

In Situ Imaging of O-Linked β-N-Acetylglucosamine Using On-Tissue Hydrolysis and MALDI Mass Spectrometry
 , , ,
, , ,
Abstract
:Simple Summary
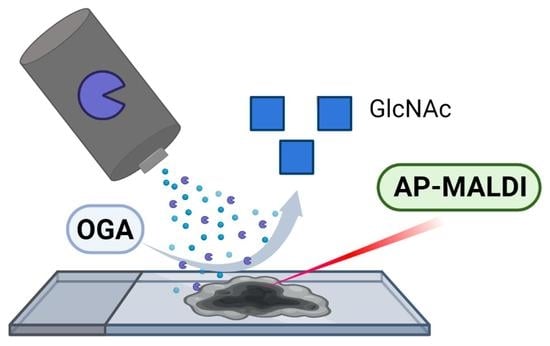
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
3. Materials & Methods
3.1. Chemicals
3.2. Sample Preparation & Analysis
3.3. LC-MS/MS Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Herrera, H.; Dilday, T.; Uber, A.; Scott, D.; Zambrano, J.N.; Wang, M.; Angel, P.M.; Mehta, A.S.; Drake, R.R.; Hill, E.G.; et al. Core-Fucosylated Tetra-Antennary N-Glycan Containing A Single N-Acetyllactosamine Branch Is Associated with Poor Survival Outcome in Breast Cancer. Int. J. Mol. Sci. 2019, 20, 2528. [Google Scholar] [CrossRef] [Green Version]
- Briggs, M.T.; Condina, M.R.; Ho, Y.Y.; Everest-Dass, A.V.; Mittal, P.; Kaur, G.; Oehler, M.K.; Packer, N.H.; Hoffmann, P. MALDI Mass Spectrometry Imaging of Early- and Late-Stage Serous Ovarian Cancer Tissue Reveals Stage-Specific N-Glycans. Proteomics 2019, 19, 1800482. [Google Scholar] [CrossRef] [Green Version]
- Powers, T.W.; Holst, S.; Wuhrer, M.; Mehta, A.S.; Drake, R.R. Two-Dimensional N-Glycan Distribution Mapping of Hepatocellular Carcinoma Tissues by MALDI-Imaging Mass Spectrometry. Biomolecules 2015, 5, 2554–2572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Shi, X.; Liu, Y.; Wang, B.; Xu, M.; Welham, N.V.; Li, L. On-Tissue Amidation of Sialic Acid with Aniline for Sensitive Imaging of Sialylated N-Glycans from FFPE Tissue Sections via MALDI Mass Spectrometry. Anal. Bioanal. Chem. 2022, 414, 5263–5274. [Google Scholar] [CrossRef] [PubMed]
- Boyaval, F.; Dalebout, H.; Van Zeijl, R.; Wang, W.; Fariña-Sarasqueta, A.; Lageveen-Kammeijer, G.S.M.; Boonstra, J.J.; McDonnell, L.A.; Wuhrer, M.; Morreau, H.; et al. High-Mannose N-Glycans as Malignant Progression Markers in Early-Stage Colorectal Cancer. Cancers 2022, 14, 1552. [Google Scholar] [CrossRef]
- West, C.A.; Liang, H.; Drake, R.R.; Mehta, A.S. New Enzymatic Approach to Distinguish Fucosylation Isomers of N-Linked Glycans in Tissues Using MALDI Imaging Mass Spectrometry. J. Proteome Res. 2020, 19, 2989–2996. [Google Scholar] [CrossRef] [PubMed]
- Ščupáková, K.; Adelaja, O.T.; Balluff, B.; Ayyappan, V.; Tressler, C.M.; Jenkinson, N.M.; Claes BS, R.; Bowman, A.P.; Cimino-Mathews, A.M.; White, M.J.; et al. Clinical Importance of High-Mannose, Fucosylated, and Complex N-Glycans in Breast Cancer Metastasis. JCI Insight 2021, 6. [Google Scholar] [CrossRef]
- West, C.A.; Wang, M.; Herrera, H.; Liang, H.; Black, A.; Angel, P.M.; Drake, R.R.; Mehta, A.S. N-Linked Glycan Branching and Fucosylation Are Increased Directly in Hcc Tissue as Determined through in Situ Glycan Imaging. J. Proteome Res. 2018, 17, 3454–3462. [Google Scholar] [CrossRef]
- McDowell, C.T.; Klamer, Z.; Hall, J.; West, C.A.; Wisniewski, L.; Powers, T.W.; Angel, P.M.; Mehta, A.S.; Lewin, D.N.; Haab, B.B.; et al. Imaging Mass Spectrometry and Lectin Analysis of N-Linked Glycans in Carbohydrate Antigen–Defined Pancreatic Cancer Tissues. Mol. Cell. Proteom. 2021, 20, 100012. [Google Scholar] [CrossRef]
