
Repurposing the Bis-Biguanide Alexidine in Combination with Tyrosine Kinase Inhibitors to Eliminate Leukemic Stem/Progenitor Cells in Chronic Myeloid Leukemia
 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagent and Antibodies
2.2. Cell Lines
2.3. Proliferation Assay
2.4. Viability Assay and Synergy Calculation Scores
2.5. Analysis of Apoptosis
2.6. May–Grünwald–Giemsa (MGG) Staining
2.7. Confocal Microscopy
2.8. Isolation of Primary Cells
2.9. Colony-Forming Cell (CFC) Assay
2.10. Cellular Division Tracking
2.11. Long-Term Culture-Initiating Cell (LTC-IC) with Limiting Dilution Assays (LDAs)
2.12. Plasmids and Transduction
2.13. Western Blot
2.14. Oxygen Consumption Rate Measurements
2.15. Statistical Analysis
2.16. In Vivo Experiments:
3. Results
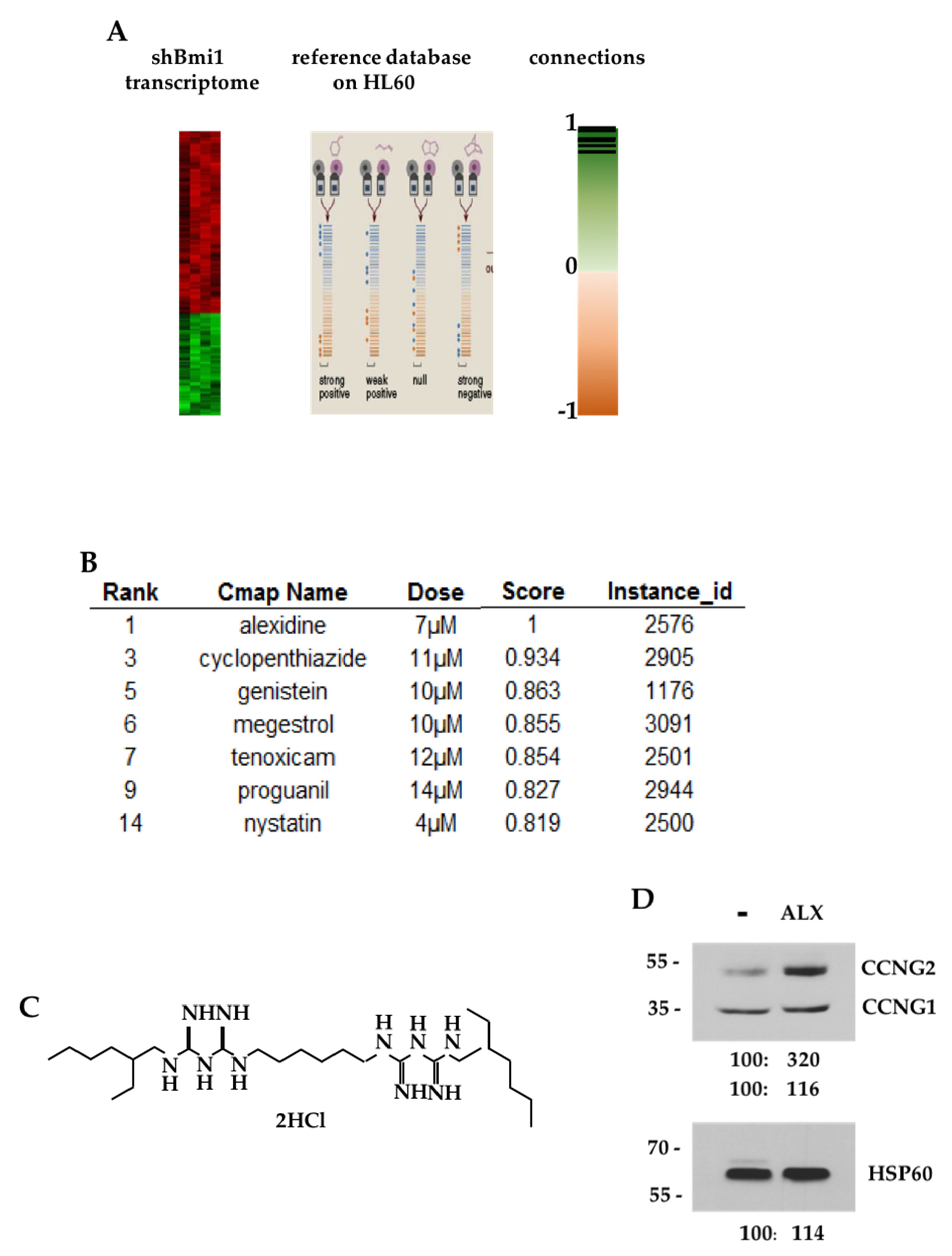
3.1. CMAP Analysis of the shBMI1-K562 Gene Signature
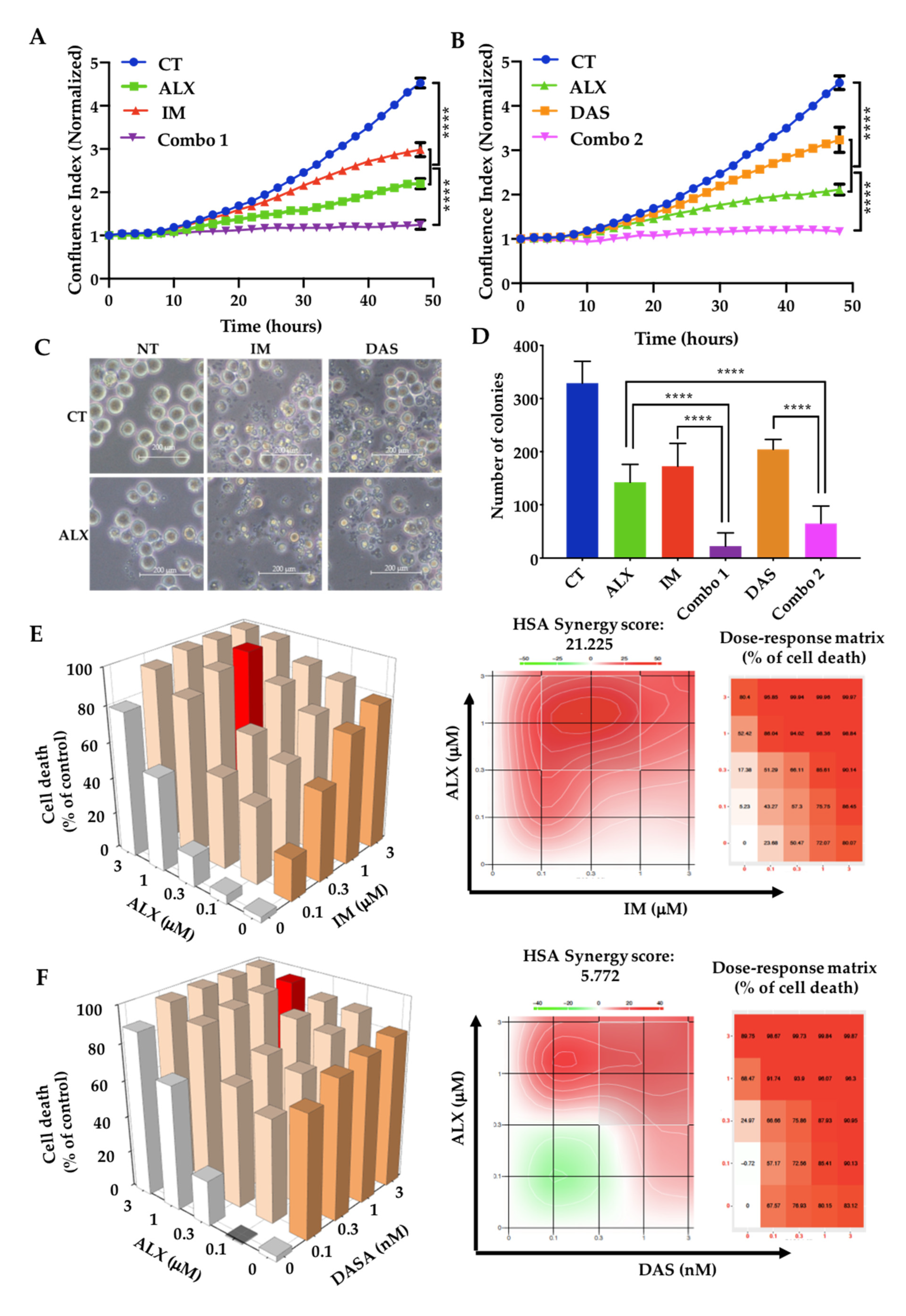
3.2. Alexidine Synergizes with TKIs to Decrease the Proliferation and Clonogenicity of CML Cells
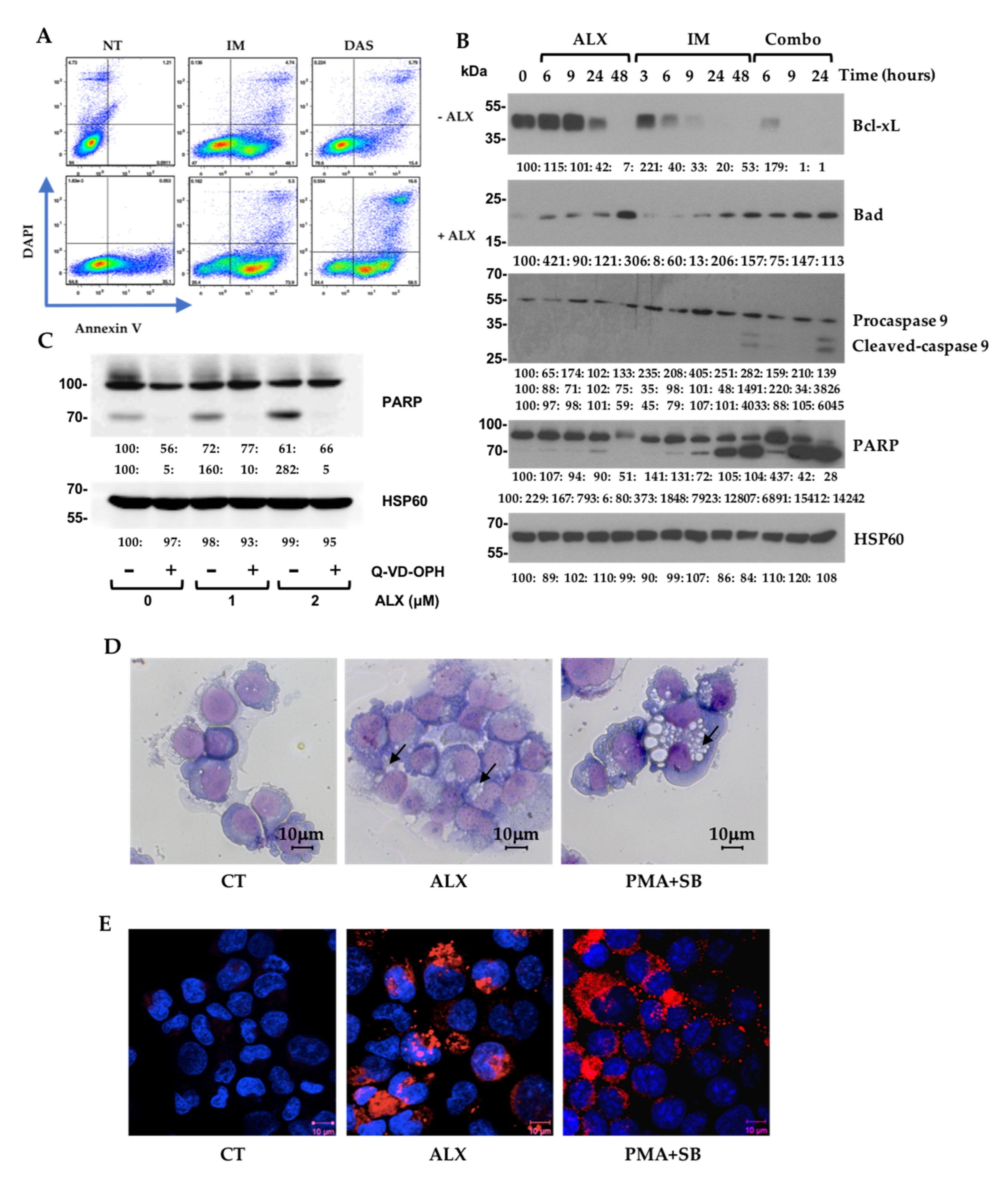
3.3. Alexidine (ALX) Induces Apoptotic and Autophagic Events
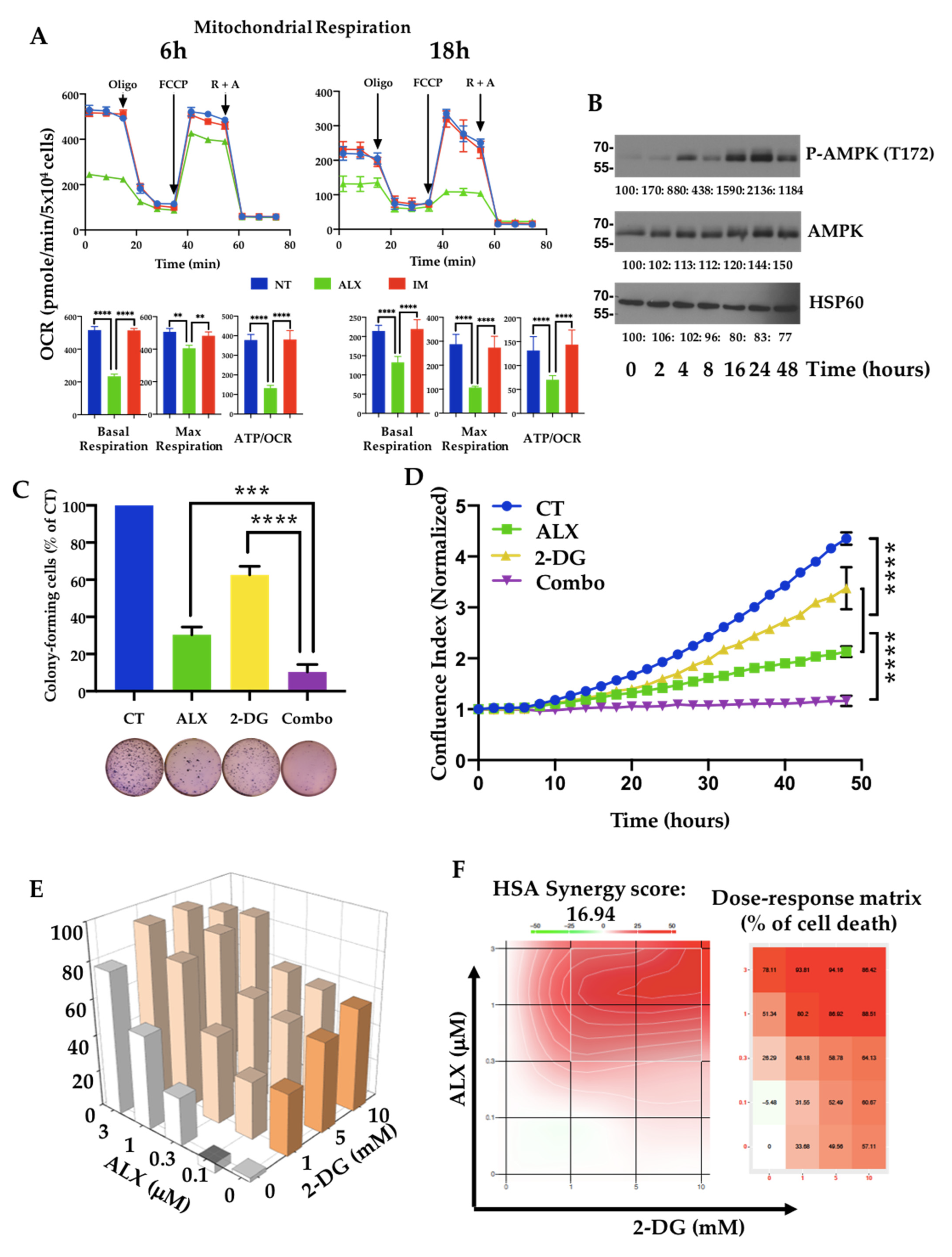
3.4. Inhibition of Mitochondrial Respiration by Alexidine (ALX) Initiates CML Cell Death
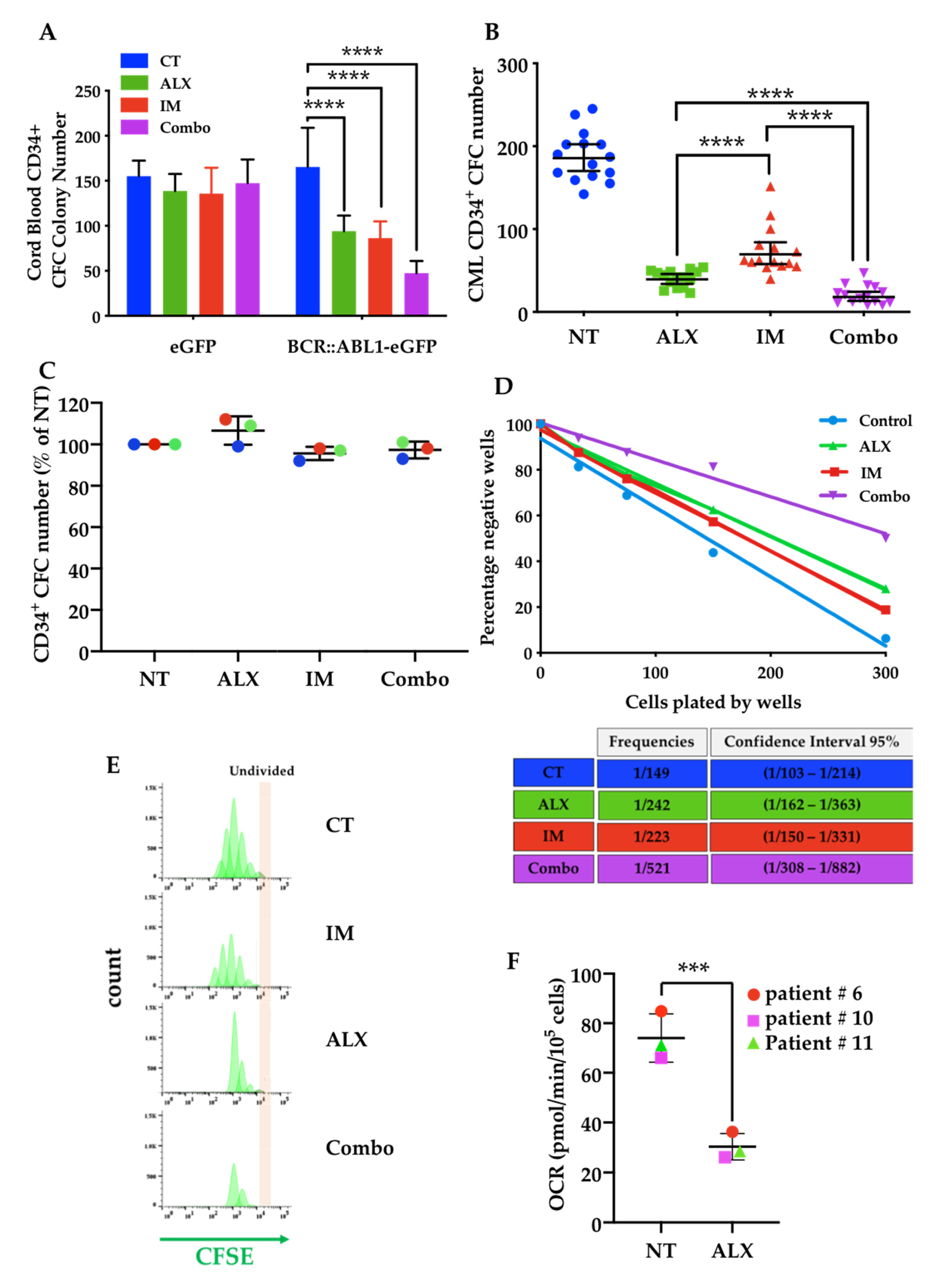
3.5. The Combination of Alexidine (ALX) with a Tyrosine Kinase Inhibitor (TKI) Affects CML Progenitor Cells
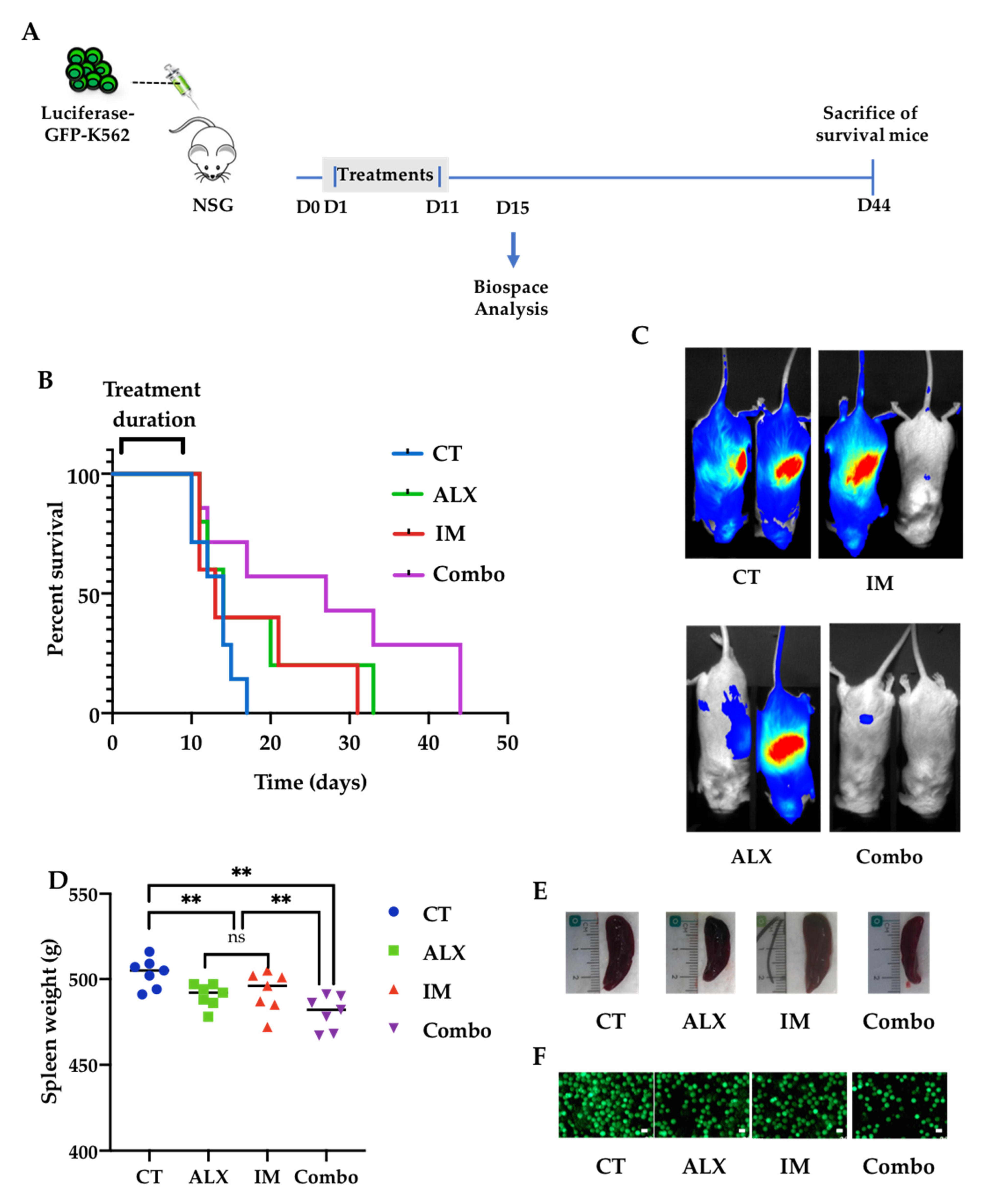
3.6. Alexidine Improves the Survival of Leukemic Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 2-DG | 2-Deoxyglucose |
| ALX | Alexidine |
| AMPK | AMP-activated protein kinase |
| ANV | Annexin V |
| ATP | Adenosine triphosphate |
| Bcl-2 | B-cell lymphoma 2 |
| Bcl-xL | B-cell lymphoma extra-large |
| BCR::ABL1 | Breakpoint cluster region-Abelson murine leukemia viral oncogene homolog 1 |
| BMI1 | B-cell-specific Moloney murine leukemia virus integration site 1 |
| BOSU | Bosutinib |
