
Combined Inhibition of IAPs and WEE1 Enhances TNFα- and Radiation-Induced Cell Death in Head and Neck Squamous Carcinoma
 and
and 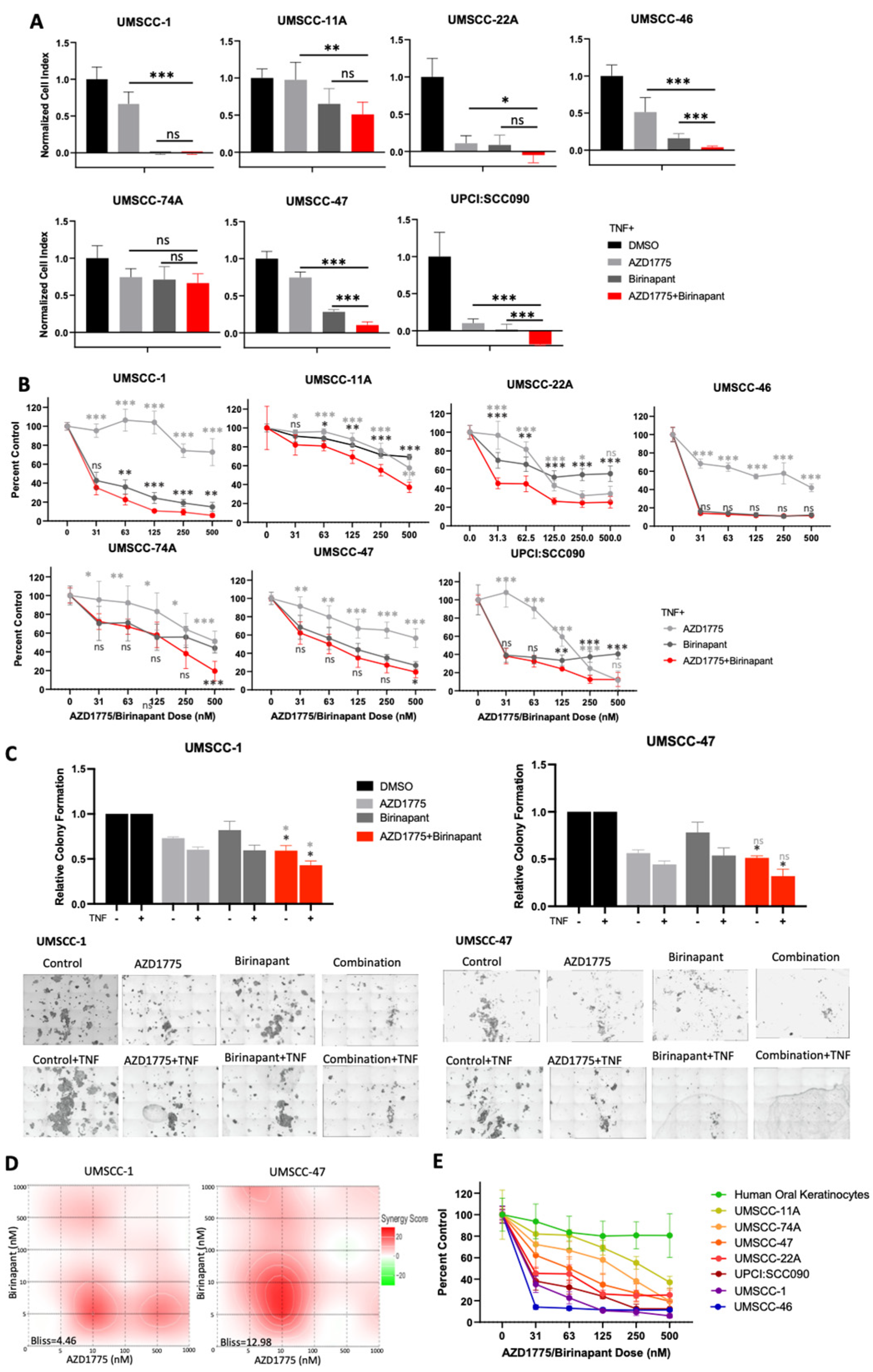{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Therapeutic Reagents
2.2. HNSCC Cell Lines
2.3. siRNA Depletion
2.4. Western Blot
2.5. Real-Time Impedance Assay
2.6. Colony Formation Assays
2.7. Cell Cycle Analysis
2.8. Flow Cytometry Analysis
2.9. Annexin V Assay
2.10. XTT Viability and Cell Death Assay
2.11. In Vitro Radiation
2.12. NFκB Reporter Analysis
2.13. qRT-PCR Analysis
2.14. ELISA
2.15. Statistical Analysis
3. Results
3.1. Combined Inhibition of IAPs and WEE1 Enhances the Inhibitory Effects of TNFα on Proliferation and Survival of HNSCC In Vitro
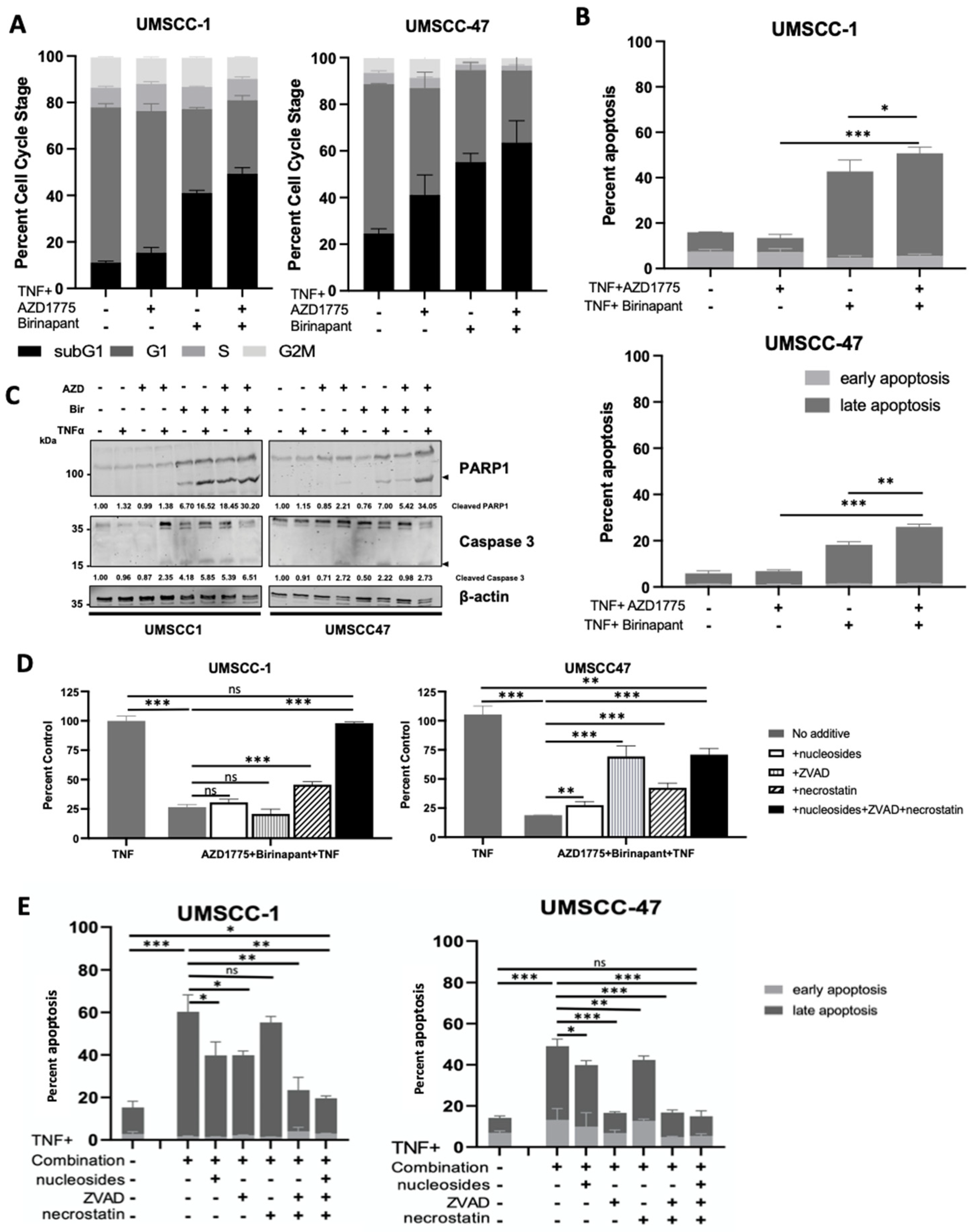
3.2. Combination Treatment Induces Cell Death through Multiple Cell Death Pathways
3.3. Depletion of WEE1 in Combination with IAP Inhibiton Also Enhances the Inhibitory Effects of TNFα on Proliferation and Survival of HNSCC In Vitro
3.4. Combination Treatment Modulates the IKK-NFκB in HNSCC Cells
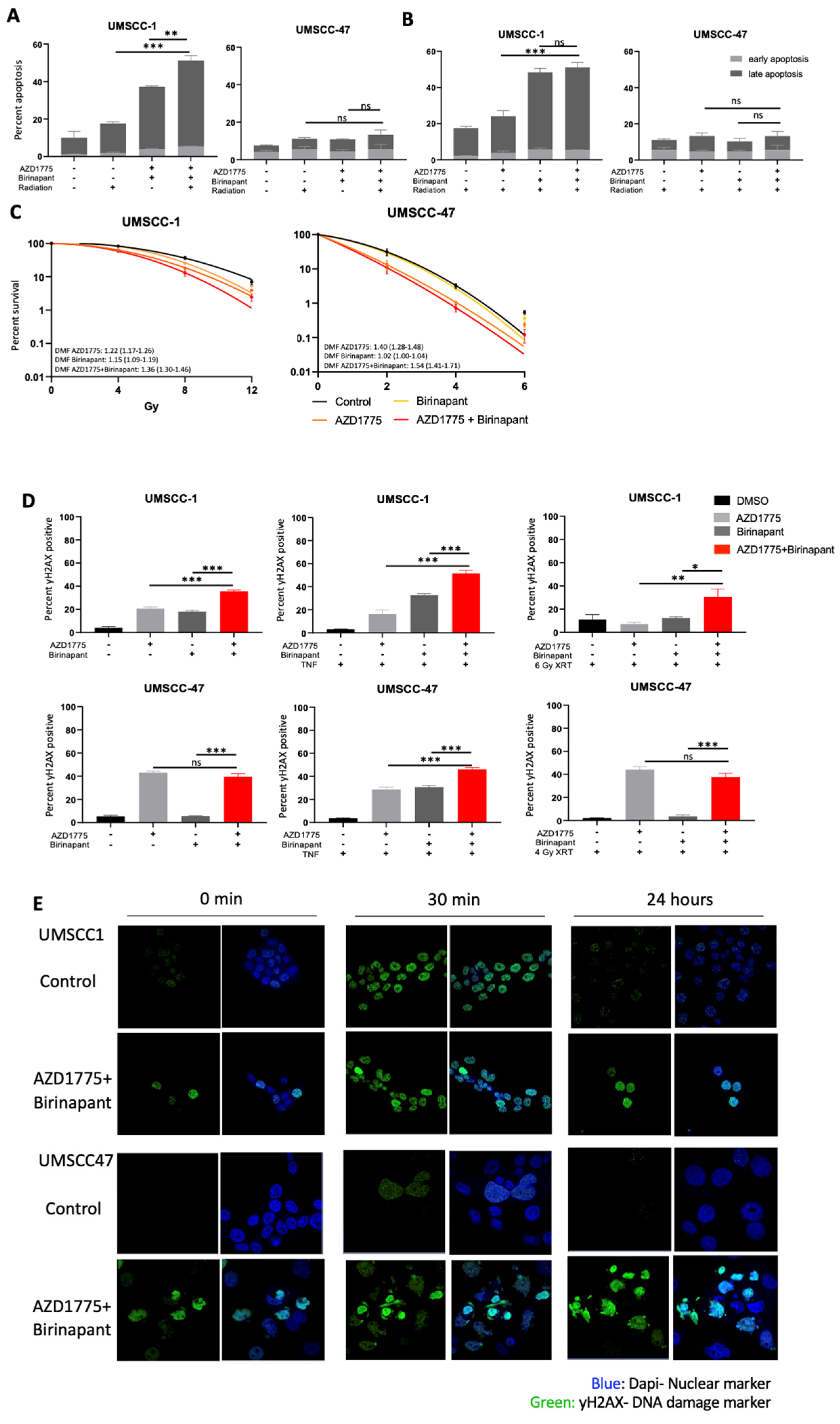
3.5. Combination Treatment Sensitizes HNSCC to Radiation Treatment and Induces DNA Damage
3.6. Mutated TP53 Is a Potential Determinant of Sensitivity to Combination Therapy in HNSCC Cells
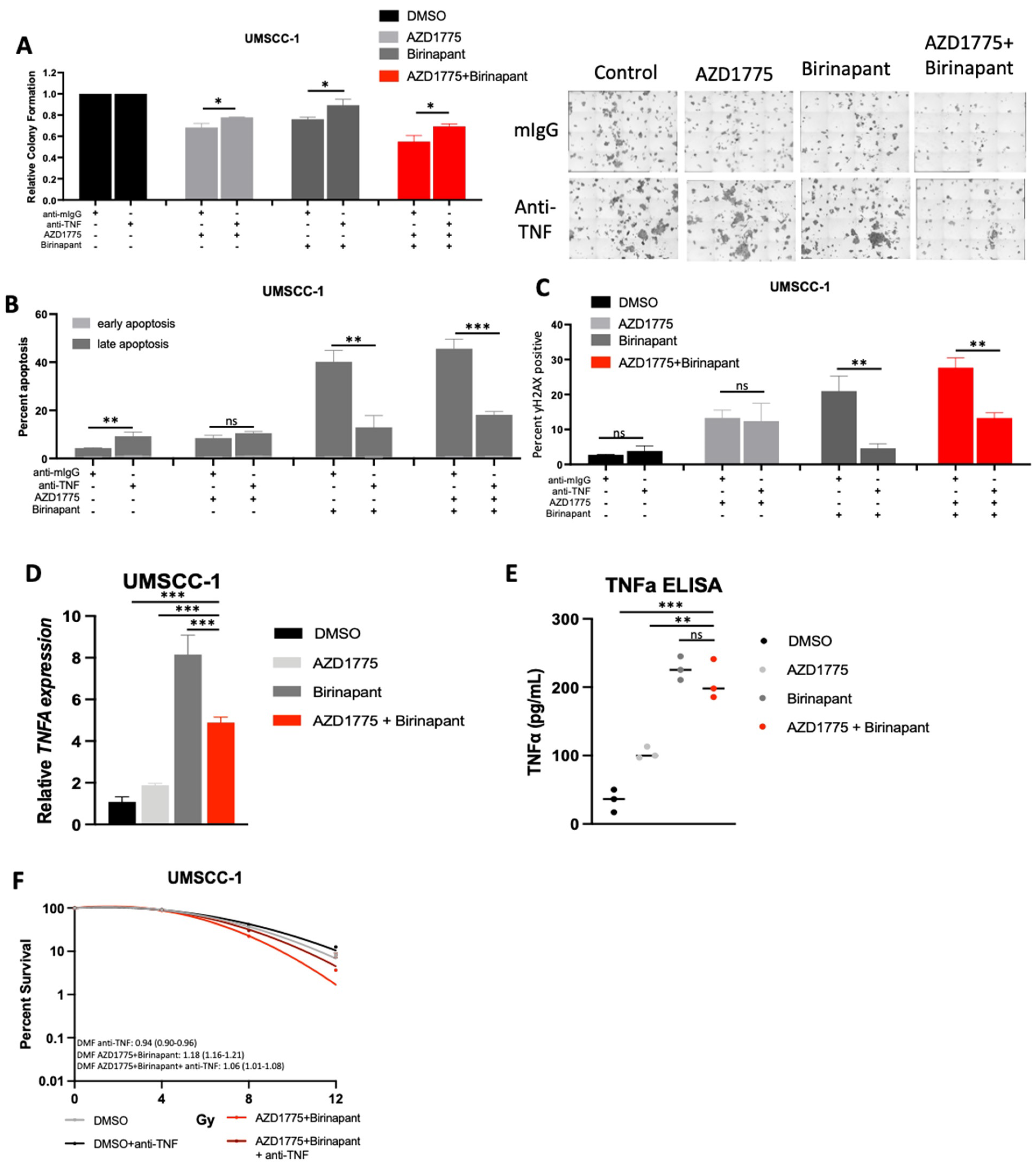
