
Challenges and Future Directions in the Management of Tumor Mutational Burden-High (TMB-H) Advanced Solid Malignancies

Abstract
:Simple Summary
Abstract
1. Background
2. Challenges of Using TMB as Biomarker of Response to ICI
3. Factors in Tumor Microenvironment (TME), TMB, and Response to ICI
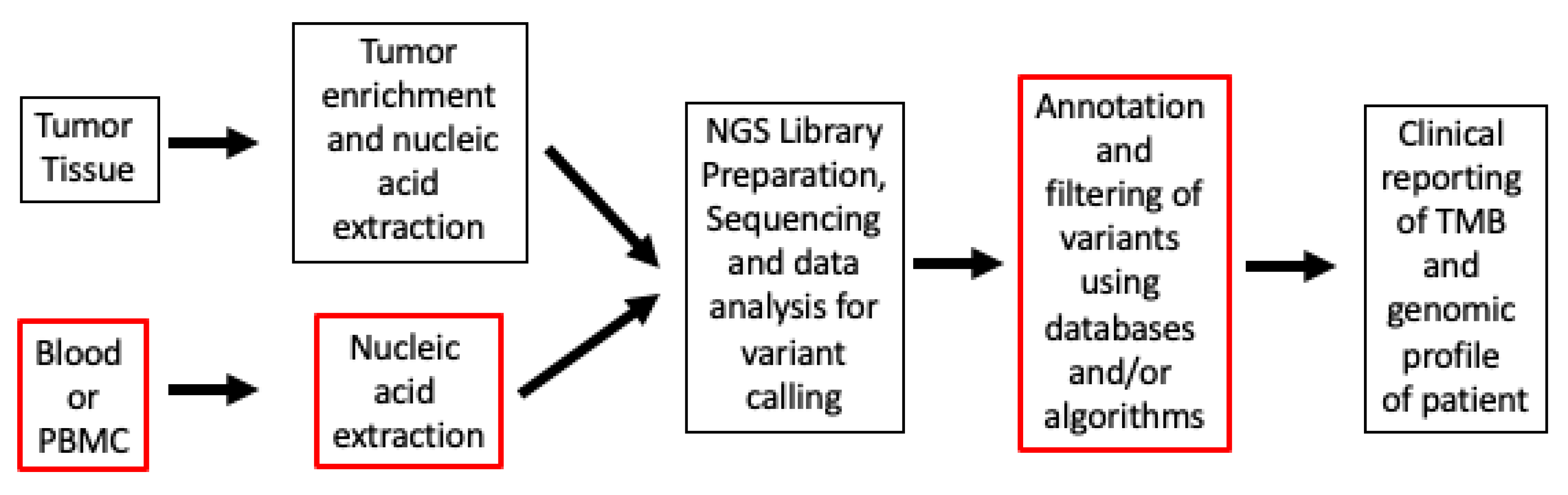
4. Efficacy and Real-World Data on TMB Testing
5. Future Directions
5.1. Standardization Efforts of TMB Reporting: Tumor Mutational Burden (TMB) Harmonization Project
5.2. Integration with Other Biomarkers and Clinical and Genomic Data
5.3. TMB in Blood
5.4. TMB as a Prognostic Biomarker
5.5. Expressed TMB (eTMB)
5.6. Additional Considerations for Future
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Yuza, K.; Nagahashi, M.; Watanabe, S.; Takabe, K.; Wakai, T. Hypermutation and microsatellite instability in gastrointestinal cancers. Oncotarget 2017, 8, 112103. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Powers, S.; Zhu, W.; Hannun, Y.A. Substantial contribution of extrinsic risk factors to cancer development. Nature 2016, 529, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Samstein, R.M.; Lee, C.-H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Heo, Y.J.; Park, S. High tumor mutational burden predicts favorable response to anti-PD-(L) 1 therapy in patients with solid tumor: A real-world pan-tumor analysis. J. Immunother. Cancer 2023, 11, e006454. [Google Scholar] [CrossRef]
- Kang, Y.-J.; O’Haire, S.; Franchini, F.; IJzerman, M.; Zalcberg, J.; Macrae, F.; Canfell, K.; Steinberg, J. A scoping review and meta-analysis on the prevalence of pan-tumour biomarkers (dMMR, MSI, high TMB) in different solid tumours. Sci. Rep. 2022, 12, 20495. [Google Scholar] [CrossRef]
- Shao, C.; Li, G.; Huang, L.; Pruitt, S.; Castellanos, E.; Frampton, G.; Carson, K.R.; Snow, T.; Singal, G.; Fabrizio, D. Prevalence of high tumor mutational burden and association with survival in patients with less common solid tumors. JAMA Netw. Open 2020, 3, e2025109. [Google Scholar] [CrossRef]
- Castle, J.C.; Uduman, M.; Pabla, S.; Stein, R.B.; Buell, J.S. Mutation-derived neoantigens for cancer immunotherapy. Front. Immunol. 2019, 10, 1856. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef]
- Marcus, L.; Fashoyin-Aje, L.A.; Donoghue, M.; Yuan, M.; Rodriguez, L.; Gallagher, P.S.; Philip, R.; Ghosh, S.; Theoret, M.R.; Beaver, J.A. FDA approval summary: Pembrolizumab for the treatment of tumor mutational burden–high solid tumors. Clin. Cancer Res. 2021, 27, 4685–4689. [Google Scholar] [CrossRef]
- Cyll, K.; Ersvær, E.; Vlatkovic, L.; Pradhan, M.; Kildal, W.; Avranden Kjær, M.; Kleppe, A.; Hveem, T.S.; Carlsen, B.; Gill, S. Tumour heterogeneity poses a significant challenge to cancer biomarker research. Br. J. Cancer 2017, 117, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Milbury, C.A.; Creeden, J.; Yip, W.-K.; Smith, D.L.; Pattani, V.; Maxwell, K.; Sawchyn, B.; Gjoerup, O.; Meng, W.; Skoletsky, J. Clinical and analytical validation of FoundationOne® CDx, a comprehensive genomic profiling assay for solid tumors. PLoS ONE 2022, 17, e0264138. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.T.; Mitchell, T.N.; Zehir, A.; Shah, R.H.; Benayed, R.; Syed, A.; Chandramohan, R.; Liu, Z.Y.; Won, H.H.; Scott, S.N. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J. Mol. Diagn. 2015, 17, 251–264. [Google Scholar] [CrossRef]
- Li, Y.; Luo, Y. Optimizing the evaluation of gene-targeted panels for tumor mutational burden estimation. Sci. Rep. 2021, 11, 21072. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, B.; Foote, M.B.; Maron, S.B.; Diplas, B.H.; Lu, S.; Argilés, G.; Cercek, A.; Diaz Jr, L.A. The spectrum of benefit from checkpoint blockade in hypermutated tumors. N. Engl. J. Med. 2021, 384, 1168–1170. [Google Scholar] [CrossRef] [PubMed]
- Schrock, A.; Fabrizio, D.; He, Y.; Chung, J.; Resnick, M.; Stephens, P.; Ross, J.; Miller, V.; Ramkissoon, S.; Elvin, J. Analysis of POLE mutation and tumor mutational burden (TMB) across 80,853 tumors: Implications for immune checkpoint inhibitors (ICPIs). Ann. Oncol. 2017, 28, v415. [Google Scholar] [CrossRef]
