossRef] [Green Version]
- Chawda, C.; McMorrow, R.; Gaspar, N.; Zambito, G.; Mezzanotte, L. Monitoring Immune Cell Function Through Optical Imaging: A Review Highlighting Transgenic Mouse Models. Mol. Imaging Biol. 2021, 24, 250–263. [Google Scholar] [CrossRef]
- Hammel, J.H.; Cook, S.R.; Belanger, M.C.; Munson, J.M.; Pompano, R.R. Modeling Immunity In Vitro: Slices, Chips, and Engineered Tissues. Annu. Rev. Biomed. Eng. 2021, 23, 461–491. [Google Scholar] [CrossRef] [PubMed]



| Preclinical Studies | |||||
|---|---|---|---|---|---|
| Experimental Model | Target | Immunotherapy | Combined Therapy | Cancer Type | Refs |
| In vitro (cell line) | CD47 | 5F9 mAb | AZA (cytotoxic analogue of the nucleoside cytidine, inhibitor of DNA methyltransferase) | AML | [251] |
| In vivo (cell-line derived xenograft) | |||||
| In vivo (patient-derived xenograft) | ROR1 | Cirmtuzumab | Ibrutinib (Bruton’s tyrosine kinase (BTK) inhibitor) | CLL | [253] |
| In vitro (cell line) | EGFR | CAR NK-92 | Cabozantinib (VEGFR-2 inhibitor) | Renal cell carcinoma | [114] |
| In vivo (cell-line derived xenograft) | |||||
| In vitro (cell line) | Carbonic Anhydrase IX (CAIX) | CAR T | Sunitinib (multi-targeted receptor kinase inhibitor) | [131] | |
| In vivo (cell-line derived xenograft) | |||||
| In vitro (cell line) | HER2 | HER2 CAR NK-92 | Apatinib (VEGFR-2 inhibitor) | Gastric cancer | [256] |
| In vivo (cell-line derived xenograft) | |||||
| In vitro (cell line) | IL-6 | Tocilizumab | MK-0752 (γ-secretase inhibitor) | Breast cancer | [257] |
| In vivo (cell-line derived xenograft) | |||||
| In vivo (patient-derived xenograft) | |||||
| In vivo (cell-line derived xenograft) | EpCAM | CAR-NK-92 | Regorafenib (multitargeted kinase inhibitor) | Colorectal cancer | [258] |
| In vivo (syngeneic models) | ALDHHigh CSCs | ALDHHigh-DC vaccine | Anti PD-L1 antibody | D5 murine melanoma and 4T1 murine breast cancer | [247] |
| In vivo (syngeneic models) | ALDHHigh CSCs | ALDHHigh-DC vaccine | Anti PD-L1 antibody; Anti CTLA-4 antibody | B16-F10 murine melanoma tumors | [248] |
| In vivo (cell-line derived xenograft) | CD133 | CAR-T | Cisplatin (DNA-binding cytotoxic drug) | Gastric cancer | [259] |
| In vitro (Cell line) | CAR NK-92 | Ovarian cancer | [260] | ||
| In vivo (cell-line derived xenograft) | CD44+/CD24− CSCs | CD44+/CD24− CSC-pulsed DC vaccine | Ehrlich carcinoma | [250] | |
| In vitro (patient-derived cell line) | OAcGD2 | 8B6 mAb | Temozolomide (alkylating agent) | GBM | [261] |
| In vivo (Patient-derived xenograft) | |||||
| In vivo (cell-line derived xenograft) | CD105 | TRC105 | Conventional fractionated RT (5 × 2 Gy) | Prostate cancer | [262] |
| In vivo (Cell-line derived xenograft) | CD47 | Anti-CD47 mAb | Conventional fractionated RT (1 × 5 Gy; 4 × 5 Gy; 1 × 10 Gy) | Small cell lung cancer, colon cancer | [263] |
| In vitro (3D model) | CD98 | UniCAR T + CD98 TM | Conventional fractionated RT (2 × 2 Gy) | HNSCC | [246] |
| In vivo (Cell-line derived xenograft) | CD25 | Anti-CD25 mAb | Conventional fractionated RT | [264] | |
| Clinical trials | |||||
| NCT number | Target | Immunotherapy | Combined therapy | Cancer type | Refs |
| NCT03248479 | CD47 | Magrolimab (Hu5F9-G4) | AZA | AML/MDS | [252] |
| NCT03088878 | ROR1 | Cirmtuzumab | Ibrutinib | CLL | [254] |
| NCT02259582 | DLL4 | Demcizumab | Carboplatin + pemetrexed | Non-squamous NSCLC (DENALI) | [265] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Köseer, A.S.; Di Gaetano, S.; Arndt, C.; Bachmann, M.; Dubrovska, A. Immunotargeting of Cancer Stem Cells. Cancers 2023, 15, 1608. https://doi.org/10.3390/cancers15051608
Köseer AS, Di Gaetano S, Arndt C, Bachmann M, Dubrovska A. Immunotargeting of Cancer Stem Cells. Cancers. 2023; 15(5):1608. https://doi.org/10.3390/cancers15051608
Chicago/Turabian StyleKöseer, Ayse Sedef, Simona Di Gaetano, Claudia Arndt, Michael Bachmann, and Anna Dubrovska. 2023. "Immunotargeting of Cancer Stem Cells" Cancers 15, no. 5: 1608. https://doi.org/10.3390/cancers15051608