3.7. Intrinsic TNFα Production Plays a Critical Role in the Response to IAP/WEE1 Inhibition in the Highly Sensitive Cell Line UMSCC-1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sacco, A.G.; Cohen, E.E. Current Treatment Options for Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma. J. Clin. Oncol. 2015, 33, 3305–3313. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Wang, M.; Liu, Y.; Wang, X.; Li, Y.; Hu, X.; Qiu, Y.; Liang, W.; Wei, Y.; Zhong, Y. HPV Positive Status Is a Favorable Prognostic Factor in Non-Nasopharyngeal Head and Neck Squamous Cell Carcinoma Patients: A Retrospective Study From the Surveillance, Epidemiology, and End Results Database. Front. Oncol. 2021, 11, 688615. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Torabi, S.; Yarbrough, W.G.; Mehra, S.; Osborn, H.A.; Judson, B. Association of Human Papillomavirus Status at Head and Neck Carcinoma Subsites With Overall Survival. JAMA Otolaryngol.–Head Neck Surg. 2018, 144, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Chitsike, L.; Duerksen-Hughes, P.J. Targeted Therapy as a Potential De-Escalation Strategy in Locally Advanced HPV-Associated Oropharyngeal Cancer: A Literature Review. Front. Oncol. 2021, 11, 730412. [Google Scholar] [CrossRef] [PubMed]
- Stuelten, C.H.; Byfield, S.D.; Arany, P.R.; Karpova, T.S. Breast cancer cells induce stromal fibroblasts to express MMP-9 via secretion of TNF-alpha and TGF-beta. J. Cell Sci. 2005, 118 Pt 10, 2143–2153. [Google Scholar] [CrossRef] [PubMed]
- Laha, D.; Grant, R.; Mishra, P.; Nilubol, N. The Role of Tumor Necrosis Factor in Manipulating the Immunological Response of Tumor Microenvironment. Front. Immunol. 2021, 12, 656908. [Google Scholar] [CrossRef]