- Cristescu, R.; Aurora-Garg, D.; Albright, A.; Xu, L.; Liu, X.Q.; Loboda, A.; Lang, L.; Jin, F.; Rubin, E.H.; Snyder, A. Tumor mutational burden predicts the efficacy of pembrolizumab monotherapy: A pan-tumor retrospective analysis of participants with advanced solid tumors. J. Immunother. Cancer 2022, 10, e003091. [Google Scholar] [CrossRef]
- Mok, T.; Lopes, G.; Cho, B.; Kowalski, D.; Kasahara, K.; Wu, Y.-L.; de Castro Jr, G.; Turna, H.; Cristescu, R.; Aurora-Garg, D. Associations of tissue tumor mutational burden and mutational status with clinical outcomes in KEYNOTE-042: Pembrolizumab versus chemotherapy for advanced PD-L1-positive NSCLC. Ann. Oncol. 2023, 34, 377–388. [Google Scholar] [CrossRef]
- Wu, Y.; Xu, J.; Du, C.; Wu, Y.; Xia, D.; Lv, W.; Hu, J. The predictive value of tumor mutation burden on efficacy of immune checkpoint inhibitors in cancers: A systematic review and meta-analysis. Front. Oncol. 2019, 9, 1161. [Google Scholar] [CrossRef]
- Ning, B.; Liu, Y.; Wang, M.; Li, Y.; Xu, T.; Wei, Y. The predictive value of tumor mutation burden on clinical efficacy of immune checkpoint inhibitors in melanoma: A systematic review and meta-analysis. Front. Pharmacol. 2022, 13, 748674. [Google Scholar] [CrossRef]
- Aggarwal, C.; Ben-Shachar, R.; Gao, Y.; Hyun, S.W.; Rivers, Z.; Epstein, C.; Kaneva, K.; Sangli, C.; Nimeiri, H.; Patel, J. Assessment of Tumor Mutational Burden and Outcomes in Patients with Diverse Advanced Cancers Treated with Immunotherapy. JAMA Netw. Open 2023, 6, e2311181. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Yao, L.; Fu, Y.; Mohiyuddin, M.; Lam, H.Y. ecTMB: A robust method to estimate and classify tumor mutational burden. Sci. Rep. 2020, 10, 4983. [Google Scholar] [CrossRef]
- Fancello, L.; Gandini, S.; Pelicci, P.G.; Mazzarella, L. Tumor mutational burden quantification from targeted gene panels: Major advancements and challenges. J. Immunother. Cancer 2019, 7, 183. [Google Scholar] [CrossRef]
- Sha, D.; Jin, Z.; Budczies, J.; Kluck, K.; Stenzinger, A.; Sinicrope, F.A. Tumor mutational burden as a predictive biomarker in solid tumors. Cancer Discov. 2020, 10, 1808–1825. [Google Scholar] [CrossRef]
- Vega, D.; Yee, L.; McShane, L.; Williams, P.; Chen, L.; Vilimas, T.; Fabrizio, D.; Funari, V.; Newberg, J.; Bruce, L. Aligning tumor mutational burden (TMB) quantification across diagnostic platforms: Phase II of the Friends of Cancer Research TMB Harmonization Project. Ann. Oncol. 2021, 32, 1626–1636. [Google Scholar] [CrossRef] [PubMed]
- Parikh, K.; Huether, R.; White, K.; Hoskinson, D.; Beaubier, N.; Dong, H.; Adjei, A.A.; Mansfield, A.S. Tumor mutational burden from tumor-only sequencing compared with germline subtraction from paired tumor and normal specimens. JAMA Netw. Open 2020, 3, e200202. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.X.; He, Y.; Sanford, E.; Montesion, M.; Frampton, G.M.; Vignot, S.; Soria, J.-C.; Ross, J.S.; Miller, V.A.; Stephens, P.J. A computational approach to distinguish somatic vs. germline origin of genomic alterations from deep sequencing of cancer specimens without a matched normal. PLoS Comput. Biol. 2018, 14, e1005965. [Google Scholar] [CrossRef] [PubMed]
- Fenizia, F.; Pasquale, R.; Abate, R.E.; Lambiase, M.; Roma, C.; Bergantino, F.; Chaudhury, R.; Hyland, F.; Allen, C.; Normanno, N. Challenges in bioinformatics approaches to tumor mutation burden analysis. Oncol. Lett. 2021, 22, 555. [Google Scholar] [CrossRef] [PubMed]
- Mankor, J.M.; Paats, M.S.; Groenendijk, F.H.; Roepman, P.; Dinjens, W.N.; Dubbink, H.J.; Sleijfer, S.; Consortium, C.; Cuppen, E.; Lolkema, M.P. Impact of panel design and cut-off on tumour mutational burden assessment in metastatic solid tumour samples. Br. J. Cancer 2020, 122, 953–956. [Google Scholar] [CrossRef]
- Ruel, L.-J.; Li, Z.; Gaudreault, N.; Henry, C.; Saavedra Armero, V.; Boudreau, D.K.; Zhang, T.; Landi, M.T.; Labbé, C.; Couture, C. Tumor mutational burden by whole-genome sequencing in resected NSCLC of never smokers. Cancer Epidemiol. Biomark. Prev. 2022, 31, 2219–2227. [Google Scholar] [CrossRef] [PubMed]
