

Targeted Therapy of Interleukin-34 as a Promising Approach to Overcome Cancer Therapy Resistance
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Mechanisms of Cancer Resistance to Standard Therapy and Immunotherapy
3. Expression of IL-34 and IL-34 Receptors in Cancer
4. IL-34 Contributes to Generating a Tumor Microenvironment That Restrains the Anti-Tumor Immunity
5. IL-34 and Cancer Immunotherapy
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Sasaki, K.; Strom, S.S.; O’Brien, S.; Jabbour, E.; Ravandi, F.; Konopleva, M.; Borthakur, G.; Pemmaraju, N.; Daver, N.; Jain, P.; et al. Relative survival in patients with chronic-phase chronic myeloid leukaemia in the tyrosine-kinase inhibitor era: Analysis of patient data from six prospective clinical trials. Lancet Haematol. 2015, 2, e186–e193. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Fu, D.; Xu, F.; Zhang, Z. Unwrapping the genomic characteristics of urothelial bladder cancer and successes with immune checkpoint blockade therapy. Oncogenesis 2018, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef]
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef]
- Mullard, A. Second CTLA4-targeted checkpoint inhibitor secures FDA approval. Nat. Rev. Drug Discov. 2022, 21, 868. [Google Scholar] [CrossRef]
- Twomey, J.D.; Zhang, B. Cancer Immunotherapy Update: FDA-Approved Checkpoint Inhibitors and Companion Diagnostics. AAPS J. 2021, 23, 39. [Google Scholar] [CrossRef]
- Weber, J.S.; D’Angelo, S.P.; Minor, D.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H., Jr.; Lao, C.D.; et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015, 16, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O’Byrne, K.; Kulasinghe, A. Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Oncol. 2022, 29, 3044–3060. [Google Scholar] [CrossRef] [PubMed]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef] [PubMed]
- Barrueto, L.; Caminero, F.; Cash, L.; Makris, C.; Lamichhane, P.; Deshmukh, R.R. Resistance to Checkpoint Inhibition in Cancer Immunotherapy. Transl. Oncol. 2020, 13, 100738. [Google Scholar] [CrossRef]
- Nedeljkovic, M.; Damjanovic, A. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer-How We Can Rise to the Challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Duan, H.; Huang, H.; Tong, X.; Han, Y.; Ru, G.; Qu, L.; Shou, C.; Zhao, Z. Cisplatin resistance in gastric cancer cells is associated with HER2 upregulation-induced epithelial-mesenchymal transition. Sci. Rep. 2016, 6, 20502. [Google Scholar] [CrossRef]
