
Suppression of Cancer Cell Stemness and Drug Resistance via MYC Destabilization by Deubiquitinase USP45 Inhibition with a Natural Small Molecule
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Cell Line and Cell Culture
2.2. Plasmids, siRNA and shRNA Information
2.3. Bioinformatics Analyses
2.4. Plasmid Transfection, RNA Interference and Lentiviral Infection
2.5. Western Blot
2.6. Ubiquitination Assays
2.7. Tumorsphere Formation Assay
2.8. Flow Cytometry Assay
2.9. Colony Formation Assay
2.10. MTT Assay
2.11. Hoechst 33,342 Assay
2.12. Virtual Docking
2.13. USP45 Expression and Purification
2.14. Alpha-Mangostin/USP45 Affinity Assay (BLI-OCTET K2 Assay)
2.15. Assessment of Tumorigenesis via Xenograft Model
2.16. The Combination of α-Mangostin and Doxorubicin Inhibits Tumorigenesis
2.17. Statistical Analysis
3. Results
3.1. USP45 Upregulates MYC Protein Level and Stability
3.2. USP45 Interacts with and Deubiquitinates MYC
3.3. USP45 Promotes Cancer Proliferation, Stemness and Drug Resistance
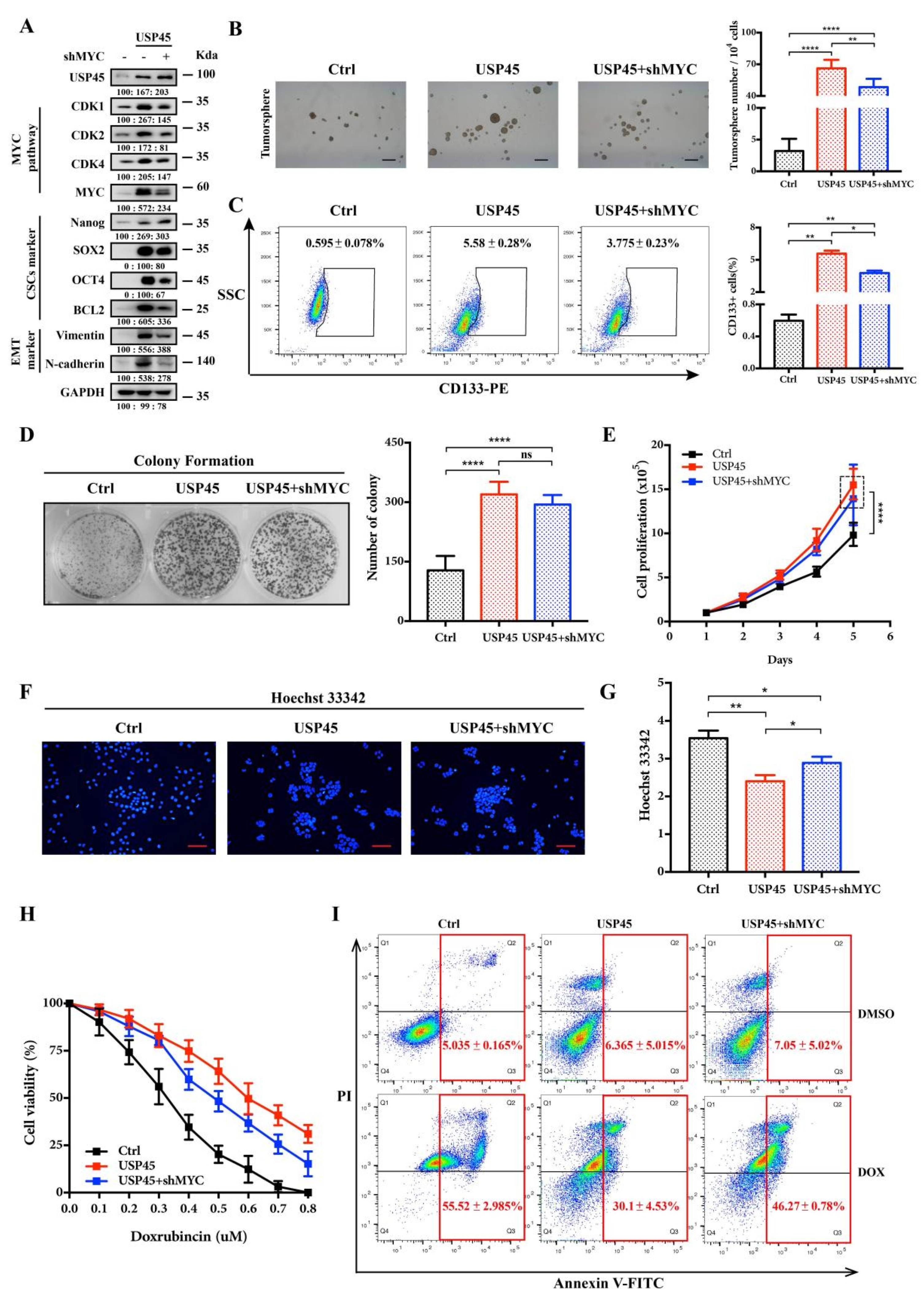
3.4. MYC Is a Downstream Effector in the USP45-Induced Cancer Stemness and Drug Resistance
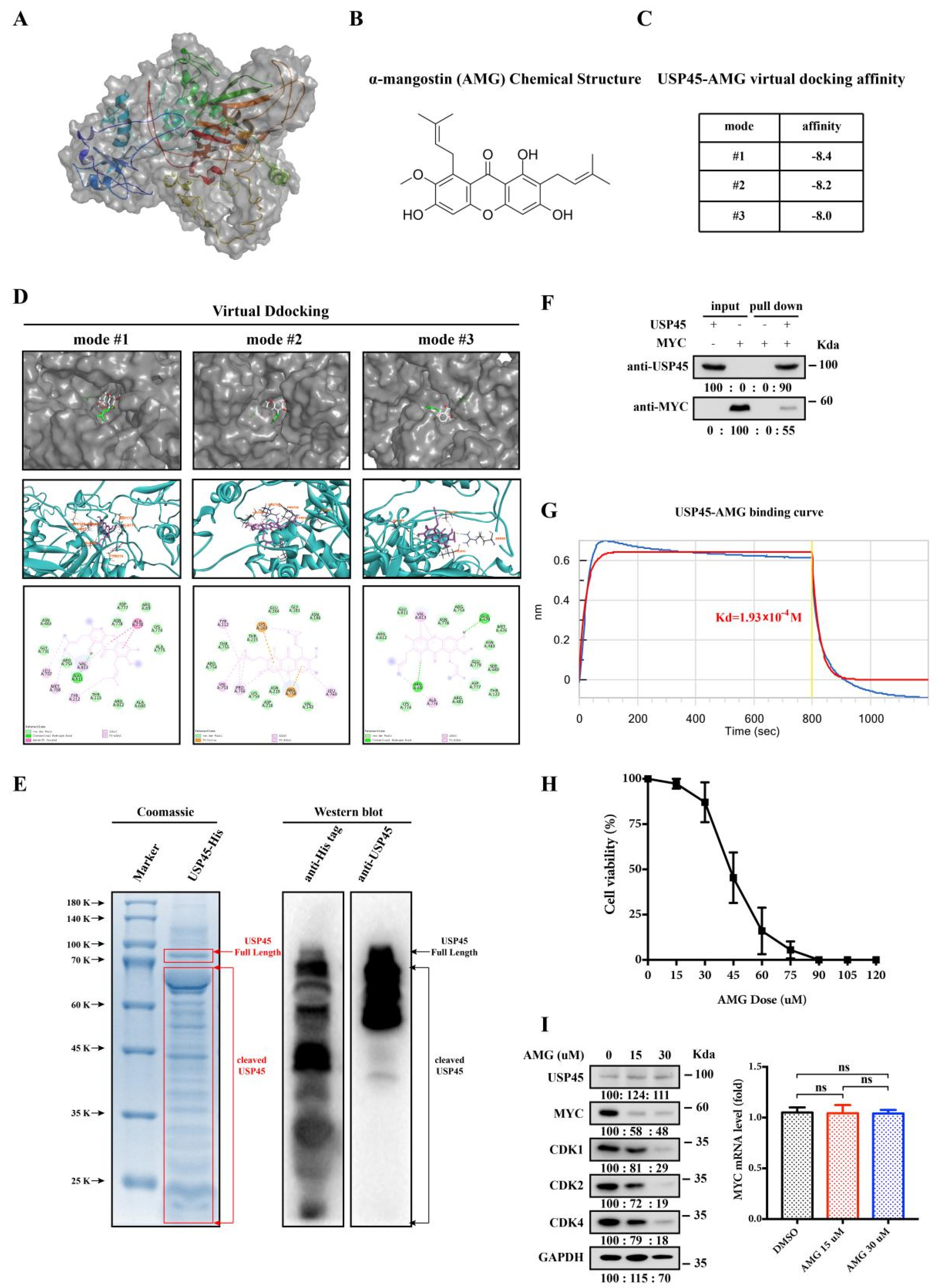
3.5. Alpha-Mangostin Is an Inhibitor of USP45
3.6. Alpha-Mangostin Inhibits the USP45-Induced Cancer Stemness, Drug Resistance and Tumorigenesis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, T.; Song, X.; Xu, D.; Tiek, D.; Goenka, A.; Wu, B.; Sastry, N.; Hu, B.; Cheng, S.-Y. Stem cell programs in cancer initiation, progression, and therapy resistance. Theranostics 2020, 10, 8721–8743. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Farhood, B.; Mortezaee, K. Cancer stem cells (CSCs) in cancer progression and therapy. J. Cell. Physiol. 2019, 234, 8381–8395. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.; Diaz, E.; Reya, T. Stem cells in cancer initiation and progression. J. Cell Biol. 2019, 219, e201911053. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.M.; Roninson, B.I. Expression and activity of P-glycoprotein, a multidrug efflux pump, in human hematopoietic stem cells. Cell 1991, 66, 85–94. [Google Scholar] [CrossRef]
- Zhou, S.; Schuetz, J.D.; Bunting, K.D.; Colapietro, A.M.; Sampath, J.; Morris, J.J.; Lagutina, I.; Grosveld, G.C.; Osawa, M.; Nakauchi, H.; et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat. Med. 2001, 7, 1028–1034. [Google Scholar] [CrossRef]