- Wang, X.; Lin, Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharm. Sin. 2008, 29, 1275–1288. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Mayo, M.W.; Baldwin, A.S. TNF- and Cancer Therapy-Induced Apoptosis: Potentiation by Inhibition of NF-kB. Science 1996, 274, 784–787. [Google Scholar] [CrossRef]
- Verzella, D.; Pescatore, A.; Capece, D.; Vecchiotti, D.; Ursini, M.V.; Franzoso, G.; Alesse, E.; Zazzeroni, F. Life, death, and autophagy in cancer: NF-κB turns up everywhere. Cell Death Dis. 2020, 11, 210. [Google Scholar] [CrossRef]
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar]
- Mayo, M.W.; Wang, C.Y.; Cogswell, P.C.; Rogers-Graham, K.S.; Lowe, S.W.; Der, C.J.; Baldwin, A.S., Jr. Requirement of NF-kB Activation to Suppress p53-Independent Apoptosis Induced by Oncogenic Ras. Science 1997, 278, 1812–1815. [Google Scholar] [CrossRef] [PubMed]
- Tchoghandjian, A.; Jennewein, C.; Eckhardt, I.; Rajalingam, K.; Fulda, S. Identification of non-canonical NF-κB signaling as a critical mediator of Smac mimetic-stimulated migration and invasion of glioblastoma cells. Cell Death Dis. 2013, 4, e564. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Molecular pathways: Targeting inhibitor of apoptosis proteins in cancer--from molecular mechanism to therapeutic application. Clin. Cancer Res. 2014, 20, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Brands, R.C.; Scheurer, M.J.; Hartmann, S.; Seher, A.; Kübler, A.; Müller-Richter, U. Apoptosis-sensitizing activity of birinapant in head and neck squamous cell carcinoma cell lines. Oncol. Lett. 2018, 15, 4010–4016. [Google Scholar] [CrossRef] [PubMed]