- Angel, P.M.; Mehta, A.; Norris-Caneda, K.; Drake, R.R. MALDI Imaging Mass Spectrometry of N-Glycans and Tryptic Peptides from the Same Formalin-Fixed, Paraffin-Embedded Tissue Section. Methods Mol. Biol. 2018, 1788, 225–241. [Google Scholar] [CrossRef] [PubMed]
- Wulff-Fuentes, E.; Berendt, R.R.; Massman, L.; Danner, L.; Malard, F.; Vora, J.; Kahsay, R.; Olivier-Van Stichelen, S. The Human O-GlcNAcome Database and Meta-Analysis. Sci. Data 2021, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Zachara, N.E.; Akimoto, Y.; Boyce, M.; Hart, G.W. The O-GlcNAc Modification. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Mohnen, D., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2022. [Google Scholar]
- Worth, M.; Li, H.; Jiang, J. Deciphering the Functions of Protein O-GlcNAcylation with Chemistry. ACS Chem. Biol. 2017, 12, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Liu, C.; Wang, X.; Xu, B.; Liu, X.; Li, Y.; Xia, J.; Li, Y.; Zhang, C.; Li, D.; et al. O-GlcNAcylation Quantification of Certain Protein by the Proximity Ligation Assay and Clostridium Perfringen OGAD298N(CpOGAD298N). ACS Chem. Biol. 2021, 16, 1040–1049. [Google Scholar] [CrossRef] [PubMed]
- Hardivillé, S.; Hart, G.W. Nutrient Regulation of Gene Expression by O-GlcNAcylation of Chromatin. Curr. Opin. Chem. Biol. 2016, 33, 88–94. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Niu, Y.; Wang, Z.; Xu, X.; Li, Y.; Ma, L.; Wang, J.; Yu, Y. Corosolic Acid Inhibits Cancer Progression by Decreasing the Level of CDK19-Mediated O-GlcNAcylation in Liver Cancer Cells. Cell Death Dis. 2021, 12, 889. [Google Scholar] [CrossRef] [PubMed]
- Ciraku, L.; Esquea, E.M.; Reginato, M.J. O-GlcNAcylation Regulation of Cellular Signaling in Cancer. Cell. Signal. 2022, 90, 110201. [Google Scholar] [CrossRef] [PubMed]
- Hanover, J.A.; Krause, M.W.; Love, D.C. Linking Metabolism to Epigenetics through O-GlcNAcylation. Nat. Rev. Mol. Cell Biol. 2012, 13, 312–321. [Google Scholar] [CrossRef]
- Fuentes-García, G.; Castañeda-Patlán, M.C.; Vercoutter-Edouart, A.-S.; Lefebvre, T.; Robles-Flores, M. O-GlcNAcylation Is Involved in the Regulation of Stem Cell Markers Expression in Colon Cancer Cells. Front. Endocrinol. 2019, 10, 289. [Google Scholar] [CrossRef]
- Hanover, J.A.; Chen, W.; Bond, M.R. O-GlcNAc in Cancer: An Oncometabolism-Fueled Vicious Cycle. J. Bioenerg. Biomembr. 2018, 50, 155–173. [Google Scholar] [CrossRef]
- Lei, L.; Xie, J.; Yu, J.; Li, Y.; Liu, Y. Parallel Study on Protein O-GlcNAcylation in Prostate Cancer Cell with a Sensitive Microarray Biochip. Anal. Biochem. 2018, 558, 53–59. [Google Scholar] [CrossRef]
- Yoo, T.Y.; Mitchison, T.J. O-GlcNAc Modification of Nuclear Pore Complexes Accelerates Bidirectional Transport. J. Cell Biol. 2021, 220, e202010141. [Google Scholar] [CrossRef]
- Wright, J.N.; Collins, H.E.; Wende, A.R.; Chatham, J.C. O-GlcNAcylation and Cardiovascular Disease. Biochem. Soc. Trans. 2017, 45, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, P.K.; Parihar, R.; Dwivedi, V.; Lakhotia, S.C.; Ganesh, S. Decreased O-Linked GlcNAcylation Protects from Cytotoxicity Mediated by Huntingtin Exon1 Protein Fragment. J. Biol. Chem. 2014, 289, 13543–13553. [Google Scholar] [CrossRef] [Green Version]