- Lin, T.; Islam, O.; Heese, K. ABC transporters, neural stem cells and neurogenesis--a different perspective. Cell Res. 2006, 16, 857–871. [Google Scholar] [CrossRef]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nature 2005, 434, 843–850. [Google Scholar] [CrossRef]
- Sari, I.N.; Phi, L.T.H.; Jun, N.; Wijaya, Y.T.; Lee, S.; Kwon, H.Y. Hedgehog Signaling in Cancer: A Prospective Therapeutic Target for Eradicating Cancer Stem Cells. Cells 2018, 7, 208. [Google Scholar] [CrossRef]
- Katoh, M.; Katoh, M. Precision medicine for human cancers with Notch signaling dysregulation (Review). Int. J. Mol. Med. 2020, 45, 279–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, Y.; Yang, H.; Levorse, J.; Yuan, S.; Polak, L.; Sribour, M.; Singh, B.; Rosenblum, M.D.; Fuchs, E. Adaptive Immune Resistance Emerges from Tumor-Initiating Stem Cells. Cell 2019, 177, 1172–1186.e14. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Bai, X.; Feng, X.; Ni, J.; Beretov, J.; Graham, P.; Li, Y. Inhibition of PI3K/Akt/mTOR signaling pathway alleviates ovarian cancer chemoresistance through reversing epithelial-mesenchymal transition and decreasing cancer stem cell marker expression. BMC Cancer 2019, 19, 618. [Google Scholar] [CrossRef]
- Van Schaijik, B.; Davis, P.F.; Wickremesekera, A.C.; Tan, S.T.; Itinteang, T. Subcellular localisation of the stem cell markers OCT4, SOX2, NANOG, KLF4 and c-MYC in cancer: A review. J. Clin. Pathol. 2017, 71, 88–91. [Google Scholar] [CrossRef]
- Xia, P.; Zhang, H.; Xu, K.; Jiang, X.; Gao, M.; Wang, G.; Liu, Y.; Yao, Y.; Chen, X.; Ma, W.; et al. MYC-targeted WDR4 promotes proliferation, metastasis, and sorafenib resistance by inducing CCNB1 translation in hepatocellular carcinoma. Cell Death Dis. 2021, 12, 691. [Google Scholar] [CrossRef]
- Luo, Q.; Wu, X.; Chang, W.; Zhao, P.; Nan, Y.; Zhu, X.; Katz, J.P.; Su, D.; Liu, Z. ARID1A prevents squamous cell carcinoma initiation and chemoresistance by antagonizing pRb/E2F1/c-Myc-mediated cancer stemness. Cell Death Differ. 2020, 27, 1981–1997. [Google Scholar] [CrossRef] [PubMed]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Nesbit, C.E.; Tersak, J.M.; Prochownik, E.V. MYC oncogenes and human neoplastic disease. Oncogene 1999, 18, 3004–3016. [Google Scholar] [CrossRef]