- Eytan, D.F.; Snow, G.E.; Carlson, S.; Derakhshan, A.; Saleh, A.; Schiltz, S.; Cheng, H.; Mohan, S.; Cornelius, S.; Coupar, J.; et al. SMAC Mimetic Birinapant plus Radiation Eradicates Human Head and Neck Cancers with Genomic Amplifications of Cell Death Genes FADD and BIRC2. Cancer Res. 2016, 76, 5442–5454. [Google Scholar] [CrossRef]
- Xiao, R.; An, Y.; Ye, W.; Derakhshan, A.; Cheng, H.; Yang, X.; Allen, C.; Chen, Z.; Schmitt, N.C.; Van Waes, C. Dual Antagonist of cIAP/XIAP ASTX660 Sensitizes HPV(−) and HPV(+) Head and Neck Cancers to TNFα, TRAIL, and Radiation Therapy. Clin. Cancer Res. 2019, 25, 6463–6474. [Google Scholar] [CrossRef]
- Xiao, R.; Allen, C.T.; Tran, L.; Patel, P.; Park, S.J.; Chen, Z.; Van Waes, C.; Schmitt, N.C. Antagonist of cIAP1/2 and XIAP enhances anti-tumor immunity when combined with radiation and PD-1 blockade in a syngeneic model of head and neck cancer. OncoImmunology 2018, 7, e1471440. [Google Scholar] [CrossRef]
- Ye, W.; Gunti, S.; Allen, C.T.; Hong, Y.; Clavijo, P.E.; Van Waes, C.; Schmitt, N.C. ASTX660, an antagonist of cIAP1/2 and XIAP, increases antigen processing machinery and can enhance radiation-induced immunogenic cell death in preclinical models of head and neck cancer. Oncoimmunology 2020, 9, 1710398. [Google Scholar] [CrossRef]
- Sun, X.-S.; Tao, Y.; Le Tourneau, C.; Pointreau, Y.; Sire, C.; Kaminsky, M.-C.; Coutte, A.; Alfonsi, M.; Boisselier, P.; Martin, L.; et al. Debio 1143 and high-dose cisplatin chemoradiotherapy in high-risk locoregionally advanced squamous cell carcinoma of the head and neck: A double-blind, multicentre, randomised, phase 2 study. Lancet Oncol. 2020, 21, 1173–1187. [Google Scholar] [CrossRef]