- Vilimas, T. Measuring tumor mutational burden using whole-exome sequencing. In Biomarkers for Immunotherapy of Cancer: Methods and Protocols; Springer: New York, NY, USA, 2020; pp. 63–91. [Google Scholar]
- Buchhalter, I.; Rempel, E.; Endris, V.; Allgäuer, M.; Neumann, O.; Volckmar, A.L.; Kirchner, M.; Leichsenring, J.; Lier, A.; von Winterfeld, M. Size matters: Dissecting key parameters for panel-based tumor mutational burden analysis. Int. J. Cancer 2019, 144, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Bertl, J.; Zhu, X.; Lam, T.C.; Wu, S.; Shih, D.J.; Wong, J.W. Tumour mutational burden is overestimated by target cancer gene panels. J. Natl. Cancer Cent. 2023, 3, 56–64. [Google Scholar] [CrossRef]
- Budczies, J.; Allgäuer, M.; Litchfield, K.; Rempel, E.; Christopoulos, P.; Kazdal, D.; Endris, V.; Thomas, M.; Fröhling, S.; Peters, S. Optimizing panel-based tumor mutational burden (TMB) measurement. Ann. Oncol. 2019, 30, 1496–1506. [Google Scholar] [CrossRef] [PubMed]
- Conroy, J.M.; Pabla, S.; Glenn, S.T.; Nesline, M.; Burgher, B.; Lenzo, F.L.; Papanicolau-Sengos, A.; Gardner, M.; Morrison, C. Tumor mutational burden (TMB): Assessment of inter-and intra-tumor heterogeneity. J. Clin. Oncol. 2019, 37, 27. [Google Scholar] [CrossRef]
- Schnidrig, D.; Turajlic, S.; Litchfield, K. Tumour mutational burden: Primary versus metastatic tissue creates systematic bias. Immuno-Oncol. Technol. 2019, 4, 8–14. [Google Scholar] [CrossRef]
- Monteran, L.; Ershaid, N.; Sabah, I.; Fahoum, I.; Zait, Y.; Shani, O.; Cohen, N.; Eldar-Boock, A.; Satchi-Fainaro, R.; Erez, N. Bone metastasis is associated with acquisition of mesenchymal phenotype and immune suppression in a model of spontaneous breast cancer metastasis. Sci. Rep. 2020, 10, 13838. [Google Scholar] [CrossRef]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 2017, 171, 934–949.e916. [Google Scholar] [CrossRef]
- Schürch, C.M.; Bhate, S.S.; Barlow, G.L.; Phillips, D.J.; Noti, L.; Zlobec, I.; Chu, P.; Black, S.; Demeter, J.; McIlwain, D.R. Coordinated cellular neighborhoods orchestrate antitumoral immunity at the colorectal cancer invasive front. Cell 2020, 182, 1341–1359.e1319. [Google Scholar] [CrossRef]
- Anagnostou, V.; Niknafs, N.; Marrone, K.; Bruhm, D.C.; White, J.R.; Naidoo, J.; Hummelink, K.; Monkhorst, K.; Lalezari, F.; Lanis, M. Multimodal genomic features predict outcome of immune checkpoint blockade in non-small-cell lung cancer. Nat. Cancer 2020, 1, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Chatrath, A.; Ratan, A.; Dutta, A. Germline variants predictive of tumor mutational burden and immune checkpoint inhibitor efficacy. Iscience 2021, 24. [Google Scholar] [CrossRef]
- Heydt, C.; Rehker, J.; Pappesch, R.; Buhl, T.; Ball, M.; Siebolts, U.; Haak, A.; Lohneis, P.; Büttner, R.; Hillmer, A.M. Analysis of tumor mutational burden: Correlation of five large gene panels with whole exome sequencing. Sci. Rep. 2020, 10, 11387. [Google Scholar] [CrossRef] [PubMed]
- Makrooni, M.A.; O’Sullivan, B.; Seoighe, C. Bias and inconsistency in the estimation of tumour mutation burden. BMC Cancer 2022, 22, 840. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yuan, Y.; Chen, X.; Chen, J.; Lin, S.; Li, X.; Du, H. Systematic comparison of somatic variant calling performance among different sequencing depth and mutation frequency. Sci. Rep. 2020, 10, 3501. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef]
- Giraldo, N.A.; Sanchez-Salas, R.; Peske, J.D.; Vano, Y.; Becht, E.; Petitprez, F.; Validire, P.; Ingels, A.; Cathelineau, X.; Fridman, W.H. The clinical role of the TME in solid cancer. Br. J. Cancer 2019, 120, 45–53. [Google Scholar] [CrossRef]
- Bejarano, L.; Jordāo, M.J.; Joyce, J.A. Therapeutic targeting of the tumor microenvironment. Cancer Discov. 2021, 11, 933–959. [Google Scholar] [CrossRef]
- Mellman, I.; Chen, D.S.; Powles, T.; Turley, S.J. The cancer-immunity cycle: Indication, genotype, and immunotype. Immunity 2023, 56, 2188–2205. [Google Scholar] [CrossRef]
- Nowicki, T.S.; Hu-Lieskovan, S.; Ribas, A. Mechanisms of resistance to PD-1 and PD-L1 blockade. Cancer J. 2018, 24, 47. [Google Scholar] [CrossRef]