- Burt, R.A.; Alghusen, I.M.; John Ephrame, S.; Villar, M.T.; Artigues, A.; Slawson, C. Mapping the O-GlcNAc Modified Proteome: Applications for Health and Disease. Front. Mol. Biosci. 2022, 9, 920727. [Google Scholar] [CrossRef]
- Lin, W.; Gao, L.; Chen, X. Protein-Specific Imaging of O-GlcNAcylation in Single Cells. ChemBioChem 2015, 16, 2571–2575. [Google Scholar] [CrossRef] [PubMed]
- Kasprowicz, A.; Spriet, C.; Terryn, C.; Rigolot, V.; Hardiville, S.; Alteen, M.G.; Lefebvre, T.; Biot, C. Exploring the Potential of β-Catenin O-GlcNAcylation by Using Fluorescence-Based Engineering and Imaging. Molecules 2020, 25, 4501. [Google Scholar] [CrossRef]
- Clark, P.M.; Dweck, J.F.; Mason, D.E.; Hart, C.R.; Buck, S.B.; Peters, E.C.; Agnew, B.J.; Hsieh-Wilson, L.C. Direct In-Gel Fluorescence Detection and Cellular Imaging of O-GlcNAc-Modified Proteins. J. Am. Chem. Soc. 2008, 130, 11576–11577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Zhang, G.; Ma, J.; Zhao, C.; Xue, Q.; Wang, J.; Liu, W.; Liu, K.; Wang, H.; Liu, N.; et al. Detection and Identification of O-GlcNAc-Modified Proteins Using 6-Azido-6-Deoxy-N-Acetyl-Galactosamine. Org. Biomol. Chem. 2019, 17, 4326–4334. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.L.; Person, A.D.; Anderson, M.; Burroughs, B.; Tatge, T.; Khatri, K.; Zou, Y.; Wang, L.; Geders, T.; Zaia, J.; et al. Imaging Specific Cellular Glycan Structures Using Glycosyltransferases via Click Chemistry. Glycobiology 2017, 28, 69–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, H.Y.; Eskandari, R.; Shen, D.; Zhu, Y.; Liu, T.-W.; Willems, L.I.; Alteen, M.G.; Madden, Z.; Vocadlo, D.J. Direct One-Step Fluorescent Labeling of O-GlcNAc-Modified Proteins in Live Cells Using Metabolic Intermediates. J. Am. Chem. Soc. 2018, 140, 15300–15308. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.L.; Tatge, T.J.; Grill, A.E.; Zou, Y. Detecting and Imaging O-GlcNAc Sites Using Glycosyltransferases: A Systematic Approach to Study O-GlcNAc. Cell Chem. Biol. 2018, 25, 1428–1435.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norris, J.L.; Caprioli, R.M. Analysis of Tissue Specimens by Matrix-Assisted Laser Desorption/Ionization Imaging Mass Spectrometry in Biological and Clinical Research. Chem. Rev. 2013, 113, 2309–2342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aichler, M.; Walch, A. MALDI Imaging Mass Spectrometry: Current Frontiers and Perspectives in Pathology Research and Practice. Lab. Invest. 2015, 95, 422–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochoa-Rios, S.; O’Connor, I.P.; Kent, L.N.; Clouse, J.M.; Hadjiyannis, Y.; Koivisto, C.; Pecot, T.; Angel, P.; Drake, R.R.; Leone, G.; et al. Imaging Mass Spectrometry Reveals Alterations in N-Linked Glycosylation That Are Associated with Histopathological Changes in Non-Alcoholic Steatohepatitis in Mouse and Human. Mol. Cell. Proteom. 2022, 21, 100225. [Google Scholar] [CrossRef] [PubMed]
- Stanback, A.E.; Conroy, L.R.; Young, L.E.A.; Hawkinson, T.R.; Markussen, K.H.; Clarke, H.A.; Allison, D.B.; Sun, R.C. Regional N-Glycan and Lipid Analysis from Tissues Using MALDI-Mass Spectrometry Imaging. STAR Protoc. 2021, 2, 100304. [Google Scholar] [CrossRef]