- Schoenfeld, J.; Cohen, E.; Nutting, C.; Licitra, L.; Burtness, B.; Omar, M.; Bouisset, F.; Nauwelaerts, H.; Urfer, Y.; Zanna, C.; et al. Trilynx: A Phase 3 Trial of Xevinapant and Concurrent Chemoradiotherapy (CRT) for Locally Advanced Head and Neck Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2022, 112, e20–e21. [Google Scholar] [CrossRef]
- Bourhis, J.; Burtness, B.; Licitra, L.F.; Nutting, C.; Schoenfeld, J.D.; Sarkouh, R.A.; Bouisset, F.; Nauwelaerts, H.; Urfer, Y.; Zanna, C.; et al. TrilynX: A phase 3 trial of xevinapant and concurrent chemoradiation for locally advanced head and neck cancer. J. Clin. Oncol. 2021, 39 (Suppl. 15), TPS6091. [Google Scholar] [CrossRef]
- Bourhis, J.; Burtness, B.; Licitra, L.F.; Nutting, C.; Schoenfeld, J.D.; Omar, M.; Bouisset, F.; Nauwelaerts, H.; Urfer, Y.; Zanna, C.; et al. Xevinapant or placebo plus chemoradiotherapy in locally advanced squamous cell carcinoma of the head and neck: TrilynX phase III study design. Future Oncol. 2022, 18, 1669–1678. [Google Scholar] [CrossRef] [PubMed]
- Debiopharm, FDA Grants Breakthrough Therapy Designation for Debiopharm’s Novel Chemo-Radio Sensitizer Debio 1143 for Front-Line Treatment of Head & Neck Cancer February 27, 2020: Lausanne, Switzerland. Available online: https://www.debiopharm.com/drug-development/press-releases/fda-grants-breakthrough-therapy-designation-for-debiopharms-novel-chemo-radio-sensitizer-debio-1143-for-front-line-treatment-of-head-neck-cancer/ (accessed on 23 June 2022).