- Chen, Z.; Shi, T.; Zhang, L.; Zhu, P.; Deng, M.; Huang, C.; Hu, T.; Jiang, L.; Li, J. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: A review of the past decade. Cancer Lett. 2016, 370, 153–164. [Google Scholar] [CrossRef]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Bracci, L.; Schiavoni, G.; Sistigu, A.; Belardelli, F. Immune-based mechanisms of cytotoxic chemotherapy: Implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ. 2014, 21, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Opzoomer, J.W.; Sosnowska, D.; Anstee, J.E.; Spicer, J.F.; Arnold, J.N. Cytotoxic Chemotherapy as an Immune Stimulus: A Molecular Perspective on Turning Up the Immunological Heat on Cancer. Front. Immunol. 2019, 10, 1654. [Google Scholar] [CrossRef]
- Neophytou, C.M.; Pierides, C.; Christodoulou, M.I.; Costeas, P.; Kyriakou, T.C.; Papageorgis, P. The Role of Tumor-Associated Myeloid Cells in Modulating Cancer Therapy. Front. Oncol. 2020, 10, 899. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Lee, E.; Hestir, K.; Leo, C.; Huang, M.; Bosch, E.; Halenbeck, R.; Wu, G.; Zhou, A.; Behrens, D.; et al. Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science 2008, 320, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Lin, W.Y.; Chen, Y.; Stawicki, S.; Mukhyala, K.; Wu, Y.; Martin, F.; Bazan, J.F.; Starovasnik, M.A. Structural basis for the dual recognition of helical cytokines IL-34 and CSF-1 by CSF-1R. Structure 2012, 20, 676–687. [Google Scholar] [CrossRef]
- Nandi, S.; Cioce, M.; Yeung, Y.G.; Nieves, E.; Tesfa, L.; Lin, H.; Hsu, A.W.; Halenbeck, R.; Cheng, H.Y.; Gokhan, S.; et al. Receptor-type protein-tyrosine phosphatase zeta is a functional receptor for interleukin-34. J. Biol. Chem. 2013, 288, 21972–21986. [Google Scholar] [CrossRef] [PubMed]
- Segaliny, A.I.; Brion, R.; Mortier, E.; Maillasson, M.; Cherel, M.; Jacques, Y.; Le Goff, B.; Heymann, D. Syndecan-1 regulates the biological activities of interleukin-34. Biochim. Biophys. Acta 2015, 1853, 1010–1021. [Google Scholar] [CrossRef]
- Chihara, T.; Suzu, S.; Hassan, R.; Chutiwitoonchai, N.; Hiyoshi, M.; Motoyoshi, K.; Kimura, F.; Okada, S. IL-34 and M-CSF share the receptor Fms but are not identical in biological activity and signal activation. Cell Death Differ. 2010, 17, 1917–1927. [Google Scholar] [CrossRef]
- Droin, N.; Solary, E. Editorial: CSF1R, CSF-1, and IL-34, a “menage a trois” conserved across vertebrates. J. Leukoc. Biol. 2010, 87, 745–747. [Google Scholar] [CrossRef] [PubMed]
- Felix, J.; Elegheert, J.; Gutsche, I.; Shkumatov, A.V.; Wen, Y.; Bracke, N.; Pannecoucke, E.; Vandenberghe, I.; Devreese, B.; Svergun, D.I.; et al. Human IL-34 and CSF-1 establish structurally similar extracellular assemblies with their common hematopoietic receptor. Structure 2013, 21, 528–539. [Google Scholar] [CrossRef]
