
Methylated Cell-Free DNA Sequencing (MeD-seq) of LpnPI Digested Fragments to Identify Early Progression in Metastatic Renal Cell Carcinoma Patients on Watchful Waiting
 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Population
2.2. Sample Collection and DNA Isolation
2.3. MeD-seq Analysis
2.4. Data Processing
2.5. RCC-Specific Methylation Score
2.6. Statistical Analysis
3. Results
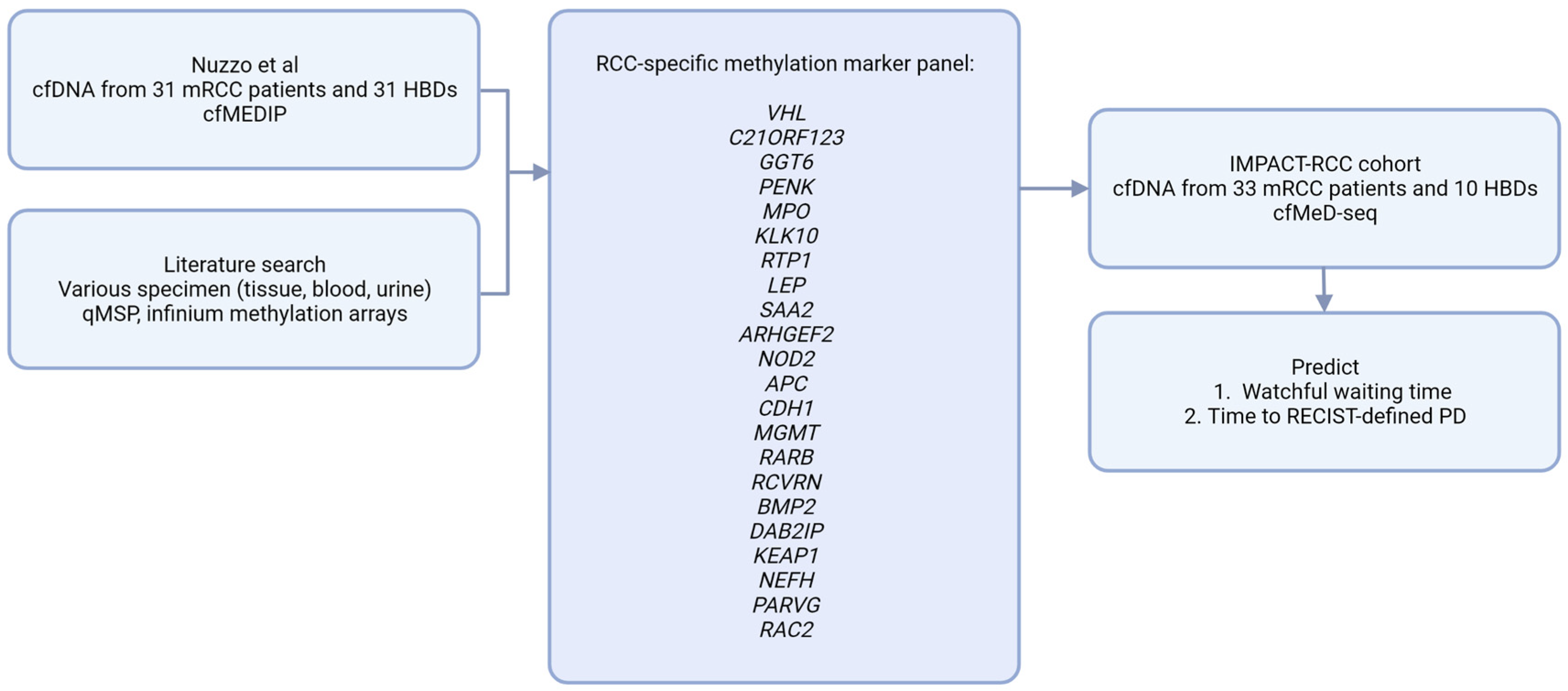
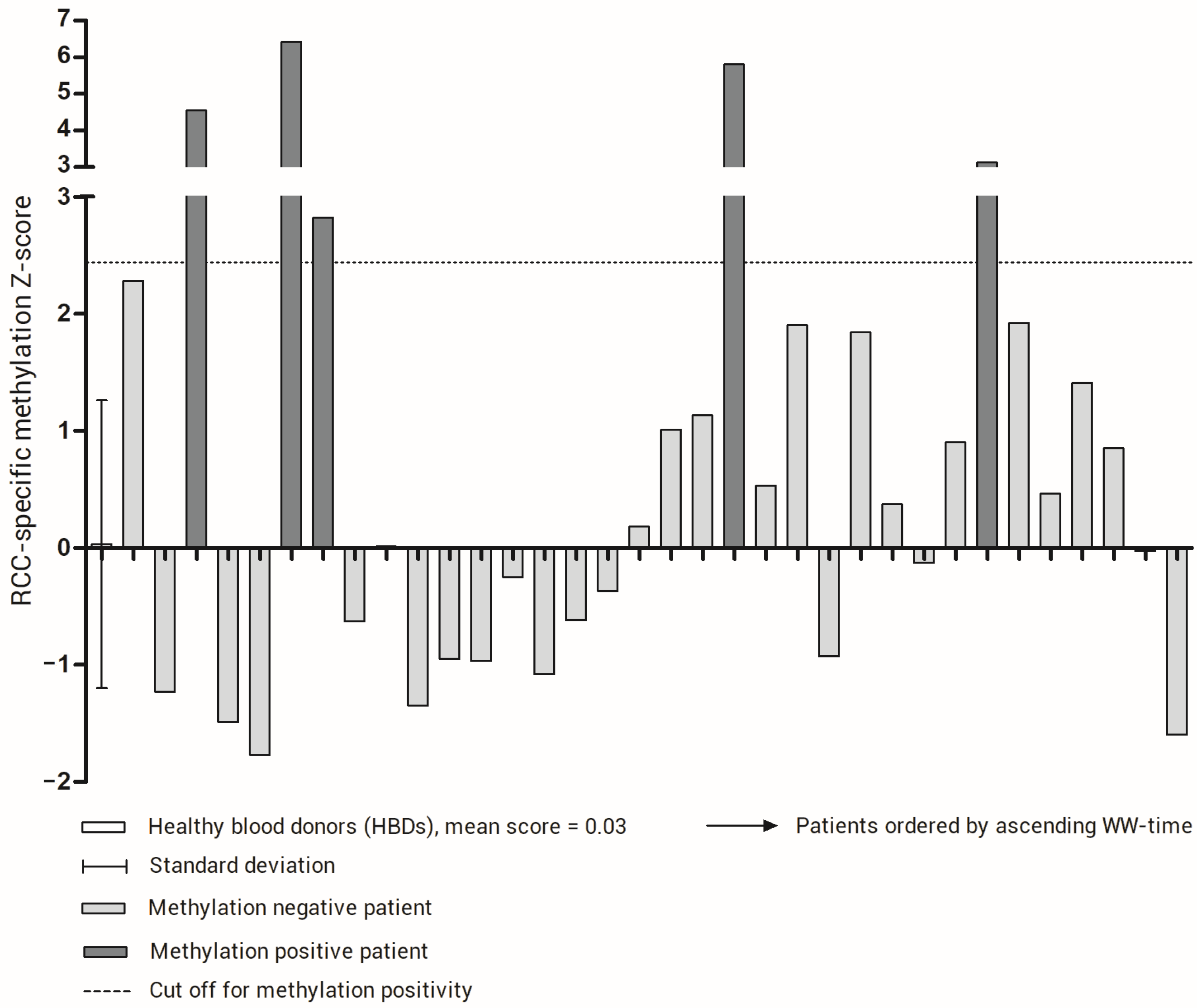
3.1. Building an RCC-Specific cfDNA Methylation Score
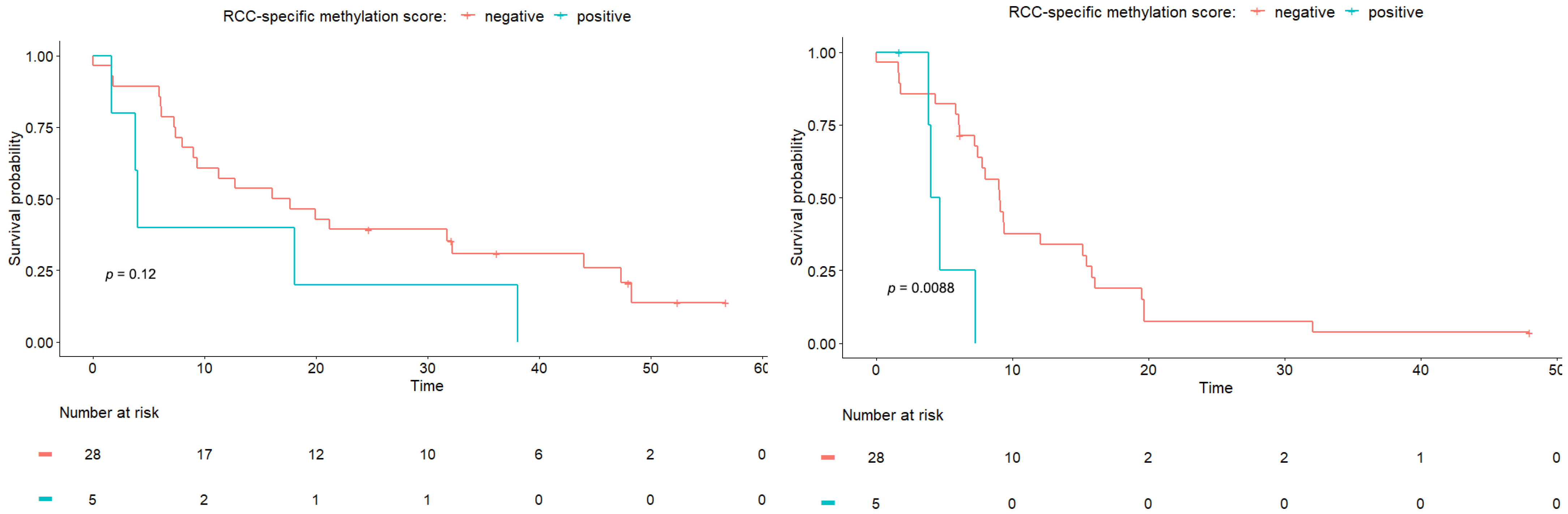
3.2. The RCC-Specific Methylation Score Associates with Clinical Outcome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sternberg, C.N.; Davis, I.D.; Mardiak, J.; Szczylik, C.; Lee, E.; Wagstaff, J.; Barrios, C.H.; Salman, P.; Gladkov, O.A.; Kavina, A.; et al. Pazopanib in Locally Advanced or Metastatic Renal Cell Carcinoma: Results of a Randomized Phase III Trial. J. Clin. Oncol. 2010, 28, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef] [PubMed]
- Heng, D.Y.C.; Xie, W.; Regan, M.M.; Harshman, L.C.; Bjarnason, G.A.; Vaishampayan, U.N.; Mackenzie, M.; Wood, L.; Donskov, F.; Tan, M.-H.; et al. External validation and comparison with other models of the International Metastatic Renal-Cell Carcinoma Database Consortium prognostic model: A population-based study. Lancet Oncol. 2013, 14, 141–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motzer, R.J.; Jonasch, E.; Agarwal, N.; Alva, A.; Baine, M.; Beckermann, K.; Carlo, M.I.; Choueiri, T.K.; Costello, B.A.; Derweesh, I.H.; et al. Kidney Cancer, Version 3.2022, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2022, 20, 71–90. [Google Scholar] [CrossRef]
- Rini, B.I.; Dorff, T.B.; Elson, P.; Rodriguez, C.S.; Shepard, D.; Wood, L.; Humbert, J.; Pyle, L.; Wong, Y.-N.; Finke, J.H.; et al. Active surveillance in metastatic renal-cell carcinoma: A prospective, phase 2 trial. Lancet Oncol. 2016, 17, 1317–1324. [Google Scholar] [CrossRef]
