
Intestinal Epithelial Cells Adapt to Chronic Inflammation through Partial Genetic Reprogramming
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Constructs and Cell Lines
2.2. Retroviral and Lentiviral Infections
2.3. TP53 Status
2.4. Oxidative Stress Induction in HCEC-hTERT Cells
2.5. Gene Expression Analysis by qRT-PCR
2.6. Gene Expression Profiling of Cell Lines
2.7. Immunoblot Analysis
2.8. In silico Analysis for Validation of the Experimental OSAP
3. Results
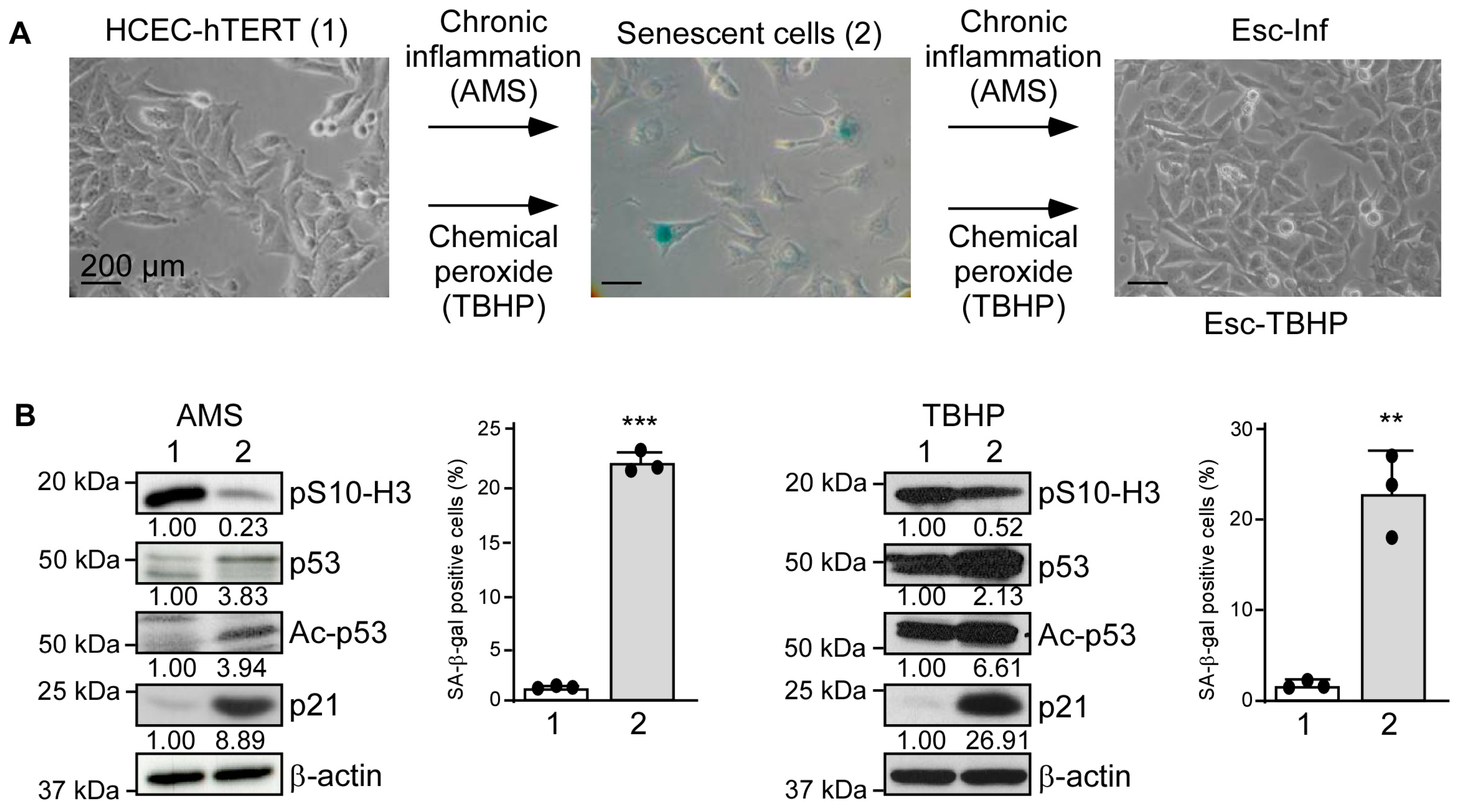
3.1. Human Colonic Epithelial Cells Exposed to Chronic Inflammation Develop a Resistance Mechanism
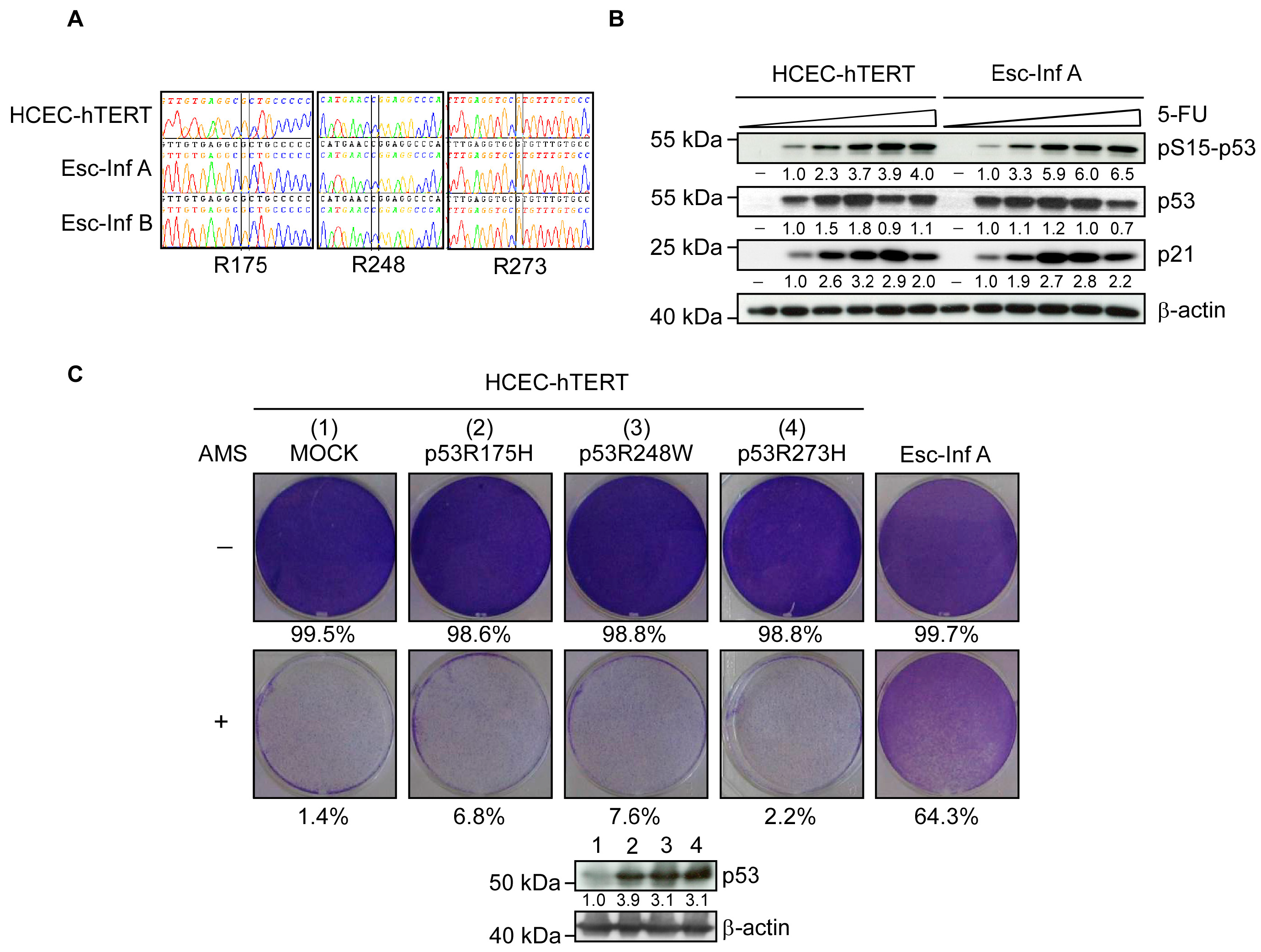
3.2. Resistance to Oxidative Stress Is p53-Independent
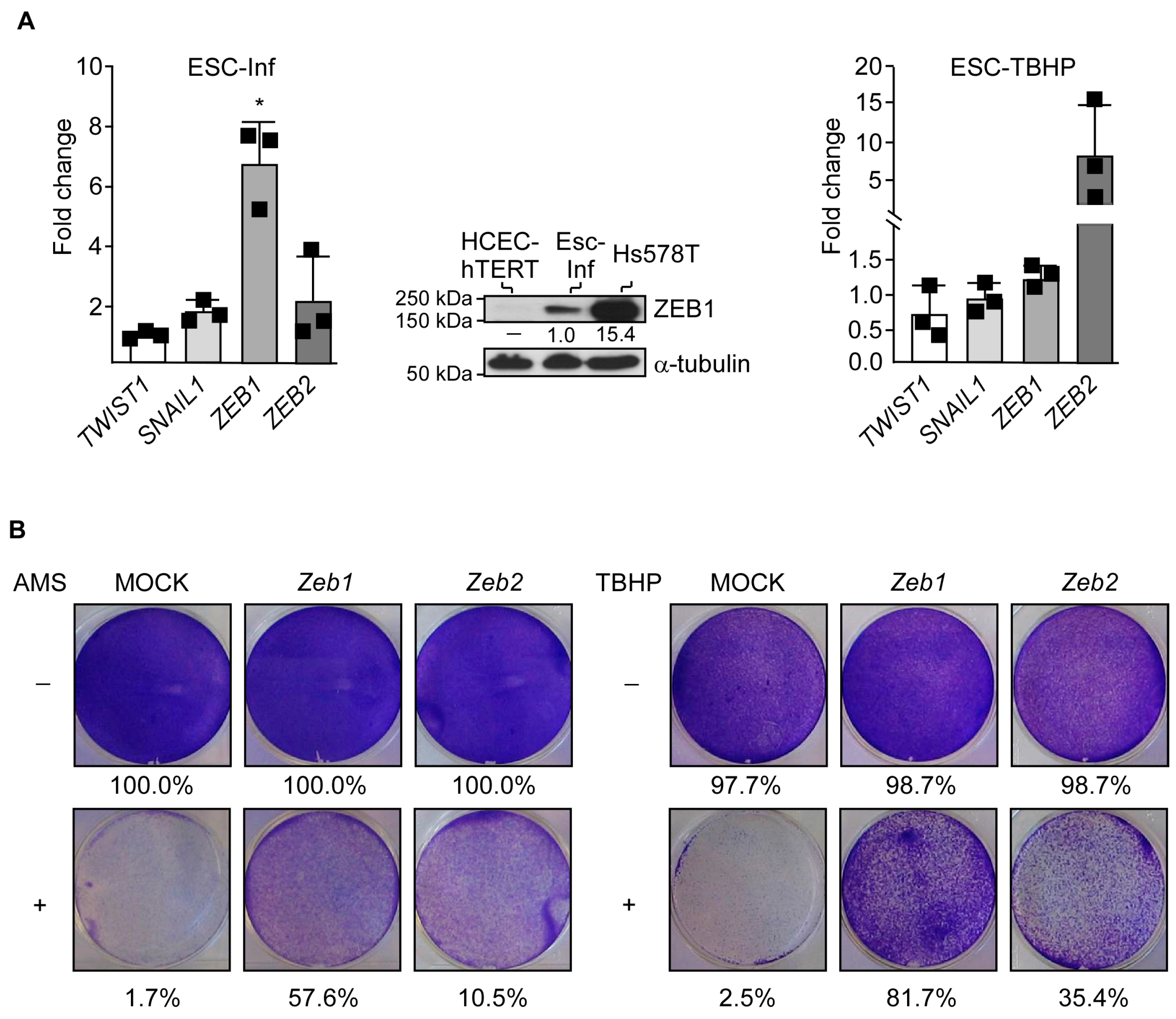
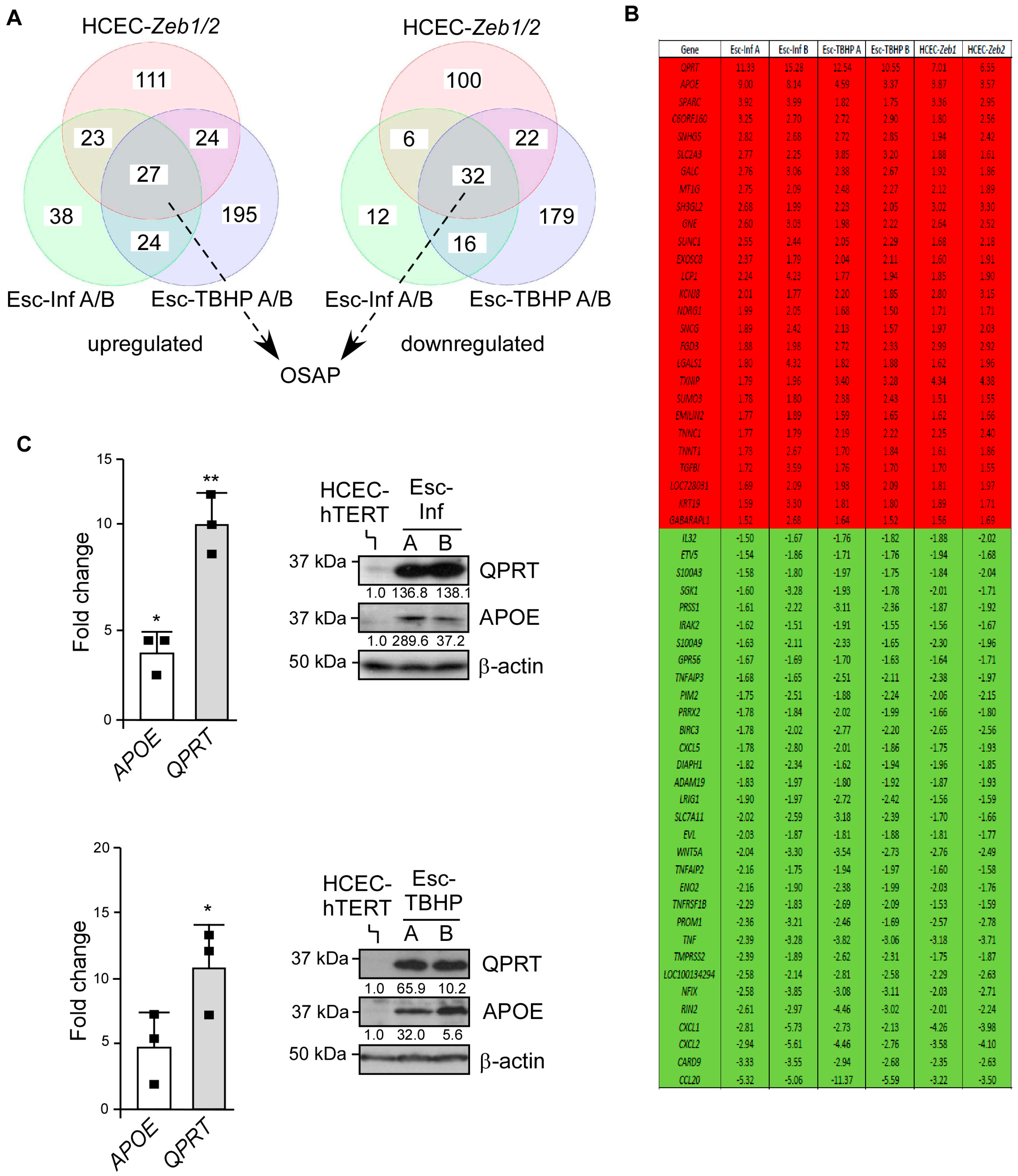
3.3. Adaptation to Oxidative Stress Relies on Partial Cell Reprogramming
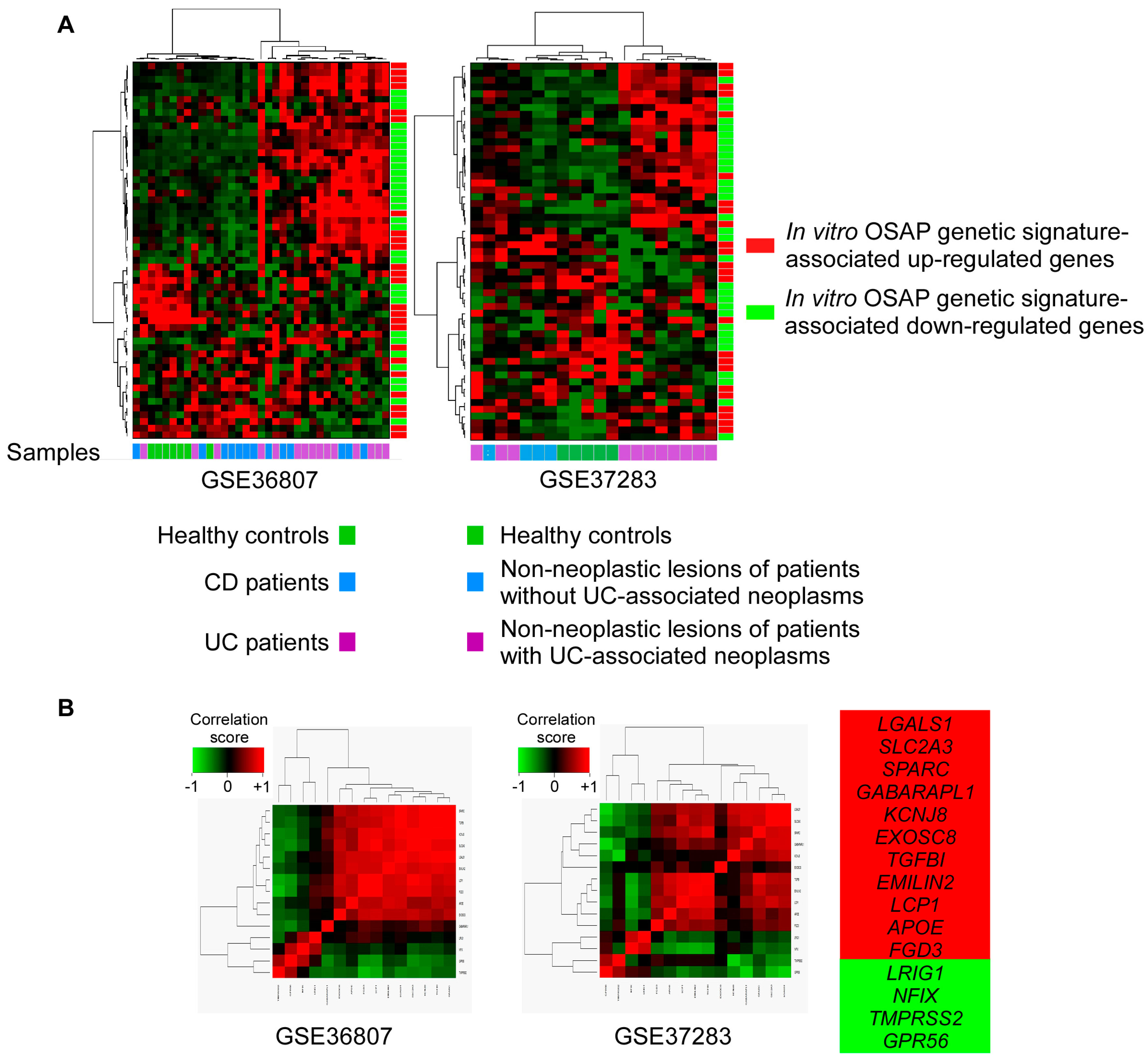
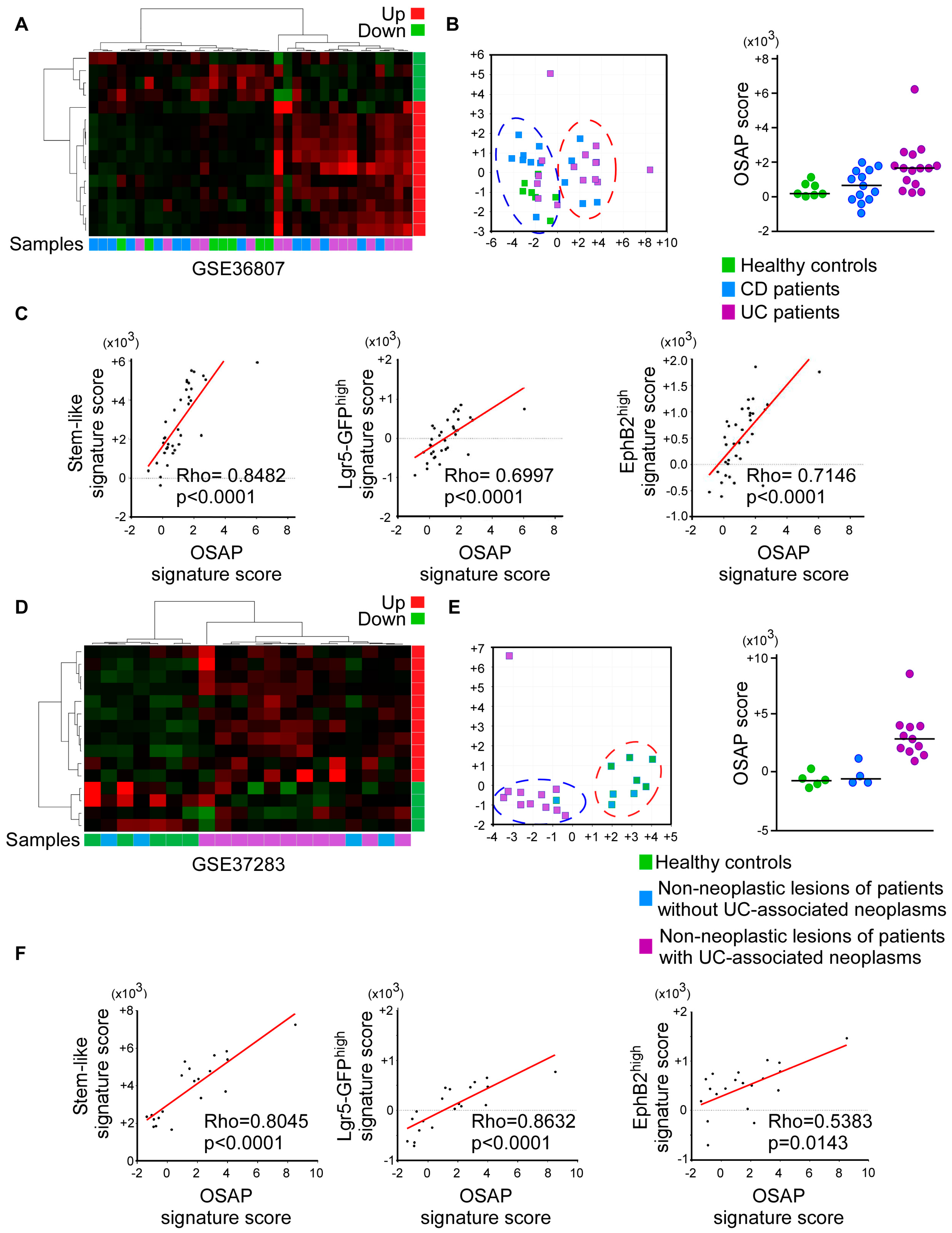
3.4. OSAP Is Activated in Ulcerative Colitis and Crohn’s Disease Mucosa
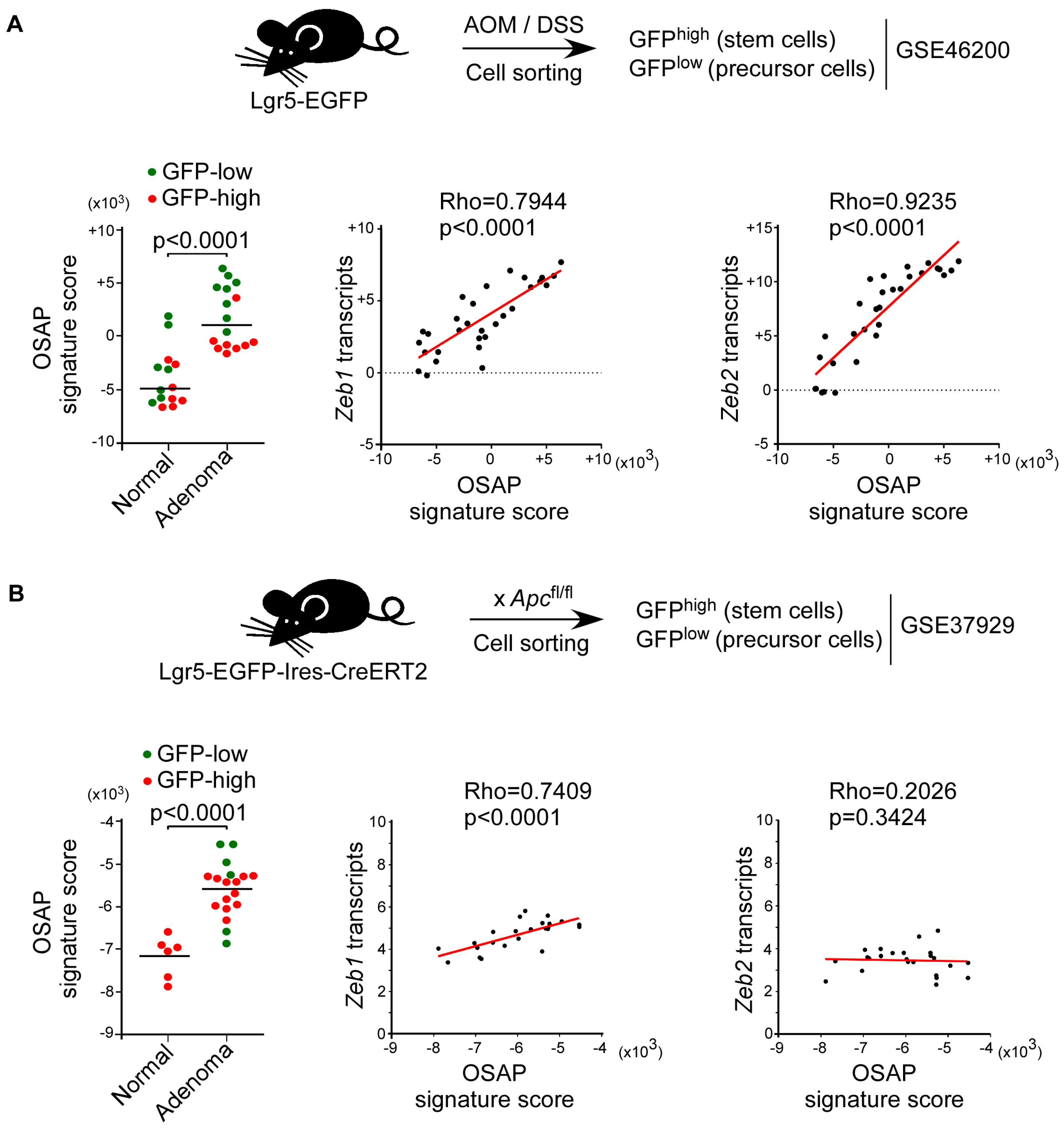
3.5. OSAP Is Induced in Intestinal Precursor Cells in Both Colitis-Associated and Sporadic Murine Colorectal Cancer Models
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roessner, A.; Kuester, D.; Malfertheiner, P.; Schneider-Stock, R. Oxidative Stress in Ulcerative Colitis-Associated Carcinogenesis. Pathol. Res. Pract. 2008, 204, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Li, Y.; Wang, R.; Ren, F.; Wang, X. Oxidative Stress and Antioxidant Nanotherapeutic Approaches for Inflammatory Bowel Disease. Biomedicines 2021, 10, 85. [Google Scholar] [CrossRef] [PubMed]
- Bourgonje, A.R.; Feelisch, M.; Faber, K.N.; Pasch, A.; Dijkstra, G.; van Goor, H. Oxidative Stress and Redox-Modulating Therapeutics in Inflammatory Bowel Disease. Trends Mol. Med. 2020, 26, 1034–1046. [Google Scholar] [CrossRef] [PubMed]
- Risques, R.A.; Lai, L.A.; Brentnall, T.A.; Li, L.; Feng, Z.; Gallaher, J.; Mandelson, M.T.; Potter, J.D.; Bronner, M.P.; Rabinovitch, P.S.; et al. Ulcerative Colitis Is a Disease of Accelerated Colon Aging: Evidence from Telomere Attrition and DNA Damage. Gastroenterology 2008, 135, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Jurk, D.; Wilson, C.; Passos, J.F.; Oakley, F.; Correia-Melo, C.; Greaves, L.; Saretzki, G.; Fox, C.; Lawless, C.; Anderson, R.; et al. Chronic Inflammation Induces Telomere Dysfunction and Accelerates Ageing in Mice. Nat. Commun. 