- García-Gutiérrez, L.; Delgado, M.D.; León, J. MYC Oncogene Contributions to Release of Cell Cycle Brakes. Genes 2019, 10, 244. [Google Scholar] [CrossRef]
- Zhong, Y.; Yang, L.; Xiong, F.; He, Y.; Tang, Y.; Shi, L.; Fan, S.; Li, Z.; Zhang, S.; Gong, Z.; et al. Long non-coding RNA AFAP1-AS1 accelerates lung cancer cells migration and invasion by interacting with SNIP1 to upregulate c-Myc. Signal Transduct. Target. Ther. 2021, 6, 240. [Google Scholar] [CrossRef]
- Park, I.-H.; Zhao, R.; West, J.A.; Yabuuchi, A.; Huo, H.; Ince, T.A.; Lerou, P.H.; Lensch, M.W.; Daley, G.Q. Reprogramming of human somatic cells to pluripotency with defined factors. Nature 2008, 451, 141–146. [Google Scholar] [CrossRef]
- Bahr, C.; Von Paleske, L.; Uslu, V.V.; Remeseiro, S.; Takayama, N.; Ng, S.W.; Murison, A.; Langenfeld, K.; Petretich, M.; Scognamiglio, R.; et al. A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature 2018, 553, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Ala, M. Target c-Myc to treat pancreatic cancer. Cancer Biol. Ther. 2022, 23, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Llombart, V.; Mansour, M.R. Therapeutic targeting of “undruggable” MYC. EBioMedicine 2022, 75, 103756. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Farrell, A.S.; Sears, R.C. MYC degradation. Cold Spring Harb. Perspect. Med. 2014, 4, a014365. [Google Scholar] [CrossRef]
- Kim, D.; Hong, A.; Park, H.I.; Shin, W.H.; Yoo, L.; Jeon, S.J.; Chung, K.C. Deubiquitinating enzyme USP22 positively regulates c-Myc stability and tumorigenic activity in mammalian and breast cancer cells. J. Cell. Physiol. 2017, 232, 3664–3676. [Google Scholar] [CrossRef]
- Popov, N.; Wanzel, M.; Madiredjo, M.; Zhang, D.; Beijersbergen, R.; Bernards, R.; Moll, R.; Elledge, S.J.; Eilers, M. The ubiquitin-specific protease USP28 is required for MYC stability. Nature 2007, 9, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Tu, R.; Kang, W.; Yang, M.; Wang, L.; Bao, Q.; Chen, Z.; Dong, Y.; Wang, J.; Jiang, J.; Liu, H.; et al. USP29 coordinates MYC and HIF1α stabilization to promote tumor metabolism and progression. Oncogene 2021, 40, 6417–6429. [Google Scholar] [CrossRef]