| CCNG2 | Cyclin G2 |
| CFC | Colony-forming cell |
| CFSE | Carboxyfluorescein succinimidyl ester |
| CSC | Cancer stem cell |
| CMAP | Connectivity Map |
| DAPI | Di Aminido Phenyl Indol |
| DAS | Dasatinib |
| ETC | Electron transport chain |
| FDA | Food and Drug Administration |
| HSC | Hematopoietic stem cell |
| IM | Imatinib |
| LIC | Leukemic-initiating cell |
| LSC | Leukemic stem cell |
| LTC-IC | Long-term culture of Initiating Cell |
| Mcl-1 | Induced myeloid leukemia cell differentiation protein |
| MGG | May–Grünwald–Giemsa |
| NILO | Nilotinib |
| NSG | Nod Scid Gamma |
| OCR | Oxygen consumption rate |
| PONA | Ponatinib |
| PRC1 | Polycomb repressive complex 1 |
| PTPMT1 | Protein tyrosine phosphatase mitochondrial 1 |
| RFP | Red fluorescent protein |
| ROS | Reactive oxygen species |
| SDH | Succinyl dehydrogenase |
| SRC | Sarc |
| TKI | Tyrosine kinase inhibitor |
| TMRE | Tetramethylrhodamine ethyl ester perchlorate |
References
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Hochhaus, A.; Saglio, G.; De Souza, C.; Flinn, I.W.; Stenke, L.; Goh, Y.T.; Rosti, G.; Nakamae, H.; Gallagher, N.J.; et al. Nilotinib versus imatinib for the treatment of patients with newly diagnosed chronic phase, Philadelphia chromosome-positive, chronic myeloid leukaemia: 24-month minimum follow-up of the phase 3 randomised ENESTnd trial. Lancet Oncol. 2011, 12, 841–851. [Google Scholar] [CrossRef]
- Tipping, A.J.; Baluch, S.; Barnes, D.J.; Veach, D.R.; Clarkson, B.M.; Bornmann, W.G.; Mahon, F.X.; Goldman, J.M.; Melo, J.V. Efficacy of dual-specific Bcr-Abl and Src-family kinase inhibitors in cells sensitive and resistant to imatinib mesylate. Leukemia 2004, 18, 1352–1356. [Google Scholar] [CrossRef]
- Keller, G.; Schafhausen, P.; Brummendorf, T.H. Bosutinib: A dual SRC/ABL kinase inhibitor for the treatment of chronic myeloid leukemia. Expert Rev. Hematol. 2009, 2, 489–497. [Google Scholar] [CrossRef]