- Yan, X.; Zhang, S.; Deng, Y.; Wang, P.; Hou, Q.; Xu, H. Prognostic factors for checkpoint inhibitor based immunotherapy: An update with new evidences. Front. Pharmacol. 2018, 9, 1050. [Google Scholar] [CrossRef]
- Blank, C.U.; Haanen, J.B.; Ribas, A.; Schumacher, T.N. The “cancer immunogram”. Science 2016, 352, 658–660. [Google Scholar] [CrossRef] [PubMed]
- Seliger, B. Combinatorial approaches with checkpoint inhibitors to enhance anti-tumor immunity. Front. Immunol. 2019, 10, 999. [Google Scholar] [CrossRef] [PubMed]
- Song, I.-W.; Vo, H.H.; Chen, Y.-S.; Baysal, M.A.; Kahle, M.; Johnson, A.; Tsimberidou, A.M. Precision Oncology: Evolving Clinical Trials across Tumor Types. Cancers 2023, 15, 1967. [Google Scholar] [CrossRef]
- Rodig, S.; Gusenleitner, D.; Jackson, D.; Gjini, E.; Giobbie-Hurder, A.; Jin, C.; Chang, H.; Lovitch, S.; Horak, C.; Weber, J.S. Association of distinct baseline tissue biomarkers with response to nivolumab (NIVO) and ipilimumab (IPI) in melanoma: CheckMate 064. J. Clin. Oncol. 2017, 35, 9515. [Google Scholar] [CrossRef]
- Chowell, D.; Morris, L.G.; Grigg, C.M.; Weber, J.K.; Samstein, R.M.; Makarov, V.; Kuo, F.; Kendall, S.M.; Requena, D.; Riaz, N. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 2018, 359, 582–587. [Google Scholar] [CrossRef] [PubMed]
- McGrail, D.; Pilié, P.; Rashid, N.; Voorwerk, L.; Slagter, M.; Kok, M.; Jonasch, E.; Khasraw, M.; Heimberger, A.; Lim, B. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann. Oncol. 2021, 32, 661–672. [Google Scholar] [CrossRef]
- Simoni, Y.; Becht, E.; Fehlings, M.; Loh, C.Y.; Koo, S.-L.; Teng, K.W.W.; Yeong, J.P.S.; Nahar, R.; Zhang, T.; Kared, H. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018, 557, 575–579. [Google Scholar] [CrossRef]
- Spranger, S.; Luke, J.J.; Bao, R.; Zha, Y.; Hernandez, K.M.; Li, Y.; Gajewski, A.P.; Andrade, J.; Gajewski, T.F. Density of immunogenic antigens does not explain the presence or absence of the T-cell–inflamed tumor microenvironment in melanoma. Proc. Natl. Acad. Sci. USA 2016, 113, E7759–E7768. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Negrao, M.V.; Skoulidis, F.; Montesion, M.; Schulze, K.; Bara, I.; Shen, V.; Xu, H.; Hu, S.; Sui, D.; Elamin, Y.Y. Oncogene-specific differences in tumor mutational burden, PD-L1 expression, and outcomes from immunotherapy in non-small cell lung cancer. J. Immunother. Cancer 2021, 9, e002891. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, Y.; Ji, Q.; Cai, H.; Liang, X.; Xie, J.; Li, H.; Wang, J.; Zhu, G.; Tian, E. Clinicopathological and molecular characteristics of patients with hypermutant lung cancer: A retrospective cohort study. Oncol. Lett. 2021, 21, 329. [Google Scholar] [CrossRef] [PubMed]
- Offin, M.; Rizvi, H.; Tenet, M.; Ni, A.; Sanchez-Vega, F.; Li, B.T.; Drilon, A.; Kris, M.G.; Rudin, C.M.; Schultz, N. Tumor mutation burden and efficacy of EGFR-tyrosine kinase inhibitors in patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2019, 25, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, C.-b.; Zheng, X.-b.; Gao, X.; Jun, L.; Xiong, J.-n.; Lin, J.; Xu, Y.; Guan, Y.-F.; Li, Y. Association of driver genes with high-tumor mutation burden and outcome in patients with head and neck cancer: Implications for immunotherapy. J. Clin. Oncol. 2020, 38, e18533. [Google Scholar] [CrossRef]
- Sholl, L.M.; Hirsch, F.R.; Hwang, D.; Botling, J.; Lopez-Rios, F.; Bubendorf, L.; Mino-Kenudson, M.; Roden, A.C.; Beasley, M.B.; Borczuk, A. The promises and challenges of tumor mutation burden as an immunotherapy biomarker: A perspective from the International Association for the Study of Lung Cancer Pathology Committee. J. Thorac. Oncol. 2020, 15, 1409–1424. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef]
- Cindy Yang, S.; Lien, S.C.; Wang, B.X.; Clouthier, D.L.; Hanna, Y.; Cirlan, I.; Zhu, K.; Bruce, J.P.; El Ghamrasni, S.; Iafolla, M.A. Pan-cancer analysis of longitudinal metastatic tumors reveals genomic alterations and immune landscape dynamics associated with pembrolizumab sensitivity. Nat. Commun. 2021, 12, 5137. [Google Scholar] [CrossRef]