- Sun, X.-X.; He, X.; Yin, L.; Komada, M.; Sears, R.C.; Dai, M.-S. The nucleolar ubiquitin-specific protease USP36 deubiquitinates and stabilizes c-Myc. Proc. Natl. Acad. Sci. USA 2015, 112, 3734–3739. [Google Scholar] [CrossRef]
- Pan, J.; Deng, Q.; Jiang, C.; Wang, X.; Niu, T.; Li, H.; Chen, T.; Jin, J.; Pan, W.; Cai, X.; et al. USP37 directly deubiquitinates and stabilizes c-Myc in lung cancer. Oncogene 2014, 34, 3957–3967. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Liu, J.; Wang, Z.; Cui, C.; Zhang, T.; Zhang, S.; Gao, P.; Hou, Z.; Liu, H.; Guo, J.; et al. Prostate-specific oncogene OTUD6A promotes prostatic tumorigenesis via deubiquitinating and stabilizing c-Myc. Cell Death Differ. 2022, 29, 1730–1743. [Google Scholar] [CrossRef]
- Clague, M.J.; Urbé, S.; Komander, D. Breaking the chains: Deubiquitylating enzyme specificity begets function. Nat. Rev. Mol. Cell Biol. 2019, 20, 338–352. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Hu, Q.; He, T.; Xu, J.; Yi, Y.; Xie, S.; Ding, L.; Fu, M.; Guo, R.; Xiao, Z.-X.J.; et al. The Deubiquitinase USP4 Stabilizes Twist1 Protein to Promote Lung Cancer Cell Stemness. Cancers 2020, 12, 1582. [Google Scholar] [CrossRef]
- Shin, S.; Kim, K.; Kim, H.-R.; Ylaya, K.; Do, S.-I.; Hewitt, S.M.; Park, H.-S.; Roe, J.-S.; Chung, J.-Y.; Song, J. Deubiquitylation and stabilization of Notch1 intracellular domain by ubiquitin-specific protease 8 enhance tumorigenesis in breast cancer. Cell Death Differ. 2019, 27, 1341–1354. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Ma, X.; Zhang, Q.; Wu, C.-J.; Liao, W.; Li, J.; Wang, H.; Zhao, J.; Zhou, X.; Guan, C.; et al. USP21 deubiquitinase promotes pancreas cancer cell stemness via Wnt pathway activation. Genes Dev. 2019, 33, 1361–1366. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, Y.; Yang, X.H.; Kang, T.; Zhao, Y.; Wang, C.; Evers, B.M.; Zhou, B.P. The Deubiquitinase USP28 Stabilizes LSD1 and Confers Stem-Cell-like Traits to Breast Cancer Cells. Cell Rep. 2013, 5, 224–236. [Google Scholar] [CrossRef]