- Greter, M.; Lelios, I.; Pelczar, P.; Hoeffel, G.; Price, J.; Leboeuf, M.; Kundig, T.M.; Frei, K.; Ginhoux, F.; Merad, M.; et al. Stroma-derived interleukin-34 controls the development and maintenance of langerhans cells and the maintenance of microglia. Immunity 2012, 37, 1050–1060. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Szretter, K.J.; Vermi, W.; Gilfillan, S.; Rossini, C.; Cella, M.; Barrow, A.D.; Diamond, M.S.; Colonna, M. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat. Immunol. 2012, 13, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Lelios, I.; Cansever, D.; Utz, S.G.; Mildenberger, W.; Stifter, S.A.; Greter, M. Emerging roles of IL-34 in health and disease. J. Exp. Med. 2020, 217, e20190290. [Google Scholar] [CrossRef] [PubMed]
- Franze, E.; Dinallo, V.; Laudisi, F.; Di Grazia, A.; Di Fusco, D.; Colantoni, A.; Ortenzi, A.; Giuffrida, P.; Di Carlo, S.; Sica, G.S.; et al. Interleukin-34 Stimulates Gut Fibroblasts to Produce Collagen Synthesis. J. Crohn’s Colitis 2020, 14, 1436–1445. [Google Scholar] [CrossRef]
- Franze, E.; Marafini, I.; De Simone, V.; Monteleone, I.; Caprioli, F.; Colantoni, A.; Ortenzi, A.; Crescenzi, F.; Izzo, R.; Sica, G.; et al. Interleukin-34 Induces Cc-chemokine Ligand 20 in Gut Epithelial Cells. J. Crohn’s Colitis 2016, 10, 87–94. [Google Scholar] [CrossRef]
- Franze, E.; Monteleone, I.; Cupi, M.L.; Mancia, P.; Caprioli, F.; Marafini, I.; Colantoni, A.; Ortenzi, A.; Laudisi, F.; Sica, G.; et al. Interleukin-34 sustains inflammatory pathways in the gut. Clin. Sci. 2015, 129, 271–280. [Google Scholar] [CrossRef]
- Preisser, L.; Miot, C.; Le Guillou-Guillemette, H.; Beaumont, E.; Foucher, E.D.; Garo, E.; Blanchard, S.; Fremaux, I.; Croue, A.; Fouchard, I.; et al. IL-34 and macrophage colony-stimulating factor are overexpressed in hepatitis C virus fibrosis and induce profibrotic macrophages that promote collagen synthesis by hepatic stellate cells. Hepatology 2014, 60, 1879–1890. [Google Scholar] [CrossRef]
- Franze, E.; Stolfi, C.; Troncone, E.; Scarozza, P.; Monteleone, G. Role of Interleukin-34 in Cancer. Cancers 2020, 12, 252. [Google Scholar] [CrossRef]
- Baghdadi, M.; Endo, H.; Tanaka, Y.; Wada, H.; Seino, K.I. Interleukin 34, from pathogenesis to clinical applications. Cytokine 2017, 99, 139–147. [Google Scholar] [CrossRef]
- Otsuka, R.; Wada, H.; Seino, K.I. IL-34, the rationale for its expression in physiological and pathological conditions. Semin. Immunol. 2021, 54, 101517. [Google Scholar] [CrossRef]
- Baud’huin, M.; Renault, R.; Charrier, C.; Riet, A.; Moreau, A.; Brion, R.; Gouin, F.; Duplomb, L.; Heymann, D. Interleukin-34 is expressed by giant cell tumours of bone and plays a key role in RANKL-induced osteoclastogenesis. J. Pathol. 2010, 221, 77–86. [Google Scholar] [CrossRef]
- Franze, E.; Dinallo, V.; Rizzo, A.; Di Giovangiulio, M.; Bevivino, G.; Stolfi, C.; Caprioli, F.; Colantoni, A.; Ortenzi, A.; Grazia, A.D.; et al. Interleukin-34 sustains pro-tumorigenic signals in colon cancer tissue. Oncotarget 2018, 9, 3432–3445. [Google Scholar] [CrossRef]