- Efremova, M.; Finotello, F.; Rieder, D.; Trajanoski, Z. Neoantigens generated by individual mutations and their role in cancer immunity and immunotherapy. Front. Immunol. 2017, 8, 1679. [Google Scholar] [CrossRef]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef]
- Gandara, D.R.; Agarwal, N.; Gupta, S.; Klempner, S.J.; Andrews, M.C.; Mahipal, A.; Subbiah, V.; Eskander, R.N.; Carbone, D.P.; Snider, J. Tumor mutational burden (TMB) measurement from an FDA-approved assay and real-world overall survival (rwOS) on single-agent immune checkpoint inhibitors (ICI) in over 8,000 patients across 24 cancer types. J. Clin. Oncol. 2023, 41, 2503. [Google Scholar] [CrossRef]
- Palmeri, M.; Mehnert, J.; Silk, A.; Jabbour, S.; Ganesan, S.; Popli, P.; Riedlinger, G.; Stephenson, R.; de Meritens, A.; Leiser, A. Real-world application of tumor mutational burden-high (TMB-high) and microsatellite instability (MSI) confirms their utility as immunotherapy biomarkers. ESMO Open 2022, 7, 100336. [Google Scholar] [CrossRef]
- Huang, R.S.; Carbone, D.P.; Li, G.; Schrock, A.; Graf, R.P.; Zhang, L.; Murugesan, K.; Ross, J.S.; Tolba, K.; Sands, J. Durable responders in advanced NSCLC with elevated TMB and treated with 1L immune checkpoint inhibitor: A real-world outcomes analysis. J. Immunother. Cancer 2023, 11, e005801. [Google Scholar] [CrossRef]
- Fernandez, E.M.; Eng, K.; Beg, S.; Beltran, H.; Faltas, B.M.; Mosquera, J.M.; Nanus, D.M.; Pisapia, D.J.; Rao, R.A.; Robinson, B.D. Cancer-specific thresholds adjust for whole exome sequencing–based tumor mutational burden distribution. JCO Precis. Oncol. 2019, 3, 1–12. [Google Scholar] [CrossRef]
- Merino, D.M.; McShane, L.M.; Fabrizio, D.; Funari, V.; Chen, S.-J.; White, J.R.; Wenz, P.; Baden, J.; Barrett, J.C.; Chaudhary, R. Establishing guidelines to harmonize tumor mutational burden (TMB): In silico assessment of variation in TMB quantification across diagnostic platforms: Phase I of the Friends of Cancer Research TMB Harmonization Project. J. Immunother. Cancer 2020, 8, e000147. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Siddiqui, B.A.; Anandhan, S.; Yadav, S.S.; Subudhi, S.K.; Gao, J.; Goswami, S.; Allison, J.P. The next decade of immune checkpoint therapy. Cancer Discov. 2021, 11, 838–857. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, K.; Reading, J.L.; Puttick, C.; Thakkar, K.; Abbosh, C.; Bentham, R.; Watkins, T.B.; Rosenthal, R.; Biswas, D.; Rowan, A. Meta-analysis of tumor-and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell 2021, 184, 596–614.e514. [Google Scholar] [CrossRef]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA damage and repair biomarkers of immunotherapy response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef]
- Pham, T.V.; Boichard, A.; Goodman, A.; Riviere, P.; Yeerna, H.; Tamayo, P.; Kurzrock, R. Role of ultraviolet mutational signature versus tumor mutation burden in predicting response to immunotherapy. Mol. Oncol. 2020, 14, 1680–1694. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Dong, L.; Liu, X.; Ou, K.; Yang, L. POLE/POLD1 mutation and tumor immunotherapy. J. Exp. Clin. Cancer Res. 2022, 41, 216. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Li, W.; Zhang, X.; Ge, M.; Song, C. Correlation between TMB and MSI in patients with solid tumors. J. Clin. Oncol. 2020, 38. [Google Scholar] [CrossRef]
- Goodman, A.M.; Sokol, E.S.; Frampton, G.M.; Lippman, S.M.; Kurzrock, R. Microsatellite-stable tumors with high mutational burden benefit from immunotherapy. Cancer Immunol. Res. 2019, 7, 1570–1573. [Google Scholar] [CrossRef] [PubMed]
- Bielska, A.A.; Chatila, W.K.; Walch, H.; Schultz, N.; Stadler, Z.K.; Shia, J.; Reidy-Lagunes, D.; Yaeger, R. Tumor mutational burden and mismatch repair deficiency discordance as a mechanism of immunotherapy resistance. J. Natl. Compr. Cancer Netw. 2021, 19, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.; Powers, E.; Wu, Y.; Datto, M.B.; Green, M.F.; Strickler, J.H.; Ready, N.E.; Zhang, T.; Clarke, J.M. Predictive value of combining biomarkers for clinical outcomes in advanced non-small cell lung cancer patients receiving immune checkpoint inhibitors. Clin. Lung Cancer 2021, 22, 500–509. [Google Scholar] [CrossRef]