- Zhang, X.-Y.; Pfeiffer, H.; Thorne, A.; McMahon, S.B. USP22, an hSAGA subunit and potential cancer stem cell marker, reverses the polycomb-catalyzed ubiquitylation of histone H2A. Cell Cycle 2008, 7, 1522–1524. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, Y.; Wang, D.; Zhang, Y.; Zhang, J.; Zhang, Y.; Xu, L.; Wang, T.; Wang, S.; Zhang, Q.; et al. USP29 enhances chemotherapy-induced stemness in non-small cell lung cancer via stabilizing Snail1 in response to oxidative stress. Cell Death Dis. 2020, 11, 796. [Google Scholar] [CrossRef] [PubMed]
- Qin, T.; Li, B.; Feng, X.; Fan, S.; Liu, L.; Liu, D.; Mao, J.; Lu, Y.; Yang, J.; Yu, X.; et al. Abnormally elevated USP37 expression in breast cancer stem cells regulates stemness, epithelial-mesenchymal transition and cisplatin sensitivity. J. Exp. Clin. Cancer Res. 2018, 37, 287. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Cao, C.-H.; Han, K.; Lv, Y.-R.; Ma, X.-D.; Cao, J.-H.; Chen, J.-W.; Li, S.; Lin, J.-L.; Fang, Y.-J.; et al. CEP63 upregulates YAP1 to promote colorectal cancer progression through stabilizing RNA binding protein FXR1. Oncogene 2022, 41, 4433–4445. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Fu, H.-L.; Wang, Z.; Huang, H.; Ni, J.; Song, J.; Xia, Y.; Jin, W.-L.; Cui, D.-X. USP22 maintains gastric cancer stem cell stemness and promotes gastric cancer progression by stabilizing BMI1 protein. Oncotarget 2017, 8, 33329–33342. [Google Scholar] [CrossRef] [PubMed]
- Cervical Cancer Clinical Data. Available online: https://portal.gdc.cancer.gov/ (accessed on 15 February 2020).
- The “GSE63514” Dataset from GEO Database. Available online: http://www.ncbi.nlm.nih.gov/geo/ (accessed on 17 February 2020).
- Conte, C.; Griffis, E.R.; Hickson, I.; Perez-Oliva, A.B. USP45 and Spindly are part of the same complex implicated in cell migration. Sci. Rep. 2018, 8, 14375. [Google Scholar] [CrossRef]
- Perez-Oliva, A.B.; Lachaud, C.; Szyniarowski, P.; Muñoz, I.; Macartney, T.; Hickson, I.; Rouse, J.; Alessi, D.R. USP45 deubiquitylase controls ERCC1-XPF endonuclease-mediated DNA damage responses. Embo J. 2015, 34, 326–343. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, X.; Gan, J.; Chen, S.; Xiao, Z.X.; Cao, Y. CB-Dock2: Improved protein-ligand blind docking by integrating cavity detection, docking and homologous template fitting. Nucleic Acids Res. 2022, 50, W159–W164. [Google Scholar] [CrossRef]
- The USP45 Expression Analysis. Available online: http://timer.cistrome.org/ (accessed on 9 May 2021).