- Verhoeff, S.R.; Oosting, S.F.; Elias, S.G.; van Es, S.C.; Gerritse, S.L.; Angus, L.; Heskamp, S.; Desar, I.M.E.; Menke-van der Houven van Oordt, C.W.; van der Veldt, A.A.M.; et al. [89Zr]Zr-DFO-girentuximab and [18F]FDG PET/CT to predict watchful waiting duration in patients with metastatic clear cell renal cell carcinoma. Clin. Cancer Res. 2022, 29, 592–601. [Google Scholar] [CrossRef]
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2019, 20, 71–88. [Google Scholar] [CrossRef]
- Lee, J.-S.; Kim, M.; Seong, M.-W.; Kim, H.-S.; Lee, Y.K.; Kang, H.J. Plasma vs. serum in circulating tumor DNA measurement: Characterization by DNA fragment sizing and digital droplet polymerase chain reaction. Clin. Chem. Lab. Med. 2020, 58, 527–532. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.G.; Moser, T.; Mouliere, F.; Field-Rayner, J.; Eldridge, M.; Riediger, A.L.; Chandrananda, D.; Heider, K.; Wan, J.C.M.; Warren, A.Y.; et al. Comprehensive characterization of cell-free tumor DNA in plasma and urine of patients with renal tumors. Genome Med. 2020, 12, 23. [Google Scholar] [CrossRef] [Green Version]
- Lasseter, K.; Nassar, A.H.; Hamieh, L.; Berchuck, J.E.; Nuzzo, P.V.; Korthauer, K.; Shinagare, A.B.; Ogorek, B.; McKay, R.; Thorner, A.R.; et al. Plasma cell-free DNA variant analysis compared with methylated DNA analysis in renal cell carcinoma. Genet. Med. 2020, 22, 1366–1373. [Google Scholar] [CrossRef]
- Nuzzo, P.V.; Berchuck, J.E.; Korthauer, K.; Spisak, S.; Nassar, A.H.; Abou Alaiwi, S.; Chakravarthy, A.; Shen, S.Y.; Bakouny, Z.; Boccardo, F.; et al. Detection of renal cell carcinoma using plasma and urine cell-free DNA methylomes. Nat. Med. 2020, 26, 1041–1043. [Google Scholar] [CrossRef]