- Franze, E.; Laudisi, F.; Di Grazia, A.; Maronek, M.; Bellato, V.; Sica, G.; Monteleone, G. Macrophages produce and functionally respond to interleukin-34 in colon cancer. Cell Death Discov. 2020, 6, 117. [Google Scholar] [CrossRef]
- Franze, E.; Di Grazia, A.; Sica, G.S.; Biancone, L.; Laudisi, F.; Monteleone, G. Interleukin-34 Enhances the Tumor Promoting Function of Colorectal Cancer-Associated Fibroblasts. Cancers 2020, 12, 3537. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, F.; Ding, X.Y.; Li, T.E.; Wang, H.Y.; Gao, Y.H.; Wang, W.J.; Liu, Y.F.; Chen, X.S.; Shen, K.W. Hippo/YAP signaling choreographs the tumor immune microenvironment to promote triple negative breast cancer progression via TAZ/IL-34 axis. Cancer Lett. 2022, 527, 174–190. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef]
- Foucher, E.D.; Blanchard, S.; Preisser, L.; Descamps, P.; Ifrah, N.; Delneste, Y.; Jeannin, P. IL-34- and M-CSF-induced macrophages switch memory T cells into Th17 cells via membrane IL-1alpha. Eur. J. Immunol. 2015, 45, 1092–1102. [Google Scholar] [CrossRef]
- Endo, H.; Hama, N.; Baghdadi, M.; Ishikawa, K.; Otsuka, R.; Wada, H.; Asano, H.; Endo, D.; Konno, Y.; Kato, T.; et al. Interleukin-34 expression in ovarian cancer: A possible correlation with disease progression. Int. Immunol. 2020, 32, 175–186. [Google Scholar] [CrossRef]
- Foucher, E.D.; Blanchard, S.; Preisser, L.; Garo, E.; Ifrah, N.; Guardiola, P.; Delneste, Y.; Jeannin, P. IL-34 induces the differentiation of human monocytes into immunosuppressive macrophages. antagonistic effects of GM-CSF and IFNgamma. PloS ONE 2013, 8, e56045. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.L.; Hu, Z.Q.; Zhou, Z.J.; Dai, Z.; Wang, Z.; Cao, Y.; Fan, J.; Huang, X.W.; Zhou, J. miR-28-5p-IL-34-macrophage feedback loop modulates hepatocellular carcinoma metastasis. Hepatology 2016, 63, 1560–1575. [Google Scholar] [CrossRef] [PubMed]
- Irie, T.; Yoshii, D.; Komohara, Y.; Fujiwara, Y.; Kadohisa, M.; Honda, M.; Suzu, S.; Matsuura, T.; Kohashi, K.; Oda, Y.; et al. IL-34 in hepatoblastoma cells potentially promote tumor progression via autocrine and paracrine mechanisms. Cancer Med. 2022, 11, 1441–1453. [Google Scholar] [CrossRef] [PubMed]
- Raggi, C.; Correnti, M.; Sica, A.; Andersen, J.B.; Cardinale, V.; Alvaro, D.; Chiorino, G.; Forti, E.; Glaser, S.; Alpini, G.; et al. Cholangiocarcinoma stem-like subset shapes tumor-initiating niche by educating associated macrophages. J. Hepatol. 2017, 66, 102–115. [Google Scholar] [CrossRef]