- Yarchoan, M.; Albacker, L.A.; Hopkins, A.C.; Montesion, M.; Murugesan, K.; Vithayathil, T.T.; Zaidi, N.; Azad, N.S.; Laheru, D.A.; Frampton, G.M. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight 2019, 4, e126908. [Google Scholar] [CrossRef]
- Zhou, K.I.; Peterson, B.; Serritella, A.; Thomas, J.; Reizine, N.; Moya, S.; Tan, C.; Wang, Y.; Catenacci, D.V. Spatial and temporal heterogeneity of PD-L1 expression and tumor mutational burden in gastroesophageal adenocarcinoma at baseline diagnosis and after chemotherapy. Clin. Cancer Res. 2020, 26, 6453–6463. [Google Scholar] [CrossRef]
- Carbone, D.P.; Reck, M.; Paz-Ares, L.; Creelan, B.; Horn, L.; Steins, M.; Felip, E.; van den Heuvel, M.M.; Ciuleanu, T.-E.; Badin, F. First-line nivolumab in stage IV or recurrent non–small-cell lung cancer. N. Engl. J. Med. 2017, 376, 2415–2426. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.-W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E. Nivolumab plus ipilimumab in advanced non–small-cell lung cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef]
- Ready, N.; Hellmann, M.D.; Awad, M.M.; Otterson, G.A.; Gutierrez, M.; Gainor, J.F.; Borghaei, H.; Jolivet, J.; Horn, L.; Mates, M. First-line nivolumab plus ipilimumab in advanced non–small-cell lung cancer (CheckMate 568): Outcomes by programmed death ligand 1 and tumor mutational burden as biomarkers. J. Clin. Oncol. 2019, 37, 992. [Google Scholar] [CrossRef]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade–based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef]
- Sautès-Fridman, C.; Petitprez, F.; Calderaro, J.; Fridman, W.H. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Trüb, M.; Zippelius, A. Tertiary lymphoid structures as a predictive biomarker of response to cancer immunotherapies. Front. Immunol. 2021, 12, 674565. [Google Scholar] [CrossRef]
- Pagliarulo, F.; Cheng, P.F.; Brugger, L.; van Dijk, N.; van den Heijden, M.; Levesque, M.P.; Silina, K.; van den Broek, M. Molecular, immunological, and clinical features associated with lymphoid neogenesis in muscle invasive bladder cancer. Front. Immunol. 2022, 12, 793992. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Berglund, A.; Zemp, L.; Dhillon, J.; Putney, R.; Kim, Y.; Jain, R.K.; Grass, G.D.; Conejo-Garcia, J.; Mulé, J.J. The 12-CK score: Global measurement of tertiary lymphoid structures. Front. Immunol. 2021, 12, 694079. [Google Scholar] [CrossRef] [PubMed]
- Brunet, M.; Vanhersecke, L.; Loarer, F.L.; Soubeyran, I.; Italiano, A. Prevalence of mature tertiary lymphoid structures and association with tumor mutational burden in patients with solid tumors. Cancer Res. 2023, 83, 2361. [Google Scholar] [CrossRef]
- Jin, J.; Yang, L.; Liu, D.; Li, W. Association of the neutrophil to lymphocyte ratio and clinical outcomes in patients with lung cancer receiving immunotherapy: A meta-analysis. BMJ Open 2020, 10, e035031. [Google Scholar] [CrossRef] [PubMed]
- Parikh, K.; Kumar, A.; Ahmed, J.; Anwar, A.; Puccio, C.; Chun, H.; Fanucchi, M.; Lim, S.H. Peripheral monocytes and neutrophils predict response to immune checkpoint inhibitors in patients with metastatic non-small cell lung cancer. Cancer Immunol. Immunother. 2018, 67, 1365–1370. [Google Scholar] [CrossRef]
- Valero, C.; Lee, M.; Hoen, D.; Weiss, K.; Kelly, D.W.; Adusumilli, P.S.; Paik, P.K.; Plitas, G.; Ladanyi, M.; Postow, M.A. Pretreatment neutrophil-to-lymphocyte ratio and mutational burden as biomarkers of tumor response to immune checkpoint inhibitors. Nat. Commun. 2021, 12, 729. [Google Scholar] [CrossRef]
- Li, M.; Gao, X.; Wang, X. Identification of tumor mutation burden-associated molecular and clinical features in cancer by analyzing multi-omics data. Front. Immunol. 2023, 14, 1090838. [Google Scholar] [CrossRef]
- Wang, W.; Liao, W.; Zhen, B.; Ji, T.; Du, C.; Cheng, S.; Chen, Y.; Yang, J. A study of tumor neoantigen burden and HLA-LOH by whole-exome sequencing to characterize immune biomarkers of lung cancer. J. Clin. Oncol. 2023, 41, 8527. [Google Scholar] [CrossRef]