- Tan, F.H.; Putoczki, T.L.; Stylli, S.S.; Luwor, R.B. Ponatinib: A novel multi-tyrosine kinase inhibitor against human malignancies. Onco Targets Ther. 2019, 12, 635–645. [Google Scholar] [CrossRef]
- Corbin, A.S.; Agarwal, A.; Loriaux, M.; Cortes, J.; Deininger, M.W.; Druker, B.J. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J. Clin. Investig. 2011, 121, 396–409. [Google Scholar] [CrossRef]
- Hamilton, A.; Helgason, G.V.; Schemionek, M.; Zhang, B.; Myssina, S.; Allan, E.K.; Nicolini, F.E.; Muller-Tidow, C.; Bhatia, R.; Brunton, V.G.; et al. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood 2012, 119, 1501–1510. [Google Scholar] [CrossRef]
- Graham, S.M.; Jørgensen, H.G.; Allan, E.; Pearson, C.; Alcorn, M.J.; Richmond, L.; Holyoake, T.L. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 2002, 99, 319–325. [Google Scholar] [CrossRef]
- Copland, M.; Hamilton, A.; Elrick, L.J.; Baird, J.W.; Allan, E.K.; Jordanides, N.; Barow, M.; Mountford, J.C.; Holyoake, T.L. Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood 2006, 107, 4532–4539. [Google Scholar] [CrossRef]
- Jørgensen, H.G.; Allan, E.K.; Jordanides, N.E.; Mountford, J.C.; Holyoake, T.L. Nilotinib exerts equipotent antiproliferative effects to imatinib and does not induce apoptosis in CD34+ CML cells. Blood 2007, 109, 4016–4019. [Google Scholar] [CrossRef] [Green Version]
- Konig, H.; Holyoake, T.L.; Bhatia, R. Effective and selective inhibition of chronic myeloid leukemia primitive hematopoietic progenitors by the dual Src/Abl kinase inhibitor SKI-606. Blood 2008, 111, 2329–2338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, M.; McDonald, T.; Holyoake, T.L.; Moon, R.T.; Campana, D.; Shultz, L.; Bhatia, R. Microenvironmental protection of CML stem and progenitor cells from tyrosine kinase inhibitors through N-cadherin and Wnt-β-catenin signaling. Blood 2013, 121, 1824–1838. [Google Scholar] [CrossRef]