- Segaliny, A.I.; Mohamadi, A.; Dizier, B.; Lokajczyk, A.; Brion, R.; Lanel, R.; Amiaud, J.; Charrier, C.; Boisson-Vidal, C.; Heymann, D. Interleukin-34 promotes tumor progression and metastatic process in osteosarcoma through induction of angiogenesis and macrophage recruitment. Int. J. Cancer 2015, 137, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Baghdadi, M.; Ishikawa, K.; Nakanishi, S.; Murata, T.; Umeyama, Y.; Kobayashi, T.; Kameda, Y.; Endo, H.; Wada, H.; Bogen, B.; et al. A role for IL-34 in osteolytic disease of multiple myeloma. Blood Adv. 2019, 3, 541–551. [Google Scholar] [CrossRef]
- Arora, H.; Panara, K.; Kuchakulla, M.; Kulandavelu, S.; Burnstein, K.L.; Schally, A.V.; Hare, J.M.; Ramasamy, R. Alterations of tumor microenvironment by nitric oxide impedes castration-resistant prostate cancer growth. Proc. Natl. Acad. Sci. USA 2018, 115, 11298–11303. [Google Scholar] [CrossRef]
- Komohara, Y.; Noyori, O.; Saito, Y.; Takeya, H.; Baghdadi, M.; Kitagawa, F.; Hama, N.; Ishikawa, K.; Okuno, Y.; Nosaka, K.; et al. Potential anti-lymphoma effect of M-CSFR inhibitor in adult T-cell leukemia/lymphoma. J. Clin. Exp. Hematop. 2018, 58, 152–160. [Google Scholar] [CrossRef]
- Li, C.H.; Chen, Z.M.; Chen, P.F.; Meng, L.; Sui, W.N.; Ying, S.C.; Xu, A.M.; Han, W.X. Interleukin-34 promotes the proliferation and epithelial-mesenchymal transition of gastric cancer cells. World J. Gastrointest. Oncol. 2022, 14, 1968–1980. [Google Scholar] [CrossRef]
- Lv, X.; Hu, Y.; Wang, L.; Zhang, D.; Wang, H.; Dai, Y.; Cui, X.; Zheng, G. DDIT4 mediates the proliferation-promotive effect of IL-34 in human monocytic leukemia cells. Blood Sci. 2021, 3, 48–56. [Google Scholar] [CrossRef]
- Arnoletti, J.P.; Reza, J.; Rosales, A.; Monreal, A.; Fanaian, N.; Whisner, S.; Srivastava, M.; Rivera-Otero, J.; Yu, G.; Phanstiel Iv, O.; et al. Pancreatic Ductal Adenocarcinoma (PDAC) circulating tumor cells influence myeloid cell differentiation to support their survival and immunoresistance in portal vein circulation. PloS ONE 2022, 17, e0265725. [Google Scholar] [CrossRef]
- Kajihara, N.; Kitagawa, F.; Kobayashi, T.; Wada, H.; Otsuka, R.; Seino, K.I. Interleukin-34 contributes to poor prognosis in triple-negative breast cancer. Breast Cancer 2020, 27, 1198–1204. [Google Scholar] [CrossRef]
- Poudel, M.; Kim, G.; Bhattarai, P.Y.; Kim, J.Y.; Choi, H.S. Interleukin-34-CSF1R Signaling Axis Promotes Epithelial Cell Transformation and Breast Tumorigenesis. Int. J. Mol. Sci. 2021, 22, 2711. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef] [PubMed]
- Cendrowicz, E.; Sas, Z.; Bremer, E.; Rygiel, T.P. The Role of Macrophages in Cancer Development and Therapy. Cancers 2021, 13, 1946. [Google Scholar] [CrossRef]
- Ruffell, B.; Coussens, L.M. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Anestakis, D.; Petanidis, S.; Domvri, K.; Tsavlis, D.; Zarogoulidis, P.; Katopodi, T. Carboplatin chemoresistance is associated with CD11b(+)/Ly6C(+) myeloid release and upregulation of TIGIT and LAG3/CD160 exhausted T cells. Mol. Immunol. 2020, 118, 99–109. [Google Scholar] [CrossRef]