- Angel, P.M.; Mehta, A.S.; Drake, R.R. Array-Based N-Glycan Profiling of Cells in Culture. In Mass Spectrometry of Glycoproteins: Methods and Protocols; Delobel, A., Ed.; Methods in Molecular Biology; Springer US: New York, NY, USA, 2021; pp. 331–342. [Google Scholar] [CrossRef]
- Blaschke, C.R.K.; Hartig, J.P.; Grimsley, G.; Liu, L.; Semmes, O.J.; Wu, J.D.; Ippolito, J.E.; Hughes-Halbert, C.; Nyalwidhe, J.O.; Drake, R.R. Direct N-Glycosylation Profiling of Urine and Prostatic Fluid Glycoproteins and Extracellular Vesicles. Front. Chem. 2021, 9, 734280. [Google Scholar] [CrossRef]
- Malaker, S.A.; Quanico, J.; Raffo-Romero, A.; Kobeissy, F.; Aboulouard, S.; Tierny, D.; Bertozzi, C.R.; Fournier, I.; Salzet, M. On-Tissue Spatially Resolved Glycoproteomics Guided by N-Glycan Imaging Reveal Global Dysregulation of Canine Glioma Glycoproteomic Landscape. Cell Chem. Biol. 2022, 29, 30–42.e4. [Google Scholar] [CrossRef] [PubMed]
- Grzeski, M.; Taube, E.T.; Braicu, E.I.; Sehouli, J.; Blanchard, V.; Klein, O. In Situ N-Glycosylation Signatures of Epithelial Ovarian Cancer Tissue as Defined by MALDI Mass Spectrometry Imaging. Cancers 2022, 14, 1021. [Google Scholar] [CrossRef]
- Drake, R.R.; McDowell, C.; West, C.; David, F.; Powers, T.W.; Nowling, T.; Bruner, E.; Mehta, A.S.; Angel, P.M.; Marlow, L.A.; et al. Defining the Human Kidney N-Glycome in Normal and Cancer Tissues Using MALDI Imaging Mass Spectrometry. J. Mass Spectrom. 2020, 55, e4490. [Google Scholar] [CrossRef] [PubMed]
- Drake, R.R.; Powers, T.W.; Jones, E.E.; Bruner, E.; Mehta, A.S.; Angel, P.M. MALDI Mass Spectrometry Imaging of N-Linked Glycans in Cancer Tissues. Adv. Cancer Res. 2017, 134, 85–116. [Google Scholar] [CrossRef]
- Rebelo, A.L.; Gubinelli, F.; Roost, P.; Jan, C.; Brouillet, E.; Van Camp, N.; Drake, R.R.; Saldova, R.; Pandit, A. Complete Spatial Characterisation of N-Glycosylation upon Striatal Neuroinflammation in the Rodent Brain. J. Neuroinflammation 2021, 18, 116. [Google Scholar] [CrossRef] [PubMed]
- Zemaitis, K.J.; Veličković, D.; Kew, W.; Fort, K.L.; Reinhardt-Szyba, M.; Pamreddy, A.; Ding, Y.; Kaushik, D.; Sharma, K.; Makarov, A.A.; et al. Enhanced Spatial Mapping of Histone Proteoforms in Human Kidney Through MALDI-MSI by High-Field UHMR-Orbitrap Detection. Anal. Chem. 2022, 94, 12604–12613. [Google Scholar] [CrossRef]
- Kunzke, T.; Balluff, B.; Feuchtinger, A.; Buck, A.; Langer, R.; Luber, B.; Lordick, F.; Zitzelsberger, H.; Aichler, M.; Walch, A. Native Glycan Fragments Detected by MALDI-FT-ICR Mass Spectrometry Imaging Impact Gastric Cancer Biology and Patient Outcome. Oncotarget 2017, 8, 68012–68025. [Google Scholar] [CrossRef] [Green Version]
- Conroy, L.R.; Stanback, A.E.; Young, L.E.A.; Clarke, H.A.; Austin, G.L.; Liu, J.; Allison, D.B.; Sun, R.C. In Situ Analysis of N-Linked Glycans as Potential Biomarkers of Clinical Course in Human Prostate Cancer. Mol. Cancer Res. 2021, 19, 1727–1738. [Google Scholar] [CrossRef]
- West, C.A.; Lu, X.; Grimsley, G.; Norris-Caneda, K.; Mehta, A.S.; Angel, P.M.; Drake, R.R. Optimization of Multiple Glycosidase and Chemical Stabilization Strategies for N-Glycan Isomer Detection by Mass Spectrometry Imaging in Formalin-Fixed, Paraffin-Embedded Tissues. In Mass Spectrometry of Glycoproteins: Methods and Protocols; Delobel, A., Ed.; Methods in Molecular Biology; Springer US: New York, NY, USA, 2021; pp. 303–316. [Google Scholar] [CrossRef]