- Dierks, C.; Beigi, R.; Guo, G.R.; Zirlik, K.; Stegert, M.R.; Manley, P.; Trussell, C.; Schmitt-Graeff, A.; Landwerlin, K.; Veelken, H.; et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell 2008, 14, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Muselli, F.; Mourgues, L.; Morcos, R.; Rochet, N.; Nebout, M.; Guerci-Bresler, A.; Faller, D.V.; William, R.M.; Mhaidly, R.; Verhoeyen, E.; et al. Combination of PKCδ Inhibition with Conventional TKI Treatment to Target CML Models. Cancers 2021, 13, 1693. [Google Scholar] [CrossRef]
- Cheloni, G.; Tanturli, M.; Tusa, I.; Ho DeSouza, N.; Shan, Y.; Gozzini, A.; Mazurier, F.; Rovida, E.; Li, S.; Dello Sbarba, P. Targeting chronic myeloid leukemia stem cells with the hypoxia-inducible factor inhibitor acriflavine. Blood 2017, 130, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Kawada, H.; Kaneko, M.; Sawanobori, M.; Uno, T.; Matsuzawa, H.; Nakamura, Y.; Matsushita, H.; Ando, K. High concentrations of L-ascorbic acid specifically inhibit the growth of human leukemic cells via downregulation of HIF-1α transcription. PLoS ONE 2013, 8, e62717. [Google Scholar] [CrossRef]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef]
- Hoelbl, A.; Schuster, C.; Kovacic, B.; Zhu, B.; Wickre, M.; Hoelzl, M.A.; Fajmann, S.; Grebien, F.; Warsch, W.; Stengl, G.; et al. Stat5 is indispensable for the maintenance of bcr/abl-positive leukaemia. EMBO Mol. Med. 2010, 2, 98–110. [Google Scholar] [CrossRef]
- Prost, S.; Relouzat, F.; Spentchian, M.; Ouzegdouh, Y.; Saliba, J.; Massonnet, G.; Beressi, J.P.; Verhoeyen, E.; Raggueneau, V.; Maneglier, B.; et al. Erosion of the chronic myeloid leukaemia stem cell pool by PPARγ agonists. Nature 2015, 525, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Mourgues, L.; Imbert, V.; Nebout, M.; Colosetti, P.; Neffati, Z.; Lagadec, P.; Verhoeyen, E.; Peng, C.; Duprez, E.; Legros, L.; et al. The BMI1 polycomb protein represses cyclin G2-induced autophagy to support proliferation in chronic myeloid leukemia cells. Leukemia 2015, 29, 1993–2002. [Google Scholar] [CrossRef]