- Joosten, S.C.; Odeh, S.N.O.; Koch, A.; Buekers, N.; Aarts, M.J.B.; Baldewijns, M.M.L.L.; Van Neste, L.; van Kuijk, S.; Schouten, L.J.; van den Brandt, P.A.; et al. Development of a prognostic risk model for clear cell renal cell carcinoma by systematic evaluation of DNA methylation markers. Clin. Epigenetics 2021, 13, 103. [Google Scholar] [CrossRef]
- Lasseigne, B.N.; Brooks, J.D. The Role of DNA Methylation in Renal Cell Carcinoma. Mol. Diagn. Ther. 2018, 22, 431–442. [Google Scholar] [CrossRef]
- Battagli, C.; Uzzo, R.G.; Dulaimi, E.; Ibanez de Caceres, I.; Krassenstein, R.; Al-Saleem, T.; Greenberg, R.E.; Cairns, P. Promoter Hypermethylation of Tumor Suppressor Genes in Urine from Kidney Cancer Patients. Cancer Res. 2003, 63, 8695–8699. [Google Scholar]
- Hoque, M.O.; Begum, S.; Topaloglu, O.; Jeronimo, C.; Mambo, E.; Westra, W.H.; Califano, J.A.; Sidransky, D. Quantitative Detection of Promoter Hypermethylation of Multiple Genes in the Tumor, Urine, and Serum DNA of Patients with Renal Cancer. Cancer Res. 2004, 64, 5511–5517. [Google Scholar] [CrossRef]
- Hauser, S.; Zahalka, T.; Fechner, G.; MÜLler, S.C.; Ellinger, J. Serum DNA Hypermethylation in Patients with Kidney Cancer: Results of a Prospective Study. Anticancer. Res. 2013, 33, 4651. [Google Scholar]
- Lasseigne, B.N.; Burwell, T.C.; Patil, M.A.; Absher, D.M.; Brooks, J.D.; Myers, R.M. DNA methylation profiling reveals novel diagnostic biomarkers in renal cell carcinoma. BMC Med. 2014, 12, 235. [Google Scholar] [CrossRef]
- Boers, R.; Boers, J.; de Hoon, B.; Kockx, C.; Ozgur, Z.; Molijn, A.; van Ijcken, W.; Laven, J.; Gribnau, J. Genome-wide DNA methylation profiling using the methylation-dependent restriction enzyme LpnPI. Genome Res. 2018, 28, 88–99. [Google Scholar] [CrossRef]