- Saito, T.; Watanabe, A.; Nakano, M.; Matsuo, K. MALDI-TOF Mass Spectrometry Imaging for N-Glycans on FFPE Tissue Sections of Mouse NASH Liver through Sialic Acid Benzylamidation. Glycoconj. J. 2021, 38, 167–175. [Google Scholar] [CrossRef]
- Clift, C.L.; Mehta, A.; Drake, R.R.; Angel, P.M. Multiplexed Imaging Mass Spectrometry of Histological Staining, N-Glycan and Extracellular Matrix from One Tissue Section: A Tool for Fibrosis Research. In Multiplexed Imaging: Methods and Protocols; Zamir, E., Ed.; Methods in Molecular Biology; Springer US: New York, NY, USA, 2021; pp. 313–329. [Google Scholar] [CrossRef]
- Clift, C.L.; Drake, R.R.; Mehta, A.; Angel, P.M. Multiplexed Imaging Mass Spectrometry of the Extracellular Matrix Using Serial Enzyme Digests from Formalin-Fixed Paraffin-Embedded Tissue Sections. Anal. Bioanal. Chem. 2021, 413, 2709–2719. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Shi, X.; Vu, N.Q.; Li, G.; Li, Z.; Shi, Y.; Li, M.; Wang, B.; Welham, N.V.; Patankar, M.S.; et al. On-Tissue Derivatization with Girard’s Reagent P Enhances N-Glycan Signals for Formalin-Fixed Paraffin-Embedded Tissue Sections in MALDI Mass Spectrometry Imaging. Anal. Chem. 2020, 92, 13361–13368. [Google Scholar] [CrossRef] [PubMed]
- Holst, S.; Heijs, B.; de Haan, N.; van Zeijl, R.J.M.; Briaire-de Bruijn, I.H.; van Pelt, G.W.; Mehta, A.S.; Angel, P.M.; Mesker, W.E.; Tollenaar, R.A.; et al. Linkage-Specific in Situ Sialic Acid Derivatization for N-Glycan Mass Spectrometry Imaging of Formalin-Fixed Paraffin-Embedded Tissues. Anal. Chem. 2016, 88, 5904–5913. [Google Scholar] [CrossRef]
- Heijs, B.; Holst, S.; Briaire-de Bruijn, I.H.; van Pelt, G.W.; de Ru, A.H.; van Veelen, P.A.; Drake, R.R.; Mehta, A.S.; Mesker, W.E.; Tollenaar, R.A.; et al. Multimodal Mass Spectrometry Imaging of N-Glycans and Proteins from the Same Tissue Section. Anal. Chem. 2016, 88, 7745–7753. [Google Scholar] [CrossRef]
- Macias, L.A.; Santos, I.C.; Brodbelt, J.S. Ion Activation Methods for Peptides and Proteins. Anal. Chem. 2020, 92, 227–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodbelt, J.S. Deciphering Combinatorial Post-Translational Modifications by Top-down Mass Spectrometry. Curr. Opin. Chem. Biol. 2022, 70, 102180. [Google Scholar] [CrossRef] [PubMed]
- Reiding, K.R.; Bondt, A.; Franc, V.; Heck, A.J.R. The Benefits of Hybrid Fragmentation Methods for Glycoproteomics. TrAC Trends Anal. Chem. 2018, 108, 260–268. [Google Scholar] [CrossRef]
- Brodbelt, J.S.; Morrison, L.J.; Santos, I. Ultraviolet Photodissociation Mass Spectrometry for Analysis of Biological Molecules. Chem. Rev. 2020, 120, 3328–3380. [Google Scholar] [CrossRef] [PubMed]
- Escobar, E.E.; Wang, S.; Goswami, R.; Lanzillotti, M.B.; Li, L.; McLellan, J.S.; Brodbelt, J.S. Analysis of Viral Spike Protein N-Glycosylation Using Ultraviolet Photodissociation Mass Spectrometry. Anal. Chem. 2022, 94, 5776–5784. [Google Scholar] [CrossRef] [PubMed]