- Pollak, M. Metformin and other biguanides in oncology: Advancing the research agenda. Cancer Prev. Res. 2010, 3, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Yip, K.W.; Ito, E.; Mao, X.; Au, P.Y.; Hedley, D.W.; Mocanu, J.D.; Bastianutto, C.; Schimmer, A.; Liu, F.F. Potential use of alexidine dihydrochloride as an apoptosis-promoting anticancer agent. Mol. Cancer Ther. 2006, 5, 2234–2240. [Google Scholar] [CrossRef] [PubMed]
- Graber, M.A. A New Introduction to American Constitutionalism; Oxford University Press: Oxford, UK, 2013; Volume xii, 292p. [Google Scholar]
- Kasikci, E.; Aydemir, E.; Yogurtcu, B.M.; Sahin, F.; Bayrak, O.F. Repurposing of Alexidine Dihydrochloride as an Apoptosis Initiator and Cell Cycle Inhibitor in Human Pancreatic Cancer. Anticancer Agents Med. Chem. 2020, 20, 1956–1965. [Google Scholar] [CrossRef] [PubMed]
- Dell’Anno, I.; Melani, A.; Martin, S.A.; Barbarino, M.; Silvestri, R.; Cipollini, M.; Giordano, A.; Mutti, L.; Nicolini, A.; Luzzi, L.; et al. A Drug Screening Revealed Novel Potential Agents against Malignant Pleural Mesothelioma. Cancers 2022, 14, 2527. [Google Scholar] [CrossRef]
- Colosetti, P.; Puissant, A.; Robert, G.; Luciano, F.; Jacquel, A.; Gounon, P.; Cassuto, J.P.; Auberger, P. Autophagy is an important event for megakaryocytic differentiation of the chronic myelogenous leukemia K562 cell line. Autophagy 2009, 5, 1092–1098. [Google Scholar] [CrossRef]
- Lévy, C.; Amirache, F.; Girard-Gagnepain, A.; Frecha, C.; Roman-Rodríguez, F.J.; Bernadin, O.; Costa, C.; Nègre, D.; Gutierrez-Guerrero, A.; Vranckx, L.S.; et al. Measles virus envelope pseudotyped lentiviral vectors transduce quiescent human HSCs at an efficiency without precedent. Blood Adv. 2017, 1, 2088–2104. [Google Scholar] [CrossRef]