- Baghdadi, M.; Wada, H.; Nakanishi, S.; Abe, H.; Han, N.; Putra, W.E.; Endo, D.; Watari, H.; Sakuragi, N.; Hida, Y.; et al. Chemotherapy-Induced IL34 Enhances Immunosuppression by Tumor-Associated Macrophages and Mediates Survival of Chemoresistant Lung Cancer Cells. Cancer Res. 2016, 76, 6030–6042. [Google Scholar] [CrossRef]
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.W. NF-kappaB in cancer: From innocent bystander to major culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar] [CrossRef]
- Nakajima, S.; Mimura, K.; Saito, K.; Thar Min, A.K.; Endo, E.; Yamada, L.; Kase, K.; Yamauchi, N.; Matsumoto, T.; Nakano, H.; et al. Neoadjuvant Chemotherapy Induces IL34 Signaling and Promotes Chemoresistance via Tumor-Associated Macrophage Polarization in Esophageal Squamous Cell Carcinoma. Mol. Cancer Res. 2021, 19, 1085–1095. [Google Scholar] [CrossRef]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.H.; Yen, W.C.; Lin, W.H.; Wang, P.C.; Lai, Y.L.; Su, Y.C.; Chang, C.Y.; Wu, C.S.; Huang, Y.C.; Yang, C.M.; et al. Discovery of BPR1R024, an Orally Active and Selective CSF1R Inhibitor that Exhibits Antitumor and Immunomodulatory Activity in a Murine Colon Tumor Model. J. Med. Chem. 2021, 64, 14477–14497. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, C.; Liu, T.; Dai, X.; Bazhin, A.V. Myeloid-Derived Suppressor Cells in Tumors: From Mechanisms to Antigen Specificity and Microenvironmental Regulation. Front. Immunol. 2020, 11, 1371. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Shi, H.; Zhang, B.; Ou, X.; Ma, Q.; Chen, Y.; Shu, P.; Li, D.; Wang, Y. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct. Target. Ther. 2021, 6, 362. [Google Scholar] [CrossRef] [PubMed]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Kajihara, N.; Kobayashi, T.; Otsuka, R.; Nio-Kobayashi, J.; Oshino, T.; Takahashi, M.; Imanishi, S.; Hashimoto, A.; Wada, H.; Seino, K.I. Tumor-derived interleukin-34 creates an immunosuppressive and chemoresistant tumor microenvironment by modulating myeloid-derived suppressor cells in triple-negative breast cancer. Cancer Immunol. Immunother. 2022, 1–14. [Google Scholar] [CrossRef]
- Bezie, S.; Picarda, E.; Ossart, J.; Tesson, L.; Usal, C.; Renaudin, K.; Anegon, I.; Guillonneau, C. IL-34 is a Treg-specific cytokine and mediates transplant tolerance. J. Clin. Investig. 2015, 125, 3952–3964. [Google Scholar] [CrossRef]
- Kim, J.I.; Turka, L.A. Transplant tolerance: A new role for IL-34. J. Clin. Investig. 2015, 125, 3751–3753. [Google Scholar] [CrossRef]
- Gorzo, A.; Galos, D.; Volovat, S.R.; Lungulescu, C.V.; Burz, C.; Sur, D. Landscape of Immunotherapy Options for Colorectal Cancer: Current Knowledge and Future Perspectives beyond Immune Checkpoint Blockade. Life 2022, 12, 229. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef]