2014, 2, 4172. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, J.N.; Bronner, M.P.; Brentnall, T.A.; Finley, J.C.; Shen, W.-T.; Emerson, S.; Emond, M.J.; Gollahon, K.A.; Moskovitz, A.H.; Crispin, D.A.; et al. Chromosomal Instability in Ulcerative Colitis Is Related to Telomere Shortening. Nat. Genet. 2002, 32, 280–284. [Google Scholar] [CrossRef]
- Chakravarti, D.; Hu, B.; Mao, X.; Rashid, A.; Li, J.; Li, J.; Liao, W.-T.; Whitley, E.M.; Dey, P.; Hou, P.; et al. Telomere Dysfunction Activates YAP1 to Drive Tissue Inflammation. Nat. Commun. 2020, 11, 4766. [Google Scholar] [CrossRef]
- Chakravarti, D.; Lee, R.; Multani, A.S.; Santoni, A.; Keith, Z.; Hsu, W.-H.; Chang, K.; Reyes, L.; Rashid, A.; Wu, C.-J.; et al. Telomere Dysfunction Instigates Inflammation in Inflammatory Bowel Disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2024853118. [Google Scholar] [CrossRef]
- Fearon, E.R. Molecular Genetics of Colorectal Cancer. Annu. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Vogelstein, B. Lessons from Hereditary Colorectal Cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Harpaz, N.; Tong, Y.; Huang, Y.; Laurin, J.; Greenwald, B.D.; Hontanosas, M.; Newkirk, C.; Meltzer, S.J. P53 Point Mutations in Dysplastic and Cancerous Ulcerative Colitis Lesions. Gastroenterology 1993, 104, 1633–1639. [Google Scholar] [CrossRef] [PubMed]
- Brentnall, T.A.; Crispin, D.A.; Rabinovitch, P.S.; Haggitt, R.C.; Rubin, C.E.; Stevens, A.C.; Burmer, G.C. Mutations in the P53 Gene: An Early Marker of Neoplastic Progression in Ulcerative Colitis. Gastroenterology 1994, 107, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; Amstad, P.; Raja, K.; Ambs, S.; Nagashima, M.; Bennett, W.P.; Shields, P.G.; Ham, A.J.; Swenberg, J.A.; Marrogi, A.J.; et al. Increased P53 Mutation Load in Noncancerous Colon Tissue from Ulcerative Colitis: A Cancer-Prone Chronic Inflammatory Disease. Cancer Res. 2000, 60, 3333–3337. [Google Scholar] [PubMed]
- Yoshida, T.; Mikami, T.; Mitomi, H.; Okayasu, I. Diverse P53 Alterations in Ulcerative Colitis-Associated Low-Grade Dysplasia: Full-Length Gene Sequencing in Microdissected Single Crypts. J. Pathol. 2003, 199, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Pribluda, A.; Elyada, E.; Wiener, Z.; Hamza, H.; Goldstein, R.E.; Biton, M.; Burstain, I.; Morgenstern, Y.; Brachya, G.; Billauer, H.; et al. A Senescence-Inflammatory Switch from Cancer-Inhibitory to Cancer-Promoting Mechanism. Cancer Cell 2013, 24, 242–256. [Google Scholar] [CrossRef]