- Rosilio, C.; Ben-Sahra, I.; Bost, F.; Peyron, J.F. Metformin: A metabolic disruptor and anti-diabetic drug to target human leukemia. Cancer Lett. 2014, 346, 188–196. [Google Scholar] [CrossRef]
- Vakana, E.; Altman, J.K.; Glaser, H.; Donato, N.J.; Platanias, L.C. Antileukemic effects of AMPK activators on BCR-ABL-expressing cells. Blood 2011, 118, 6399–6402. [Google Scholar] [CrossRef]
- Chen, Z.X.; Zou, X.P.; Yan, H.Q.; Zhang, R.; Pang, J.S.; Qin, X.G.; He, R.Q.; Ma, J.; Feng, Z.B.; Chen, G.; et al. Identification of putative drugs for gastric adenocarcinoma utilizing differentially expressed genes and connectivity map. Mol. Med. Rep. 2019, 19, 1004–1015. [Google Scholar] [CrossRef]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.C.; Fang, Y.F.; Yamaguchi, H.; Wang, W.J.; Chen, T.C.; Hong, X.; Ke, B.; Xia, W.; Wei, Y.; Zha, Z.; et al. Targeting PKCdelta as a Therapeutic Strategy against Heterogeneous Mechanisms of EGFR Inhibitor Resistance in EGFR-Mutant Lung Cancer. Cancer Cell 2018, 34, 954–969.E4. [Google Scholar] [CrossRef] [PubMed]
- Gräber, M.; Hell, M.; Gröst, C.; Friberg, A.; Sperl, B.; Sattler, M.; Berg, T. Oral disinfectants inhibit protein-protein interactions mediated by the anti-apoptotic protein Bcl-xL and induce apoptosis in human oral tumor cells. Angew. Chem. Int. Ed. Engl. 2013, 52, 4487–4491. [Google Scholar] [CrossRef]
- Harb, J.G.; Neviani, P.; Chyla, B.J.; Ellis, J.J.; Ferenchak, G.J.; Oaks, J.J.; Walker, C.J.; Hokland, P.; Roy, D.C.; Caligiuri, M.A.; et al. Bcl-xL anti-apoptotic network is dispensable for development and maintenance of CML but is required for disease progression where it represents a new therapeutic target. Leukemia 2013, 27, 1996–2005. [Google Scholar] [CrossRef]
- Chen, S.; Dai, Y.; Harada, H.; Dent, P.; Grant, S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res. 2007, 67, 782–791. [Google Scholar] [CrossRef] [PubMed]