- Shim, J.; Kim, H.; Cha, H.; Kim, S.; Kim, T.; Anagnostou, V.; Choi, Y.-L.; Jung, H.; Sun, J.-M.; Ahn, J. HLA-corrected tumor mutation burden and homologous recombination deficiency for the prediction of response to PD-(L) 1 blockade in advanced non-small-cell lung cancer patients. Ann. Oncol. 2020, 31, 902–911. [Google Scholar] [CrossRef] [PubMed]
- Rempel, E.; Kluck, K.; Beck, S.; Ourailidis, I.; Kazdal, D.; Neumann, O.; Volckmar, A.; Kirchner, M.; Goldschmid, H.; Pfarr, N. Pan-cancer analysis of genomic scar patterns caused by homologous repair deficiency (HRD). NPJ Precis. Oncol. 2022, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Xiong, D.; Wang, X.; Liu, H.; Wang, T. Mapping the functional landscape of T cell receptor repertoires by single-T cell transcriptomics. Nat. Methods 2021, 18, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Aran, A.; Garrigós, L.; Curigliano, G.; Cortés, J.; Martí, M. Evaluation of the TCR repertoire as a predictive and prognostic biomarker in cancer: Diversity or clonality? Cancers 2022, 14, 1771. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Huang, J.; Ao, X.; Guo, W.; Chen, Y.; Lu, D.; Lv, Z.; Tan, X.; He, W.; Jiang, M. TMB and TCR are correlated indicators predictive of the efficacy of neoadjuvant chemotherapy in breast cancer. Front. Oncol. 2021, 11, 740427. [Google Scholar] [CrossRef]
- Sun, J.; Chen, F.; Wu, G. Potential effects of gut microbiota on host cancers: Focus on immunity, DNA damage, cellular pathways, and anticancer therapy. ISME J. 2023, 17, 1535–1551. [Google Scholar] [CrossRef]
- Chang, A.E.; Golob, J.L.; Schmidt, T.M.; Peltier, D.C.; Lao, C.D.; Tewari, M. Targeting the gut microbiome to mitigate immunotherapy-induced colitis in cancer. Trends Cancer 2021, 7, 583–593. [Google Scholar] [CrossRef]
- Li, X.; Zhang, S.; Guo, G.; Han, J.; Yu, J. Gut microbiome in modulating immune checkpoint inhibitors. EBioMedicine 2022, 82, 104163. [Google Scholar] [CrossRef]
- Bartha, Á.; Győrffy, B. Comprehensive outline of whole exome sequencing data analysis tools available in clinical oncology. Cancers 2019, 11, 1725. [Google Scholar] [CrossRef]
- Schwarze, K.; Buchanan, J.; Taylor, J.C.; Wordsworth, S. Are whole-exome and whole-genome sequencing approaches cost-effective? A systematic review of the literature. Genet. Med. 2018, 20, 1122–1130. [Google Scholar] [CrossRef]
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N. Evolution of neoantigen landscape during immune checkpoint blockade in non–small cell lung cancer. Cancer Discov. 2017, 7, 264–276. [Google Scholar] [CrossRef]
- Garassino, M.C.; Gadgeel, S.; Novello, S.; Halmos, B.; Felip, E.; Speranza, G.; Hui, R.; Garon, E.B.; Horinouchi, H.; Sugawara, S. Associations of tissue tumor mutational burden and mutational status with clinical outcomes with pembrolizumab plus chemotherapy versus chemotherapy for metastatic NSCLC. JTO Clin. Res. Rep. 2023, 4, 100431. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Schilling, B.; Liu, D.; Sucker, A.; Livingstone, E.; Jerby-Arnon, L.; Zimmer, L.; Gutzmer, R.; Satzger, I.; Loquai, C. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat. Med. 2019, 25, 1916–1927. [Google Scholar] [CrossRef] [PubMed]
- Okamura, R.; Kato, S.; Lee, S.; Jimenez, R.E.; Sicklick, J.K.; Kurzrock, R. ARID1A alterations function as a biomarker for longer progression-free survival after anti-PD-1/PD-L1 immunotherapy. J. Immunother. Cancer 2020, 8, e000438. [Google Scholar] [CrossRef]
- Goswami, S.; Chen, Y.; Anandhan, S.; Szabo, P.M.; Basu, S.; Blando, J.M.; Liu, W.; Zhang, J.; Natarajan, S.M.; Xiong, L. ARID1A mutation plus CXCL13 expression act as combinatorial biomarkers to predict responses to immune checkpoint therapy in mUCC. Sci. Transl. Med. 2020, 12, eabc4220. [Google Scholar] [CrossRef]
- Romero, J.M.; Titmuss, E.; Wang, Y.; Vafiadis, J.; Pacis, A.; Jang, G.H.; Zhang, A.; Golesworthy, B.; Lenko, T.; Williamson, L.M. Chemokine expression predicts T cell-inflammation and improved survival with checkpoint inhibition across solid cancers. NPJ Precis. Oncol. 2023, 7, 73. [Google Scholar] [CrossRef]