- Dennis, R.J.; Taylor, E.J.; Macauley, M.S.; Stubbs, K.A.; Turkenburg, J.P.; Hart, S.J.; Black, G.N.; Vocadlo, D.J.; Davies, G.J. Structure and Mechanism of a Bacterial β-Glucosaminidase Having O-GlcNAcase Activity. Nat. Struct. Mol. Biol. 2006, 13, 365–371. [Google Scholar] [CrossRef]
- Woo, C.M.; Lund, P.J.; Huang, A.C.; Davis, M.M.; Bertozzi, C.R.; Pitteri, S.J. Mapping and Quantification of Over 2000 O-Linked Glycopeptides in Activated Human T Cells with Isotope-Targeted Glycoproteomics (Isotag). Mol. Cell. Proteom. 2018, 17, 764–775. [Google Scholar] [CrossRef] [Green Version]
- Young, L.E.A.; Conroy, L.R.; Clarke, H.A.; Hawkinson, T.R.; Bolton, K.E.; Sanders, W.C.; Chang, J.E.; Webb, M.B.; Alilain, W.J.; Kooi, C.W.V.; et al. In Situ Mass Spectrometry Imaging Reveals Heterogeneous Glycogen Stores in Human Normal and Cancerous Tissues. EMBO Mol. Med. 2022, 14, e16029. [Google Scholar] [CrossRef]
- Scott, D.A.; Norris-Caneda, K.; Spruill, L.; Bruner, E.; Kono, Y.; Angel, P.M.; Mehta, A.S.; Drake, R.R. Specific N-Linked Glycosylation Patterns in Areas of Necrosis in Tumor Tissues. Int. J. Mass Spectrom. 2019, 437, 69–76. [Google Scholar] [CrossRef]
- Lee, J.B.; Pyo, K.-H.; Kim, H.R. Role and Function of O-GlcNAcylation in Cancer. Cancers 2021, 13, 5365. [Google Scholar] [CrossRef]
- Bern, M.; Kil, Y.J.; Becker, C. Byonic: Advanced Peptide and Protein Identification Software. Curr. Protoc. Bioinform. 2012. [Google Scholar] [CrossRef] [Green Version]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and Recalibrations. Nucleic. Acids. Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, D.; Carberry, S.; Murphy, Á.C.; Lindner, A.U.; Fay, J.; Hector, S.; McCawley, N.; Bacon, O.; Concannon, C.G.; Kay, E.W.; et al. Calnexin, an ER-Induced Protein, Is a Prognostic Marker and Potential Therapeutic Target in Colorectal Cancer. J. Transl. Med. 2016, 14, 196. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.J.; Mook-Jung, I. O-GlcNAcylation Regulates Endoplasmic Reticulum Exit Sites through Sec31A Modification in Conventional Secretory Pathway. FASEB J. 2018, 32, 4641–4657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.J.; Mook-Jung, I. Amyloid Beta Regulates ER Exit Sites Formation through O-GlcNAcylation Triggered by Disrupted Calcium Homeostasis. Biol. Cell 2020, 112, 439–451. [Google Scholar] [CrossRef]
- Wang, Z.; Park, K.; Comer, F.; Hsieh-Wilson, L.C.; Saudek, C.D.; Hart, G.W. Site-Specific GlcNAcylation of Human Erythrocyte Proteins: Potential Biomarker(s) for Diabetes. Diabetes 2009, 58, 309–317. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Guo, H.; Song, Y.; Zhao, X.; Shi, Y.; Lu, Y.; Hu, S.; Nie, Y.; Fan, D.; Wu, K. Loss of Vinculin and Membrane-Bound β-Catenin Promotes Metastasis and Predicts Poor Prognosis in Colorectal Cancer. Mol. Cancer 2014, 13, 263. [Google Scholar] [CrossRef] [Green Version]
- Goldmann, W.H.; Auernheimer, V.; Thievessen, I.; Fabry, B. Vinculin, Cell Mechanics and Tumour Cell Invasion. Cell Biol. Int. 2013, 37, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Bays, J.L.; DeMali, K.A. Vinculin in Cell-Cell and Cell-Matrix Adhesions. Cell. Mol. Life Sci. 2017, 74, 2999–3009. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Yu, Q.; Li, J.; Hu, B.; Zhao, Q.; Ma, C.; Huang, W.; Zhuo, L.; Fang, H.; Liao, L.; et al. O-GlcNAcylation of Fumarase Maintains Tumour Growth under Glucose Deficiency. Nat. Cell Biol. 2017, 19, 833–843. [Google Scholar] [CrossRef]