- González-Herrero, I.; Vicente-Dueñas, C.; Orfao, A.; Flores, T.; Jiménez, R.; Cobaleda, C.; Sánchez-García, I. Bcl2 is not required for the development and maintenance of leukemia stem cells in mice. Carcinogenesis 2010, 31, 1292–1297. [Google Scholar] [CrossRef] [PubMed]
- Doughty-Shenton, D.; Joseph, J.D.; Zhang, J.; Pagliarini, D.J.; Kim, Y.; Lu, D.; Dixon, J.E.; Casey, P.J. Pharmacological targeting of the mitochondrial phosphatase PTPMT1. J. Pharmacol. Exp. Ther. 2010, 333, 584–592. [Google Scholar] [CrossRef]
- Yu, W.M.; Liu, X.; Shen, J.; Jovanovic, O.; Pohl, E.E.; Gerson, S.L.; Finkel, T.; Broxmeyer, H.E.; Qu, C.K. Metabolic regulation by the mitochondrial phosphatase PTPMT1 is required for hematopoietic stem cell differentiation. Cell Stem Cell 2013, 12, 62–74. [Google Scholar] [CrossRef]
- Liu, X.; Zheng, H.; Yu, W.M.; Cooper, T.M.; Bunting, K.D.; Qu, C.K. Maintenance of mouse hematopoietic stem cells ex vivo by reprogramming cellular metabolism. Blood 2015, 125, 1562–1565. [Google Scholar] [CrossRef]
- Zhang, J.; Guan, Z.; Murphy, A.N.; Wiley, S.E.; Perkins, G.A.; Worby, C.A.; Engel, J.L.; Heacock, P.; Nguyen, O.K.; Wang, J.H.; et al. Mitochondrial phosphatase PTPMT1 is essential for cardiolipin biosynthesis. Cell Metab. 2011, 13, 690–700. [Google Scholar] [CrossRef]
- Nath, A.K.; Ryu, J.H.; Jin, Y.N.; Roberts, L.D.; Dejam, A.; Gerszten, R.E.; Peterson, R.T. PTPMT1 Inhibition Lowers Glucose through Succinate Dehydrogenase Phosphorylation. Cell Rep. 2015, 10, 694–701. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Gao, J.; Ng, P.Y.; Qin, A.; Steer, J.H.; Pavlos, N.J.; Zheng, M.H.; Dong, Y.; Cheng, T.S. Alexidine Dihydrochloride Attenuates Osteoclast Formation and Bone Resorption and Protects Against LPS-Induced Osteolysis. J. Bone Miner. Res. 2016, 31, 560–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Targets | Antibodies | Ref. | Supplier |