- Deger, T.; Boers, R.G.; de Weerd, V.; Angus, L.; van der Put, M.M.J.; Boers, J.B.; Azmani, Z.; van Ijcken, W.F.J.; Grünhagen, D.J.; van Dessel, L.F.; et al. High-throughput and affordable genome-wide methylation profiling of circulating cell-free DNA by methylated DNA sequencing (MeD-seq) of LpnPI digested fragments. Clin. Epigenetics 2021, 13, 196. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Belic, J.; Koch, M.; Ulz, P.; Auer, M.; Gerhalter, T.; Mohan, S.; Fischereder, K.; Petru, E.; Bauernhofer, T.; Geigl, J.B.; et al. Rapid Identification of Plasma DNA Samples with Increased ctDNA Levels by a Modified FAST-SeqS Approach. Clin. Chem. 2015, 61, 838–849. [Google Scholar] [CrossRef] [Green Version]
- Harrison, M.R.; Costello, B.A.; Bhavsar, N.A.; Vaishampayan, U.; Pal, S.K.; Zakharia, Y.; Jim, H.S.L.; Fishman, M.N.; Molina, A.M.; Kyriakopoulos, C.E.; et al. Active surveillance of metastatic renal cell carcinoma: Results from a prospective observational study (MaRCC). Cancer 2021, 127, 2204–2212. [Google Scholar] [CrossRef]
- Wan, J.; Zhu, L.; Jiang, Z.; Cheng, K. Monitoring of Plasma Cell-Free DNA in Predicting Postoperative Recurrence of Clear Cell Renal Cell Carcinoma. Urol. Int. 2013, 91, 273–278. [Google Scholar] [CrossRef]
- Feng, G.; Ye, X.; Fang, F.; Pu, C.; Huang, H.; Li, G. Quantification of Plasma Cell-Free DNA1 in Predicting Therapeutic Efficacy of Sorafenib on Metastatic Clear Cell Renal Cell Carcinoma. Dis. Markers 2013, 34, 651323. [Google Scholar] [CrossRef]
- de Martino, M.; Klatte, T.; Haitel, A.; Marberger, M. Serum cell-free DNA in renal cell carcinoma. Cancer 2012, 118, 82–90. [Google Scholar] [CrossRef]
- Mouliere, F.; Chandrananda, D.; Piskorz, A.M.; Moore, E.K.; Morris, J.; Ahlborn, L.B.; Mair, R.; Goranova, T.; Marass, F.; Heider, K.; et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 2018, 10, eaat4921. [Google Scholar] [CrossRef] [Green Version]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [Green Version]
- Zill, O.A.; Banks, K.C.; Fairclough, S.R.; Mortimer, S.A.; Vowles, J.V.; Mokhtari, R.; Gandara, D.R.; Mack, P.C.; Odegaard, J.I.; Nagy, R.J.; et al. The Landscape of Actionable Genomic Alterations in Cell-Free Circulating Tumor DNA from 21,807 Advanced Cancer Patients. Clin. Cancer Res. 2018, 24, 3528–3538. [Google Scholar] [CrossRef] [Green Version]