- Cooks, T.; Pateras, I.S.; Tarcic, O.; Solomon, H.; Schetter, A.J.; Wilder, S.; Lozano, G.; Pikarsky, E.; Forshew, T.; Rosenfeld, N.; et al. Mutant P53 Prolongs NF-ΚB Activation and Promotes Chronic Inflammation and Inflammation-Associated Colorectal Cancer. Cancer Cell 2013, 23, 634–646. [Google Scholar] [CrossRef]
- Schwitalla, S.; Fingerle, A.A.; Cammareri, P.; Nebelsiek, T.; Göktuna, S.I.; Ziegler, P.K.; Canli, O.; Heijmans, J.; Huels, D.J.; Moreaux, G.; et al. Intestinal Tumorigenesis Initiated by Dedifferentiation and Acquisition of Stem-Cell-like Properties. Cell 2013, 152, 25–38. [Google Scholar] [CrossRef]
- Sebastian, C.; Ferrer, C.; Serra, M.; Choi, J.-E.; Ducano, N.; Mira, A.; Shah, M.S.; Stopka, S.A.; Perciaccante, A.J.; Isella, C.; et al. A Non-Dividing Cell Population with High Pyruvate Dehydrogenase Kinase Activity Regulates Metabolic Heterogeneity and Tumorigenesis in the Intestine. Nat. Commun. 2022, 13, 1503. [Google Scholar] [CrossRef]
- Morel, A.-P.; Hinkal, G.W.; Thomas, C.; Fauvet, F.; Courtois-Cox, S.; Wierinckx, A.; Devouassoux-Shisheboran, M.; Treilleux, I.; Tissier, A.; Gras, B.; et al. EMT Inducers Catalyze Malignant Transformation of Mammary Epithelial Cells and Drive Tumorigenesis towards Claudin-Low Tumors in Transgenic Mice. PLoS Genet. 2012, 8, e1002723. [Google Scholar] [CrossRef]
- Gras, B.; Jacqueroud, L.; Wierinckx, A.; Lamblot, C.; Fauvet, F.; Lachuer, J.; Puisieux, A.; Ansieau, S. Snail Family Members Unequally Trigger EMT and Thereby Differ in Their Ability to Promote the Neoplastic Transformation of Mammary Epithelial Cells. PLoS ONE 2014, 9, e92254. [Google Scholar] [CrossRef] [Green Version]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A Biomarker That Identifies Senescent Human Cells in Culture and in Aging Skin in Vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Sayan, A.E.; Griffiths, T.R.; Pal, R.; Browne, G.J.; Ruddick, A.; Yagci, T.; Edwards, R.; Mayer, N.J.; Qazi, H.; Goyal, S.; et al. SIP1 Protein Protects Cells from DNA Damage-Induced Apoptosis and Has Independent Prognostic Value in Bladder Cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 14884–14889. [Google Scholar] [CrossRef] [PubMed]