|---|---|---|---|
| AMPK | AMPKα | 2532 S | CST |
| Bad | D24A9 | 9239 | CST |
| BCR | BCR | 3902 S | CST |
| Bcl-xL | Bcl-xL | 2762 | CST |
| BMI1 | Bmi1 (F6) | 05-637 | Merck Millipore (Upstate) |
| BCR | BCR | 3902 S | CST |
| Caspase-3 | Caspase-3 | 9662 S | CST |
| Caspase-9 | Caspase-9 | 9502 T | CST |
| CCNG2 | FL-249 | sc-851 | SCB |
| CCNG2 | N19 | sc-7266 | SCB |
| HSP60 | HSP60 (K-19) | sc-1722 | SCB |
| HSP90 | HSP 90α/β Antibody (F-8) | sc-13119 | SCB |
| P-AMPK | P-AMPK (T172) | 2535 | SCB |
| PARP | PARP | 9542 S | CST |
| P-c Abl | P-c Abl (Y245) (73E5) | 2868 S | CST |
| PTPMT1 | PTPMT1 | sc-390901 | SCB |
| P-STAT5 | P-STAT5 (Tyr694) (C11C5) | 9359 S | CST |
| STAT5 | STAT5 (D2O6Y) | 94205 | CST |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muselli, F.; Mourgues, L.; Rochet, N.; Nebout, M.; Guerci, A.; Verhoeyen, E.; Krug, A.; Legros, L.; Peyron, J.-F.; Mary, D. Repurposing the Bis-Biguanide Alexidine in Combination with Tyrosine Kinase Inhibitors to Eliminate Leukemic Stem/Progenitor Cells in Chronic Myeloid Leukemia. Cancers 2023, 15, 995. https://doi.org/10.3390/cancers15030995
Muselli F, Mourgues L, Rochet N, Nebout M, Guerci A, Verhoeyen E, Krug A, Legros L, Peyron J-F, Mary D. Repurposing the Bis-Biguanide Alexidine in Combination with Tyrosine Kinase Inhibitors to Eliminate Leukemic Stem/Progenitor Cells in Chronic Myeloid Leukemia. Cancers. 2023; 15(3):995. https://doi.org/10.3390/cancers15030995
Chicago/Turabian StyleMuselli, Fabien, Lucas Mourgues, Nathalie Rochet, Marielle Nebout, Agnès Guerci, Els Verhoeyen, Adrien Krug, Laurence Legros, Jean-François Peyron, and Didier Mary. 2023. "Repurposing the Bis-Biguanide Alexidine in Combination with Tyrosine Kinase Inhibitors to Eliminate Leukemic Stem/Progenitor Cells in Chronic Myeloid Leukemia" Cancers 15, no. 3: 995. https://doi.org/10.3390/cancers15030995