- Akella, N.M.; Ciraku, L.; Reginato, M.J. Fueling the Fire: Emerging Role of the Hexosamine Biosynthetic Pathway in Cancer. BMC Biol. 2019, 17, 52. [Google Scholar] [CrossRef] [Green Version]
- Adam, I.; Dewi, D.L.; Mooiweer, J.; Sadik, A.; Mohapatra, S.R.; Berdel, B.; Keil, M.; Sonner, J.K.; Thedieck, K.; Rose, A.J.; et al. Upregulation of Tryptophanyl-TRNA Synthethase Adapts Human Cancer Cells to Nutritional Stress Caused by Tryptophan Degradation. Oncoimmunology 2018, 7, e1486353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, Y.H.; Oh, S.-C.; Zhou, S.; Kim, T.-D. Tryptophanyl-TRNA Synthetase as a Potential Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 4523. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Caprioli, R.M. Matrix Sublimation/Recrystallization for Imaging Proteins by Mass Spectrometry at High Spatial Resolution. Anal. Chem. 2011, 83, 5728–5734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.-H.; Weng, C.-L.; Lin, K.-I. O-GlcNAcylation and Its Role in the Immune System. J. Biomed. Sci. 2020, 27, 57. [Google Scholar] [CrossRef]
- Escobar, E.E.; King, D.T.; Serrano-Negrón, J.E.; Alteen, M.G.; Vocadlo, D.J.; Brodbelt, J.S. Precision Mapping of O-Linked N-Acetylglucosamine Sites in Proteins Using Ultraviolet Photodissociation Mass Spectrometry. J. Am. Chem. Soc. 2020, 142, 11569–11577. [Google Scholar] [CrossRef] [PubMed]
- Fonville, J.M.; Carter, C.; Cloarec, O.; Nicholson, J.K.; Lindon, J.C.; Bunch, J.; Holmes, E. Robust Data Processing and Normalization Strategy for MALDI Mass Spectrometric Imaging. Anal. Chem. 2012, 84, 1310–1319. [Google Scholar] [CrossRef] [Green Version]
- Taverna, D.; Norris, J.L.; Caprioli, R.M. Histology-Directed Microwave Assisted Enzymatic Protein Digestion for MALDI MS Analysis of Mammalian Tissue. Anal. Chem. 2015, 87, 670–676. [Google Scholar] [CrossRef]
- Fata, C.R.; Seeley, E.H.; Desouki, M.M.; Du, L.; Gwin, K.; Hanley, K.Z.; Hecht, J.L.; Jarboe, E.A.; Liang, S.X.; Parkash, V.; et al. Are Clear Cell Carcinomas of the Ovary and Endometrium Phenotypically Identical? A Proteomic Analysis. Hum. Pathol. 2015, 46, 1427–1436. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Escobar, E.E.; Seeley, E.H.; Serrano-Negrón, J.E.; Vocadlo, D.J.; Brodbelt, J.S. In Situ Imaging of O-Linked β-N-Acetylglucosamine Using On-Tissue Hydrolysis and MALDI Mass Spectrometry. Cancers 2023, 15, 1224. https://doi.org/10.3390/cancers15041224
Escobar EE, Seeley EH, Serrano-Negrón JE, Vocadlo DJ, Brodbelt JS. In Situ Imaging of O-Linked β-N-Acetylglucosamine Using On-Tissue Hydrolysis and MALDI Mass Spectrometry. Cancers. 2023; 15(4):1224. https://doi.org/10.3390/cancers15041224
Chicago/Turabian StyleEscobar, Edwin E., Erin H. Seeley, Jesús E. Serrano-Negrón, David J. Vocadlo, and Jennifer S. Brodbelt. 2023. "In Situ Imaging of O-Linked β-N-Acetylglucosamine Using On-Tissue Hydrolysis and MALDI Mass Spectrometry" Cancers 15, no. 4: 1224. https://doi.org/10.3390/cancers15041224