- Pittella-Silva, F.; Chin, Y.M.; Chan, H.T.; Nagayama, S.; Miyauchi, E.; Low, S.-K.; Nakamura, Y. Plasma or Serum: Which Is Preferable for Mutation Detection in Liquid Biopsy? Clin. Chem. 2020, 66, 946–957. [Google Scholar] [CrossRef]
- Bacon, J.V.W.; Annala, M.; Soleimani, M.; Lavoie, J.-M.; So, A.; Gleave, M.E.; Fazli, L.; Wang, G.; Chi, K.N.; Kollmannsberger, C.K.; et al. Plasma Circulating Tumor DNA and Clonal Hematopoiesis in Metastatic Renal Cell Carcinoma. Clin. Genitourin. Cancer 2020, 18, 322–331.e2. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Characteristics | Patients in IMPACT-RCC Study (n = 42) | Patients with Serum Samples Successfully Analyzed (n = 33) | p-Value |
|---|---|---|---|
| Gender | |||
| Male, n (%) | 31 (74%) | 26 (76%) | |
| Female, n (%) | 11 (26%) | 7 (21%) | 0.786 |
| Median age in years (range) | 66 (44–86) | 66 (44–86) | 0.790 |
| Nephrectomy | |||
| Yes, n (%) | 36 (86%) | 28 (85%) | |
| No, n (%) | 6 (14%) | 5 (15%) | 1 |
| Histology | |||
| Pure clear cell, n (%) | 32 (76%) | 27 (79%) | |
| Mixed, n (%) | 10 (24%) | 6 (18%) | 0.586 |
| Time from diagnosis to first metastasis | |||
| <1 year, n (%) | 23 (55%) | 19 (58%) | |
| ≥1 year, n (%) | 19 (45%) | 14 (42%) | 0.820 |
| IMDC risk factors | |||
| 0 (favorable) | 14 (33%) | 10 (30%) | |
| 1 (intermediate) | 13 (31%) | 10 (30%) | |
| 2 (intermediate) | 15 (36%) | 13 (39%) | 0.941 |
| Variables | HR | 95% CI HR | p-Value | |
|---|---|---|---|---|
| (a) Cox proportional hazards regression analysis results for watchful waiting time. | ||||
| Clinical | Age | 1.00 | 0.96–1.024 | 0.99 |
| IMDC criteria, per unit increase in score | 2.01 | 1.19–3.41 | 0.01 | |
| Number of affected organ sites per patient | 1.43 | 0.96–2.11 | 0.07 | |