- Marisa, L.; de Reyniès, A.; Duval, A.; Selves, J.; Gaub, M.P.; Vescovo, L.; Etienne-Grimaldi, M.-C.; Schiappa, R.; Guenot, D.; Ayadi, M.; et al. Gene Expression Classification of Colon Cancer into Molecular Subtypes: Characterization, Validation, and Prognostic Value. PLoS Med. 2013, 10, e1001453. [Google Scholar] [CrossRef] [PubMed]
- Pekow, J.; Dougherty, U.; Huang, Y.; Gometz, E.; Nathanson, J.; Cohen, G.; Levy, S.; Kocherginsky, M.; Venu, N.; Westerhoff, M.; et al. Gene Signature Distinguishes Patients with Chronic Ulcerative Colitis Harboring Remote Neoplastic Lesions. Inflamm. Bowel Dis. 2013, 19, 461–470. [Google Scholar] [CrossRef]
- Montero-Meléndez, T.; Llor, X.; García-Planella, E.; Perretti, M.; Suárez, A. Identification of Novel Predictor Classifiers for Inflammatory Bowel Disease by Gene Expression Profiling. PLoS ONE 2013, 8, e76235. [Google Scholar] [CrossRef]
- Arijs, I.; de Hertogh, G.; Lemaire, K.; Quintens, R.; van Lommel, L.; van Steen, K.; Leemans, P.; Cleynen, I.; van Assche, G.; Vermeire, S.; et al. Mucosal Gene Expression of Antimicrobial Peptides in Inflammatory Bowel Disease before and after First Infliximab Treatment. PLoS ONE 2009, 4, e7984. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia Enables Predictive Modelling of Anticancer Drug Sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Hirsch, D.; Barker, N.; McNeil, N.; Hu, Y.; Camps, J.; McKinnon, K.; Clevers, H.; Ried, T.; Gaiser, T. LGR5 Positivity Defines Stem-like Cells in Colorectal Cancer. Carcinogenesis 2014, 35, 849–858. [Google Scholar] [CrossRef]
- Schepers, A.G.; Snippert, H.J.; Stange, D.E.; van den Born, M.; van Es, J.H.; van de Wetering, M.; Clevers, H. Lineage Tracing Reveals Lgr5+ Stem Cell Activity in Mouse Intestinal Adenomas. Science 2012, 337, 730–735. [Google Scholar] [CrossRef]
- Risques, R.A.; Lai, L.A.; Himmetoglu, C.; Ebaee, A.; Li, L.; Feng, Z.; Bronner, M.P.; Al-Lahham, B.; Kowdley, K.V.; Lindor, K.D.; et al. Ulcerative Colitis-Associated Colorectal Cancer Arises in a Field of Short Telomeres, Senescence, and Inflammation. Cancer Res. 2011, 71, 1669–1679. [Google Scholar] [CrossRef] [Green Version]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A Reciprocal Repression between ZEB1 and Members of the MiR-200 Family Promotes EMT and Invasion in Cancer Cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of P53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. The Many Faces of P53: Something for Everyone. J. Mol. Cell Biol. 2019, 11, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Okayasu, I. Development of