- Bukhari, A.B.; Chan, G.K.; Gamper, A.M. Targeting the DNA Damage Response for Cancer Therapy by Inhibiting the Kinase Wee1. Front. Oncol. 2022, 12, 828684. [Google Scholar] [CrossRef] [PubMed]
- Ghelli Luserna di Rorà, A.; Cerchione, C.; Martinelli, G.; Simonetti, G. A WEE1 family business: Regulation of mitosis, cancer progression, and therapeutic target. J. Hematol. Oncol. 2020, 13, 126. [Google Scholar] [CrossRef] [PubMed]
- Bi, S.; Wei, Q.; Zhao, Z.; Chen, L.; Wang, C.; Xie, S. Wee1 Inhibitor AZD1775 Effectively Inhibits the Malignant Phenotypes of Esophageal Squamous Cell Carcinoma In Vitro and In Vivo. Front. Pharmacol. 2019, 10, 864. [Google Scholar] [CrossRef] [PubMed]
- Oza, A.M.; Estevez-Diz, M.D.P.; Grischke, E.-M.; Hall, M.; Marmé, F.; Provencher, D.M.; Uyar, D.S.; Weberpals, J.I.; Wenham, R.M.; Laing, N.; et al. A Biomarker-enriched, Randomized Phase II Trial of Adavosertib (AZD1775) Plus Paclitaxel and Carboplatin for Women with Platinum-sensitive TP53-mutant Ovarian Cancer. Clin. Cancer Res. 2020, 26, 4767–4776. [Google Scholar] [CrossRef]
- Cole, K.A.; Pal, S.; Kudgus, R.A.; Ijaz, H.; Liu, X.; Minard, C.G.; Pawel, B.R.; Maris, J.M.; Haas-Kogan, D.A.; Voss, S.D.; et al. Phase I Clinical Trial of the Wee1 Inhibitor Adavosertib (AZD1775) with Irinotecan in Children with Relapsed Solid Tumors: A COG Phase I Consortium Report (ADVL1312). Clin. Cancer Res. 2020, 26, 1213–1219. [Google Scholar] [CrossRef]
- Hu, Z.; Viswanathan, R.; Cheng, H.; Chen, J.; Yang, X.; Huynh, A.; Clavijo, P.; An, Y.; Robbins, Y.; Silvin, C.; et al. Inhibiting WEE1 and IKK-RELA Crosstalk Overcomes TNFα Resistance in Head and Neck Cancers. Mol. Cancer Res. 2022, 20, 867–882. [Google Scholar] [CrossRef]
- Cheng, H.; Yang, X.; Si, H.; Saleh, A.D.; Xiao, W.; Coupar, J.; Gollin, S.M.; Ferris, R.L.; Issaeva, N.; Yarbrough, W.G.; et al. Genomic and Transcriptomic Characterization Links Cell Lines with Aggressive Head and Neck Cancers. Cell Rep. 2018, 25, 1332–1345.e5. [Google Scholar] [CrossRef]
- Mita, M.M.; LoRusso, P.M.; Papadopoulos, K.P.; Gordon, M.S.; Mita, A.C.; Ferraldeschi, R.; Keer, H.; Oganesian, A.; Su, X.Y.; Jueliger, S.; et al. A Phase I Study of ASTX660, an Antagonist of Inhibitors of Apoptosis Proteins, in Adults with Advanced Cancers or Lymphoma. Clin. Cancer Res. 2020, 26, 2819–2826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bliss, C.I. The Toxicity of Poisons Applied Jointly. Ann. Appl. Biol. 1939, 26, 585–615. [Google Scholar] [CrossRef]
- Plesca, D.; Mazumder, S.; Almasan, A. DNA damage response and apoptosis. Methods Enzym. 2008, 446, 107–122. [Google Scholar]
- Patel, P.; Sun, L.; Robbins, Y.; Clavijo, P.E.; Friedman, J.; Silvin, C.; Van Waes, C.; Cook, J.; Mitchell, J.; Allen, C. Enhancing direct cytotoxicity and response to immune checkpoint blockade following ionizing radiation with Wee1 kinase inhibition. Oncoimmunology 2019, 8, e1638207. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.; Golubeva, V.; Rix, L.L.R.; Berndt, N.; Luo, Y.; Ward, G.A.; Gray, J.E.; Schonbrunn, E.; Lawrence, H.R.; Monteiro, A.N.; et al. Dual Targeting of WEE1 and PLK1 by AZD1775 Elicits Single Agent Cellular Anticancer Activity. ACS Chem. Biol. 2017, 12, 1883–1892. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.L.; Kamata, H.; Karin, M. IKK/NF-kappaB signaling: Balancing life and death--a new approach to cancer therapy. J. Clin. Invest. 2005, 115, 2625–2632. [Google Scholar] [CrossRef]