- Hallmark Gene Enrichment Analysis. Available online: http://guotosky.vip:13838/GTBA (accessed on 28 November 2021).
- The Survival Analysis. Available online: http://gepia2.cancer-pku.cn/-index (accessed on 15 November 2021).
- den Boon, J.A.; Pyeon, D.; Wang, S.S.; Horswill, M.; Schiffman, M.; Sherman, M.; Zuna, R.E.; Wang, Z.; Hewitt, S.M.; Pearson, R.; et al. Molecular transitions from papillomavirus infection to cervical precancer and cancer: Role of stromal estrogen receptor signaling. Proc. Natl. Acad. Sci. USA 2015, 112, E3255–E3264. [Google Scholar] [CrossRef]
- Choi, S.H.; Mahankali, M.; Lee, S.J.; Hull, M.; Petrassi, H.M.; Chatterjee, A.K.; Schultz, P.G.; Jones, K.A.; Shen, W. Targeted Disruption of Myc–Max Oncoprotein Complex by a Small Molecule. ACS Chem. Biol. 2017, 12, 2715–2719. [Google Scholar] [CrossRef]
- Huang, H.-L.; Weng, H.-Y.; Wang, L.-Q.; Yu, C.-H.; Huang, Q.-J.; Zhao, P.-P.; Wen, J.-Z.; Zhou, H.; Qu, L.-H. Triggering Fbw7-Mediated Proteasomal Degradation of c-Myc by Oridonin Induces Cell Growth Inhibition and Apoptosis. Mol. Cancer Ther. 2012, 11, 1155–1165. [Google Scholar] [CrossRef]
- Ruiz, E.J.; Pinto-Fernandez, A.; Turnbull, A.P.; Lan, L.; Charlton, T.M.; Scott, H.C.; Damianou, A.; Vere, G.; Riising, E.M.; Da Costa, C.; et al. USP28 deletion and small-molecule inhibition destabilizes c-MYC and elicits regression of squamous cell lung carcinoma. Elife 2021, 10, e71596. [Google Scholar] [CrossRef]
- Kim, H.-J.; Fei, X.; Cho, S.-C.; Choi, B.Y.; Ahn, H.-C.; Lee, K.; Seo, S.-Y.; Keum, Y.-S. Discovery of α-mangostin as a novel competitive inhibitor against mutant isocitrate dehydrogenase-1. Bioorganic Med. Chem. Lett. 2015, 25, 5625–5631. [Google Scholar] [CrossRef] [PubMed]
- Chavan, T.; Muth, A. The diverse bioactivity of α-mangostin and its therapeutic implications. Futur. Med. Chem. 2021, 13, 1679–1694. [Google Scholar] [CrossRef] [PubMed]
- Chien, H.J.; Ying, T.H.; Hsieh, S.C.; Lin, C.L.; Yu, Y.L.; Kao, S.H.; Hsieh, Y.H. α-Mangostin attenuates stemness and enhances cisplatin-induced cell death in cervical cancer stem-like cells through induction of mitochondrial-mediated apoptosis. J. Cell. Physiol. 2020, 235, 5590–5601. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tu, X.; Li, C.; Sun, W.; Tian, X.; Li, Q.; Wang, S.; Ding, X.; Huang, Z. Suppression of Cancer Cell Stemness and Drug Resistance via MYC Destabilization by Deubiquitinase USP45 Inhibition with a Natural Small Molecule. Cancers 2023, 15, 930. https://doi.org/10.3390/cancers15030930
Tu X, Li C, Sun W, Tian X, Li Q, Wang S, Ding X, Huang Z. Suppression of Cancer Cell Stemness and Drug Resistance via MYC Destabilization by Deubiquitinase USP45 Inhibition with a Natural Small Molecule. Cancers. 2023; 15(3):930. https://doi.org/10.3390/cancers15030930
Chicago/Turabian StyleTu, Xiao, Chuncheng Li, Wen Sun, Xi Tian, Qiufu Li, Shaoxin Wang, Xiaoling Ding, and Zhen Huang. 2023. "Suppression of Cancer Cell Stemness and Drug Resistance via MYC Destabilization by Deubiquitinase USP45 Inhibition with a Natural Small Molecule" Cancers 15, no. 3: 930. https://doi.org/10.3390/cancers15030930