Ulcerative Colitis and Its Associated Colorectal Neoplasia as a Model of the Organ-Specific Chronic Inflammation-Carcinoma Sequence. Pathol. Int. 2012, 62, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Noffsinger, A.E.; Belli, J.M.; Miller, M.A.; Fenoglio-Preiser, C.M. A Unique Basal Pattern of P53 Expression in Ulcerative Colitis Is Associated with Mutation in the P53 Gene. Histopathology 2001, 39, 482–492. [Google Scholar] [CrossRef]
- Leedham, S.J.; Graham, T.A.; Oukrif, D.; McDonald, S.A.C.; Rodriguez-Justo, M.; Harrison, R.F.; Shepherd, N.A.; Novelli, M.R.; Jankowski, J.A.Z.; Wright, N.A.; et al. Clonality, Founder Mutations, and Field Cancerization in Human Ulcerative Colitis-Associated Neoplasia. Gastroenterology 2009, 136, 542–550.e6. [Google Scholar] [CrossRef]
- Ansieau, S.; Bastid, J.; Doreau, A.; Morel, A.-P.; Bouchet, B.P.; Thomas, C.; Fauvet, F.; Puisieux, I.; Doglioni, C.; Piccinin, S.; et al. Induction of EMT by Twist Proteins as a Collateral Effect of Tumor-Promoting Inactivation of Premature Senescence. Cancer Cell 2008, 14, 79–89. [Google Scholar] [CrossRef]
- Tran, P.T.; Shroff, E.H.; Burns, T.F.; Thiyagarajan, S.; Das, S.T.; Zabuawala, T.; Chen, J.; Cho, Y.-J.; Luong, R.; Tamayo, P.; et al. Twist1 Suppresses Senescence Programs and Thereby Accelerates and Maintains Mutant Kras-Induced Lung Tumorigenesis. PLoS Genet. 2012, 8, e1002650. [Google Scholar] [CrossRef]
- Furuya, S.; Endo, K.; Takahashi, A.; Miyazawa, K.; Saitoh, M. Snail Suppresses Cellular Senescence and Promotes Fibroblast-Led Cancer Cell Invasion. FEBS Open Bio 2017, 7, 1586–1597. [Google Scholar] [CrossRef]
- Liu, Y.; El-Naggar, S.; Darling, D.S.; Higashi, Y.; Dean, D.C. Zeb1 Links Epithelial-Mesenchymal Transition and Cellular Senescence. Development 2008, 135, 579–588. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lu, X.; Huang, L.; Wang, W.; Jiang, G.; Dean, K.C.; Clem, B.; Telang, S.; Jenson, A.B.; Cuatrecasas, M.; et al. Different Thresholds of ZEB1 Are Required for Ras-Mediated Tumour Initiation and Metastasis. Nat. Commun. 2014, 5, 5660. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sánchez-Tilló, E.; Lu, X.; Huang, L.; Clem, B.; Telang, S.; Jenson, A.B.; Cuatrecasas, M.; Chesney, J.; Postigo, A.; et al. Sequential Inductions of the ZEB1 Transcription Factor Caused by Mutation of Rb and Then Ras Proteins Are Required for Tumor Initiation and Progression. J. Biol. Chem. 2013, 288, 11572–11580. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Chaturvedi, R.; Barry, D.P.; Coburn, L.A.; Asim, M.; Lewis, N.D.; Piazuelo, M.B.; Washington, M.K.; Vitek, M.P.; Wilson, K.T.; et al. The Apolipoprotein E-Mimetic Peptide COG112 Inhibits NF-KappaB Signaling, Proinflammatory Cytokine Expression, and Disease Activity in Murine Models of Colitis. J. Biol. Chem. 2011, 286, 3839–3850. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.J.; Ohkubo, N.; Oddo, J.; van Kanegan, M.J.; Neil, J.; Li, F.; Colton, C.A.; Vitek, M.P. Apolipoprotein E and Peptide Mimetics Modulate Inflammation by Binding the SET Protein and Activating Protein Phosphatase 2A. J. Immunol. 2011, 186, 2535–2542. [Google Scholar] [CrossRef]
- Kemp, S.B.; Carpenter, E.S.; Steele, N.G.; Donahue, K.L.; Nwosu, Z.C.; Pacheco, A.; Velez-Delgado, A.; Menjivar, R.E.; Lima, F.; The, S.; et al. Apolipoprotein E Promotes Immune Suppression in Pancreatic Cancer through NF-ΚB-Mediated Production of CXCL1. Cancer Res. 2021, 81, 4305–4318. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Merlos-Suárez, A.; Barriga, F.M.; Jung, P.; Iglesias, M.; Céspedes, M.V.; Rossell, D.; Sevillano, M.; Hernando-Momblona, X.; da Silva-Diz, V.; Muñoz, P.; et al. The Intestinal Stem Cell Signature Identifies Colorectal Cancer Stem Cells and Predicts Disease Relapse. Cell Stem Cell 2011, 8, 511–524. [Google Scholar] [CrossRef]
- Sadanandam, A.; Lyssiotis, C.A.; Homicsko, K.; Collisson, E.A.; Gibb, W.J.; Wullschleger, S.; Ostos, L.C.G.; Lannon, W.A.; Grotzinger, C.; Del Rio, M.; et al. A Colorectal Cancer Classification System That Associates Cellular Phenotype and Responses to Therapy. Nat. Med. 2013, 19, 619–625. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative Stress, Inflammation, and Cancer: How Are They Linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef]
- Romanov, S.R.; Kozakiewicz, B.K.; Holst, C.R.; Stampfer, M.R.; Haupt, L.M.; Tlsty, T.D. Normal Human Mammary Epithelial Cells Spontaneously Escape Senescence and Acquire Genomic Changes. Nature 2001, 409, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Galanos, P.; Vougas, K.; Walter, D.; Polyzos, A.; Maya-Mendoza, A.; Haagensen, E.J.; Kokkalis, A.; Roumelioti, F.