- Varfolomeev, E.; Blankenship, J.W.; Wayson, S.M.; Fedorova, A.V.; Kayagaki, N.; Garg, P.; Zobel, K.; Dynek, J.N.; Elliott, L.O.; Wallweber, H.J.A.; et al. IAP Antagonists Induce Autoubiquitination of c-IAPs, NF-κB Activation, and TNFα-Dependent Apoptosis. Cell 2007, 131, 669–681. [Google Scholar]
- Vince, J.E.; Wong, W.W.-L.; Khan, N.; Feltham, R.; Chau, D.; Ahmed, A.U.; Benetatos, C.A.; Chunduru, S.K.; Condon, S.M.; McKinlay, M.; et al. IAP Antagonists Target cIAP1 to Induce TNFα-Dependent Apoptosis. Cell 2007, 131, 682–693. [Google Scholar] [CrossRef]
- Grénman, R.; Carey, T.E.; McClatchey, K.D.; Wagner, J.G.; Pekkola-Heino, K.; Ms, D.R.S.; Wolf, G.T.; Lacivita, L.P.; Ho, L.; Baker, S.R.; et al. In vitro radiation resistance among cell lines established from patients with squamous cell carcinoma of the head and neck. Cancer 1991, 67, 2741–2747. [Google Scholar] [CrossRef]
- Kinner, A.; Wu, W.; Staudt, C.; Iliakis, G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008, 36, 5678–5694. [Google Scholar] [CrossRef]
- Spary, L.; Al-Taei, S.; Salimu, J.; Cook, A.D.; Ager, A.; Watson, H.A.; Clayton, A.; Staffurth, J.; Mason, M.D.; Tabi, Z. Enhancement of T Cell Responses as a Result of Synergy between Lower Doses of Radiation and T Cell Stimulation. J. Immunol. 2014, 192, 3101–3110. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Zhao, J.; Hu, K.; Hou, X.; Sun, X.; Pan, X.; Wang, X.; Li, N.; Yang, Z.; Zhang, F.; et al. Single High-Dose Radiation Enhances Dendritic Cell Homing and T Cell Priming by Promoting Reactive Oxygen Species-Induced Cytoskeletal Reorganization. Int. J. Radiat. Oncol. Biol. Phys. 2021, 109, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Ziemann, F.; Arenz, A.; Preising, S.; Wittekindt, C.; Klussmann, J.P.; Engenhart-Cabillic, R.; Wittig, A. Increased sensitivity of HPV-positive head and neck cancer cell lines to x-irradiation ± Cisplatin due to decreased expression of E6 and E7 oncoproteins and enhanced apoptosis. Am. J. Cancer Res. 2015, 5, 1017–1031. [Google Scholar] [PubMed]
- Kimple, R.J.; Smith, M.A.; Blitzer, G.C.; Torres, A.D.; Martin, J.A.; Yang, R.Z.; Peet, C.R.; Lorenz, L.D.; Nickel, K.P.; Klingelhutz, A.J.; et al. Enhanced radiation sensitivity in HPV-positive head and neck cancer. Cancer Res. 2013, 73, 4791–4800. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Pim, D.; Banks, L. The role of the E6-p53 interaction in the molecular pathogenesis of HPV. Oncogene 1999, 18, 7690–7700. [Google Scholar] [CrossRef] [PubMed]
- Diab, A.; Gem, H.; Swanger, J.; Kim, H.Y.; Smith, K.; Zou, G.; Raju, S.; Kao, M.; Fitzgibbon, M.; Loeb, K.R.; et al. FOXM1 drives HPV+ HNSCC sensitivity to WEE1 inhibition. Proc. Natl. Acad. Sci. USA 2020, 117, 28287–28296. [Google Scholar] [CrossRef] [PubMed]
- Eytan, D.F.; Snow, G.E.; Carlson, S.G.; Schiltz, S.; Chen, Z.; Van Waes, C. Combination effects of SMAC mimetic birinapant with TNFα, TRAIL, and docetaxel in preclinical models of HNSCC. Laryngoscope 2015, 125, E118–E124. [Google Scholar] [CrossRef]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Prim. 2020, 6, 92. [Google Scholar] [CrossRef]
- Derakhshan, A.; Chen, Z.; Van Waes, C. Therapeutic Small Molecules Target Inhibitor of Apoptosis Proteins in Cancers with Deregulation of Extrinsic and Intrinsic Cell Death Pathways. Clin. Cancer Res. 2017, 23, 1379–1387. [Google Scholar] [CrossRef]
- Marquard, F.E.; Jücker, M. PI3K/AKT/mTOR signaling as a molecular target in head and neck cancer. Biochem. Pharm. 2020, 172, 113729. [Google Scholar] [CrossRef]