- Prima, V.; Kaliberova, L.N.; Kaliberov, S.; Curiel, D.T.; Kusmartsev, S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc. Natl. Acad. Sci. USA 2017, 114, 1117–1122. [Google Scholar] [CrossRef] [Green Version]
- Ugel, S.; De Sanctis, F.; Mandruzzato, S.; Bronte, V. Tumor-induced myeloid deviation: When myeloid-derived suppressor cells meet tumor-associated macrophages. J. Clin. Investig. 2015, 125, 3365–3376. [Google Scholar] [CrossRef] [PubMed]
- Han, N.; Baghdadi, M.; Ishikawa, K.; Endo, H.; Kobayashi, T.; Wada, H.; Imafuku, K.; Hata, H.; Seino, K.I. Enhanced IL-34 expression in Nivolumab-resistant metastatic melanoma. Inflamm. Regen. 2018, 38, 3. [Google Scholar] [CrossRef] [PubMed]
- Hama, N.; Kobayashi, T.; Han, N.; Kitagawa, F.; Kajihara, N.; Otsuka, R.; Wada, H.; Lee, H.K.; Rhee, H.; Hasegawa, Y.; et al. Interleukin-34 Limits the Therapeutic Effects of Immune Checkpoint Blockade. iScience 2020, 23, 101584. [Google Scholar] [CrossRef] [PubMed]
- Han, N.; Jang, H.Y.; Hama, N.; Kobayashi, T.; Otsuka, R.; Wada, H.; Seino, K.I. An optimized protocol for patient-derived xenograft in humanized mice to evaluate the role of IL-34 in immunotherapeutic resistance. STAR Protoc. 2021, 2, 100460. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Yang, Q.; Zhang, Y.; Jiang, Q.; Lin, Y.; Yang, S.; Wang, H.; Cheng, L.; Zhang, X.; Li, Y.; et al. Modulating the Tumor Microenvironment via Oncolytic Viruses and CSF-1R Inhibition Synergistically Enhances Anti-PD-1 Immunotherapy. Mol. Ther. J. Am. Soc. Gene Ther. 2019, 27, 244–260. [Google Scholar] [CrossRef]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef]
- Booker, B.E.; Clark, R.S.; Pellom, S.T.; Adunyah, S.E. Interleukin-34 induces monocytic-like differentiation in leukemia cell lines. Int. J. Biochem. Mol. Biol. 2015, 6, 1–16. [Google Scholar]
- Wang, Z.; Zhu, J.; Wang, T.; Zhou, H.; Wang, J.; Huang, Z.; Zhang, H.; Shi, J. Loss of IL-34 Expression Indicates Poor Prognosis in Patients with Lung Adenocarcinoma. Front. Oncol. 2021, 11, 639724. [Google Scholar] [CrossRef]
- Zins, K.; Heller, G.; Mayerhofer, M.; Schreiber, M.; Abraham, D. Differential prognostic impact of interleukin-34 mRNA expression and infiltrating immune cell composition in intrinsic breast cancer subtypes. Oncotarget 2018, 9, 23126–23148. [Google Scholar] [CrossRef] [Green Version]


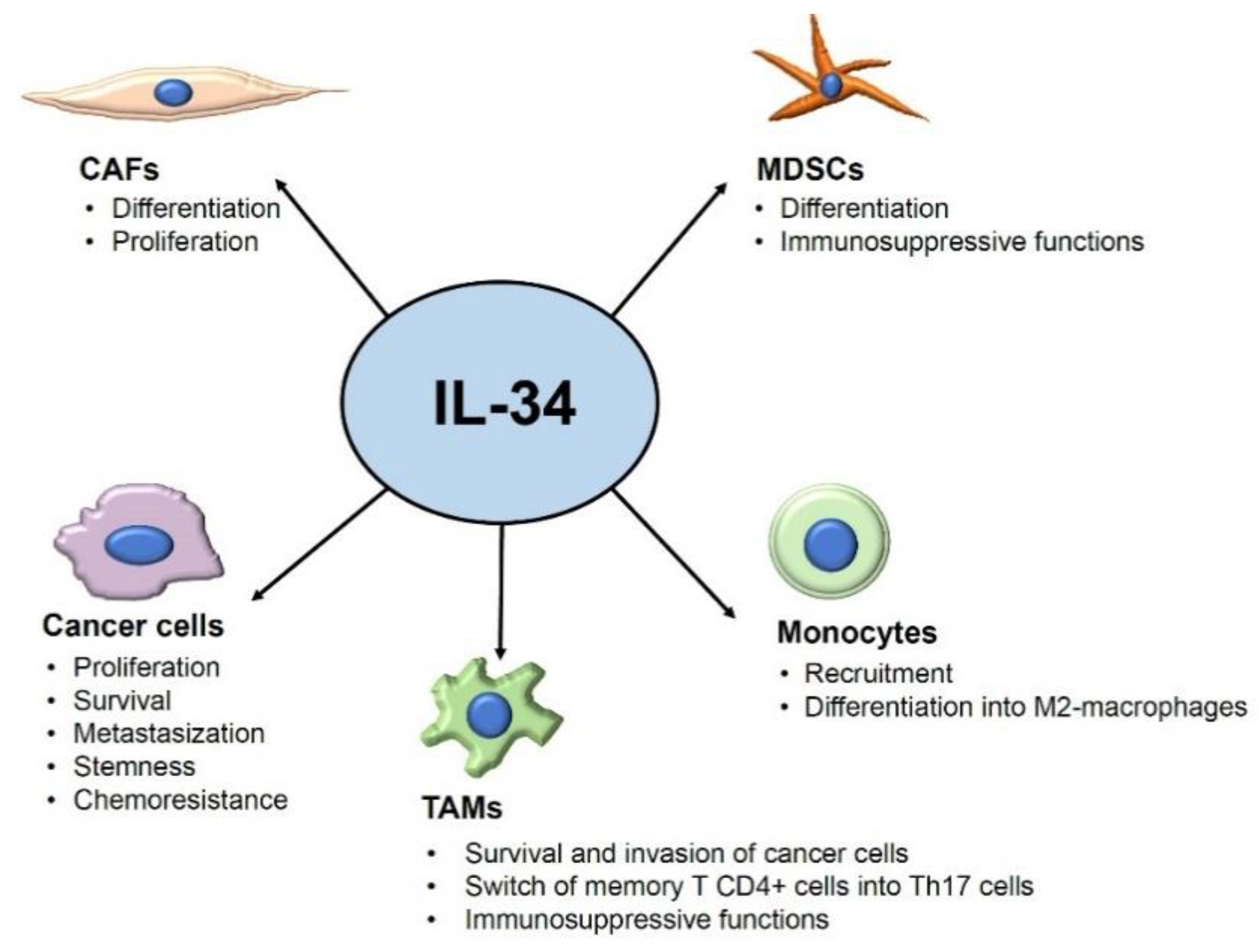{kind=link}
| Cancer | System | IL-34 Functions | References |
|---|---|---|---|
| Colorectal | Cancer cell line | -Increases cell proliferation and invasion -Enhances resistance to oxaliplatin-induced death | [43] |