-M.; Gagos, S.; Tzetis, M.; et al. Chronic P53-Independent P21 Expression Causes Genomic Instability by Deregulating Replication Licensing. Nat. Cell Biol. 2016, 18, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.L.; Suram, A.; Mirani, N.; Bischof, O.; Herbig, U. Derepression of HTERT Gene Expression Promotes Escape from Oncogene-Induced Cellular Senescence. Proc. Natl. Acad. Sci. USA 2016, 113, E5024–E5033. [Google Scholar] [CrossRef] [PubMed]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-Associated Reprogramming Promotes Cancer Stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Saleh, T.; Tyutyunyk-Massey, L.; Murray, G.F.; Alotaibi, M.R.; Kawale, A.S.; Elsayed, Z.; Henderson, S.C.; Yakovlev, V.; Elmore, L.W.; Toor, A.; et al. Tumor Cell Escape from Therapy-Induced Senescence. Biochem. Pharmacol. 2019, 162, 202–212. [Google Scholar] [CrossRef]
- Milanovic, M.; Yu, Y.; Schmitt, C.A. The Senescence-Stemness Alliance—A Cancer-Hijacked Regeneration Principle. Trends Cell Biol. 2018, 28, 1049–1061. [Google Scholar] [CrossRef]
- Krizhanovsky, V.; Lowe, S.W. Stem Cells: The Promises and Perils of P53. Nature 2009, 460, 1085–1086. [Google Scholar] [CrossRef]
- Sahm, F.; Oezen, I.; Opitz, C.A.; Radlwimmer, B.; von Deimling, A.; Ahrendt, T.; Adams, S.; Bode, H.B.; Guillemin, G.J.; Wick, W.; et al. The Endogenous Tryptophan Metabolite and NAD+ Precursor Quinolinic Acid Confers Resistance of Gliomas to Oxidative Stress. Cancer Res. 2013, 73, 3225–3234. [Google Scholar] [CrossRef]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine Signaling via the CXCR2 Receptor Reinforces Senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- De Barrios, O.; Sanchez-Moral, L.; Cortés, M.; Ninfali, C.; Profitós-Pelejà, N.; Martínez-Campanario, M.C.; Siles, L.; del Campo, R.; Fernández-Aceñero, M.J.; Darling, D.S.; et al. ZEB1 Promotes Inflammation and Progression towards Inflammation-Driven Carcinoma through Repression of the DNA Repair Glycosylase MPG in Epithelial Cells. Gut 2019, 68, 2129–2141. [Google Scholar] [CrossRef] [PubMed]
- Iyama, T.; Wilson, D.M. DNA Repair Mechanisms in Dividing and Non-Dividing Cells. DNA Repair 2013, 12, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Puisieux, A.; Brabletz, T.; Caramel, J. Oncogenic Roles of EMT-Inducing Transcription Factors. Nat. Cell. Biol. 2014, 16, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; Osborne, J.; Hutchinson, P.; Tse, G.; et al. A Switch in the Expression of Embryonic EMT-Inducers Drives the Development of Malignant Melanoma. Cancer Cell 2013, 24, 466–480. [Google Scholar] [CrossRef]