- van Harten, A.M.; de Boer, D.V.; Martens-de Kemp, S.R.; Buijze, M.; Ganzevles, S.H.; Hunter, K.D.; Leemans, C.R.; van Beusechem, V.W.; Wolthuis, R.M.; de Menezes, R.X.; et al. Chemopreventive targeted treatment of head and neck precancer by Wee1 inhibition. Sci. Rep. 2020, 10, 2330. [Google Scholar] [CrossRef] [PubMed]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.L.; Chen, Z.; Van Waes, C. Regulation of NFκB Signalling by Ubiquitination: A Potential Therapeutic Target in Head and Neck Squamous Cell Carcinoma? Cancers 2020, 12, 2877. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, M.-K.N.D.; Mierzwa, M.; D’Silva, N.J. Radiation resistance in head and neck squamous cell carcinoma: Dire need for an appropriate sensitizer. Oncogene 2020, 39, 3638–3649. [Google Scholar] [CrossRef]
- Tanimoto, T.; Tsuda, H.; Imazeki, N.; Ohno, Y.; Imoto, I.; Inazawa, J.; Matsubara, O. Nuclear expression of cIAP-1, an apoptosis inhibiting protein, predicts lymph node metastasis and poor patient prognosis in head and neck squamous cell carcinomas. Cancer Lett. 2005, 224, 141–151. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Regulation of NF-κB by TNF family cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef]
- Roberts, N.J.; Zhou, S.; Diaz, L.A.; Holdhoff, M. Systemic use of tumor necrosis factor alpha as an anticancer agent. Oncotarget 2011, 2, 739–751. [Google Scholar] [CrossRef]
- Citrin, D.E.; Hitchcock, Y.J.; Chung, E.J.; Frandsen, J.; Urick, M.E.; Shield, W.; Gaffney, D. Determination of cytokine protein levels in oral secretions in patients undergoing radiotherapy for head and neck malignancies. Radiat. Oncol. 2012, 7, 64. [Google Scholar] [CrossRef]
- Benetatos, C.A.; Mitsuuchi, Y.; Burns, J.M.; Neiman, E.M.; Condon, S.M.; Yu, G.; Seipel, M.E.; Kapoor, G.S.; LaPorte, M.G.; Rippin, S.R.; et al. Birinapant (TL32711), a bivalent SMAC mimetic, targets TRAF2-associated cIAPs, abrogates TNF-induced NF-κB activation, and is active in patient-derived xenograft models. Mol. Cancer Ther. 2014, 13, 867–879. [Google Scholar] [CrossRef]
- Apu, E.H.; Akram, S.U.; Rissanen, J.; Wan, H.; Salo, T. Desmoglein 3–Influence on oral carcinoma cell migration and invasion. Exp. Cell Res. 2018, 370, 353–364. [Google Scholar]
- Engelmann, L.; Thierauf, J.; Laureano, N.K.; Stark, H.-J.; Prigge, E.-S.; Horn, D.; Freier, K.; Grabe, N.; Rong, C.; Federspil, P.; et al. Organotypic co-cultures as a novel 3D model for head and neck squamous cell carcinoma. Cancers 2020, 12, 2330. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toni, T.; Viswanathan, R.; Robbins, Y.; Gunti, S.; Yang, X.; Huynh, A.; Cheng, H.; Sowers, A.L.; Mitchell, J.B.; Allen, C.T.; et al. Combined Inhibition of IAPs and WEE1 Enhances TNFα- and Radiation-Induced Cell Death in Head and Neck Squamous Carcinoma. Cancers 2023, 15, 1029. https://doi.org/10.3390/cancers15041029
Toni T, Viswanathan R, Robbins Y, Gunti S, Yang X, Huynh A, Cheng H, Sowers AL, Mitchell JB, Allen CT, et al. Combined Inhibition of IAPs and WEE1 Enhances TNFα- and Radiation-Induced Cell Death in Head and Neck Squamous Carcinoma. Cancers. 2023; 15(4):1029. https://doi.org/10.3390/cancers15041029
Chicago/Turabian StyleToni, Tiffany, Ramya Viswanathan, Yvette Robbins, Sreenivasulu Gunti, Xinping Yang, Angel Huynh, Hui Cheng, Anastasia L. Sowers, James B. Mitchell, Clint T. Allen, and et al. 2023. "Combined Inhibition of IAPs and WEE1 Enhances TNFα- and Radiation-Induced Cell Death in Head and Neck Squamous Carcinoma" Cancers 15, no. 4: 1029. https://doi.org/10.3390/cancers15041029