| Sum of CT measured diameter of lesions per patient (cm) | 1.02 | 1.00–1.05 | 0.08 | |
| Molecular | RCC-specific methylation score | 2.18 | 0.81–5.87 | 0.12 |
| (b). Cox proportional hazards regression analysis results for time until RECIST-defined PD | ||||
| Clinical | Age | 1.01 | 0.96–1.05 | 0.82 |
| IMDC criteria, per unit increase in score | 1.35 | 0.86–2.10 | 0.19 | |
| Number of affected organ sites per patient | 1.07 | 0.73–1.57 | 0.74 | |
| Sum of CT measured diameter of lesions per patient (cm) | 1.01 | 0.99–1.04 | 0.30 | |
| Molecular | RCC-specific methylation score | 4.45 | 1.32–15.05 | 0.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bos, M.K.; Verhoeff, S.R.; Oosting, S.F.; Menke-van der Houven van Oordt, W.C.; Boers, R.G.; Boers, J.B.; Gribnau, J.; Martens, J.W.M.; Sleijfer, S.; van Herpen, C.M.L.; et al. Methylated Cell-Free DNA Sequencing (MeD-seq) of LpnPI Digested Fragments to Identify Early Progression in Metastatic Renal Cell Carcinoma Patients on Watchful Waiting. Cancers 2023, 15, 1374. https://doi.org/10.3390/cancers15051374
Bos MK, Verhoeff SR, Oosting SF, Menke-van der Houven van Oordt WC, Boers RG, Boers JB, Gribnau J, Martens JWM, Sleijfer S, van Herpen CML, et al. Methylated Cell-Free DNA Sequencing (MeD-seq) of LpnPI Digested Fragments to Identify Early Progression in Metastatic Renal Cell Carcinoma Patients on Watchful Waiting. Cancers. 2023; 15(5):1374. https://doi.org/10.3390/cancers15051374
Chicago/Turabian StyleBos, Manouk K., Sarah R. Verhoeff, Sjoukje F. Oosting, Willemien C. Menke-van der Houven van Oordt, Ruben G. Boers, Joachim B. Boers, Joost Gribnau, John W. M. Martens, Stefan Sleijfer, Carla M. L. van Herpen, and et al. 2023. "Methylated Cell-Free DNA Sequencing (MeD-seq) of LpnPI Digested Fragments to Identify Early Progression in Metastatic Renal Cell Carcinoma Patients on Watchful Waiting" Cancers 15, no. 5: 1374. https://doi.org/10.3390/cancers15051374