- Denecker, G.; Vandamme, N.; Akay, O.; Koludrovic, D.; Taminau, J.; Lemeire, K.; Gheldof, A.; De Craene, B.; Van Gele, M.; Brochez, L.; et al. Identification of a ZEB2-MITF-ZEB1 Transcriptional Network That Controls Melanogenesis and Melanoma Progression. Cell Death Differ. 2014, 21, 1250–1261. [Google Scholar] [CrossRef]
- Ungaro, R.; Mehandru, S.; Allen, P.B.; Peyrin-Biroulet, L.; Colombel, J.-F. Ulcerative Colitis. Lancet 2017, 389, 1756–1770. [Google Scholar] [CrossRef]
- Cai, Z.; Wang, S.; Li, J. Treatment of Inflammatory Bowel Disease: A Comprehensive Review. Front. Med. 2021, 8, 765474. [Google Scholar] [CrossRef]
- Sphyris, N.; Hodder, M.C.; Sansom, O.J. Subversion of Niche-Signalling Pathways in Colorectal Cancer: What Makes and Breaks the Intestinal Stem Cell. Cancers 2021, 13, 1000. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef]
- Dai, M.; Wang, P.; Boyd, A.D.; Kostov, G.; Athey, B.; Jones, E.G.; Bunney, W.E.; Myers, R.M.; Speed, T.P.; Akil, H.; et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005, 33, e175. [Google Scholar] [CrossRef] [Green Version]
- Sandberg, R.; Larsson, O. Improved precision and accuracy for microarrays using updated probe set definitions. BMC Bioinform. 2007, 8, 48. [Google Scholar] [CrossRef] [PubMed]
- Maglott, D.; Ostell, J.; Pruitt, K.D.; Tatusova, T. Entrez Gene: Gene-centered information at NCBI. Nucleic Acids Res. 2005, 33, D54–D58. [Google Scholar] [CrossRef]
- Reich, M.; Liefeld, T.; Gould, J.; Lerner, J.; Tamayo, P.; Mesirov, J.P. GenePattern 2.0. Nat. Genet 2006, 38, 500–501. [Google Scholar] [CrossRef] [PubMed]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Collin, G.; Foy, J.-P.; Aznar, N.; Rama, N.; Wierinckx, A.; Saintigny, P.; Puisieux, A.; Ansieau, S. Intestinal Epithelial Cells Adapt to Chronic Inflammation through Partial Genetic Reprogramming. Cancers 2023, 15, 973. https://doi.org/10.3390/cancers15030973
Collin G, Foy J-P, Aznar N, Rama N, Wierinckx A, Saintigny P, Puisieux A, Ansieau S. Intestinal Epithelial Cells Adapt to Chronic Inflammation through Partial Genetic Reprogramming. Cancers. 2023; 15(3):973. https://doi.org/10.3390/cancers15030973
Chicago/Turabian StyleCollin, Guillaume, Jean-Philippe Foy, Nicolas Aznar, Nicolas Rama, Anne Wierinckx, Pierre Saintigny, Alain Puisieux, and Stéphane Ansieau. 2023. "Intestinal Epithelial Cells Adapt to Chronic Inflammation through Partial Genetic Reprogramming" Cancers 15, no. 3: 973. https://doi.org/10.3390/cancers15030973