| TAMs | -Induces type 2 macrophage markers -Enhances IL-6 production | [44] | |
| CAFs | -Promotes CAFs differentiation and proliferation | [45] | |
| Breast | TAMs | -Enhances proliferation, Chemotaxis, and tumor infiltration | [46] |
| Cancer cells | -Enhances metastatic properties | [47] | |
| Ovarian | Macrophages and TAMs | -Promotes the switch of non-Th17 committed memory T cells into conventional Th17 cells | [48] |
| Cancer cells | -Promotes survival of chemoresistant cancer cells | [49] | |
| TAMs | -Enhances the tumorigenic and immunosuppressive functions | [50] | |
| Hepatocellular carcinoma | Cancer cell line | -Increases growth and metastatic properties | [51] |
| TAMs | -Increases TGF-β production | [51] | |
| Hepatoblastoma | Cancer cell line | -Increases cell growth and chemoresistance | [52] |
| TAMs | -Promotes M2 polarization -Stimulates production of IL-6 -Enhances chemotaxis and tumor infiltration | [52] | |
| Cholangiocarcinoma | TAMs | -Induces differentiation and activation and promotes tumor infiltration | [53] |
| Cancer stem cells | -Promotes stemness features | [53] | |
| Osteosarcoma | Cancer cells | -Induces cell proliferation and metastasis | [54] |
| TAMs | -Increases recruitment into tumor tissue | [54] | |
| Endothelial cells | -Stimulates proliferation and vascular cord formation | [54] | |
| Multiple myeloma | CD141+Monocytes | -Accelerates multiple myeloma-induced osteoclast formation | [55] |
| Castration-resistant prostate cancer | TAMs | -Induces differentiation and chemotaxis and tumor infiltration | [56] |
| Adult T-cell leukemia/lymphoma | Cancer cell line | -Increases proliferation | [57] |
| Gastric cancer | Cancer cells line | -Increases proliferation, clone formation, migration, and invasion | [58] |
| Acute monocytic leukemia | Cancer cell line | -Increases proliferation and colony formation | [59] |
| Pancreatic ductal adenocarcinoma | Portal blood MDSCs | -Promotes differentiation and immune suppressive functions | [60] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monteleone, G.; Franzè, E.; Maresca, C.; Colella, M.; Pacifico, T.; Stolfi, C. Targeted Therapy of Interleukin-34 as a Promising Approach to Overcome Cancer Therapy Resistance. Cancers 2023, 15, 971. https://doi.org/10.3390/cancers15030971
Monteleone G, Franzè E, Maresca C, Colella M, Pacifico T, Stolfi C. Targeted Therapy of Interleukin-34 as a Promising Approach to Overcome Cancer Therapy Resistance. Cancers. 2023; 15(3):971. https://doi.org/10.3390/cancers15030971
Chicago/Turabian StyleMonteleone, Giovanni, Eleonora Franzè, Claudia Maresca, Marco Colella, Teresa Pacifico, and Carmine Stolfi. 2023. "Targeted Therapy of Interleukin-34 as a Promising Approach to Overcome Cancer Therapy Resistance" Cancers 15, no. 3: 971. https://doi.org/10.3390/cancers15030971