- Wang, X.; Li, M. Correlate tumor mutation burden with immune signatures in human cancers. BMC Immunol. 2019, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Park, C.-K.; Jun, H.R.; Oh, H.-J.; Lee, J.-Y.; Cho, H.-J.; Kim, Y.-C.; Lee, J.E.; Yoon, S.H.; Choi, C.M.; Lee, J.C. Evaluation of Blood Tumor Mutation Burden for the Efficacy of Second-Line Atezolizumab Treatment in Non-Small Cell Lung Cancer: Buddy Trial. Cells 2023, 12, 1246. [Google Scholar] [CrossRef]
- Chae, Y.K.; Davis, A.A.; Agte, S.; Pan, A.; Simon, N.I.; Iams, W.T.; Cruz, M.R.; Tamragouri, K.; Rhee, K.; Mohindra, N. Clinical implications of circulating tumor DNA tumor mutational burden (ctDNA TMB) in non-small cell lung cancer. Oncologist 2019, 24, 820–828. [Google Scholar] [CrossRef]
- Sturgill, E.G.; Misch, A.; Jones, C.C.; Luckett, D.; Fu, X.; Schlauch, D.; Jones, S.F.; Burris III, H.A.; Spigel, D.R.; McKenzie, A.J. Discordance in tumor mutation burden from blood and tissue affects association with response to immune checkpoint inhibition in real-world settings. Oncologist 2022, 27, 175–182. [Google Scholar] [CrossRef]
- Fehrenbacher, L.; Spira, A.; Ballinger, M.; Kowanetz, M.; Vansteenkiste, J.; Mazieres, J.; Park, K.; Smith, D.; Artal-Cortes, A.; Lewanski, C. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): A multicentre, open-label, phase 2 randomised controlled trial. Lancet 2016, 387, 1837–1846. [Google Scholar] [CrossRef] [PubMed]
- Gandara, D.R.; Paul, S.M.; Kowanetz, M.; Schleifman, E.; Zou, W.; Li, Y.; Rittmeyer, A.; Fehrenbacher, L.; Otto, G.; Malboeuf, C. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat. Med. 2018, 24, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; Von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Mazieres, J.; Rittmeyer, A.; Gadgeel, S.; Hida, T.; Gandara, D.R.; Cortinovis, D.L.; Barlesi, F.; Yu, W.; Matheny, C.; Ballinger, M. Atezolizumab versus docetaxel in pretreated patients with NSCLC: Final results from the randomized phase 2 POPLAR and phase 3 OAK clinical trials. J. Thorac. Oncol. 2021, 16, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Dziadziuszko, R.; Morabito, A.; Felip, E.; Gadgeel, S.M.; Cheema, P.; Cobo, M.; Andric, Z.; Barrios, C.H.; Yamaguchi, M. Atezolizumab versus chemotherapy in advanced or metastatic NSCLC with high blood-based tumor mutational burden: Primary analysis of BFAST cohort C randomized phase 3 trial. Nat. Med. 2022, 28, 1831–1839. [Google Scholar] [CrossRef]
- Kim, E.S.; Velcheti, V.; Mekhail, T.; Yun, C.; Shagan, S.M.; Hu, S.; Chae, Y.K.; Leal, T.A.; Dowell, J.E.; Tsai, M.L. Blood-based tumor mutational burden as a biomarker for atezolizumab in non-small cell lung cancer: The phase 2 B-F1RST trial. Nat. Med. 2022, 28, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Si, H.; Kuziora, M.; Quinn, K.J.; Helman, E.; Ye, J.; Liu, F.; Scheuring, U.; Peters, S.; Rizvi, N.A.; Brohawn, P.Z. A blood-based assay for assessment of tumor mutational burden in first-line metastatic NSCLC treatment: Results from the MYSTIC study. Clin. Cancer Res. 2021, 27, 1631–1640. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Cho, B.C.; Reinmuth, N.; Lee, K.H.; Luft, A.; Ahn, M.-J.; Van Den Heuvel, M.M.; Cobo, M.; Vicente, D.; Smolin, A. Durvalumab with or without tremelimumab vs standard chemotherapy in first-line treatment of metastatic non–small cell lung cancer: The MYSTIC phase 3 randomized clinical trial. JAMA Oncol. 2020, 6, 661–674. [Google Scholar] [CrossRef]
- Zhang, N.; Zhang, J.; Wang, G.; He, X.; Mi, Y.; Cao, Y.; Yu, X. Predictive Efficacy of Blood-Based Tumor Mutation Burden Assay for Immune Checkpoint Inhibitors Therapy in Non-Small Cell Lung Cancer: A Systematic Review and Meta-Analysis. Front. Oncol. 2022, 12, 795933. [Google Scholar] [CrossRef]
- Baden, J.; Chang, H.; Greenawalt, D.; Kirov, S.; Pant, S.; Seminara, A.; Srinivasan, S.; Green, G. Comparison of platforms for determining tumor mutational burden (TMB) from blood samples in patients with non-small cell lung cancer (NSCLC). Ann. Oncol. 2019, 30, v28. [Google Scholar] [CrossRef]
- Fridland, S.; Choi, J.; Nam, M.; Schellenberg, S.J.; Kim, E.; Lee, G.; Yoon, N.; Chae, Y.K. Assessing tumor heterogeneity: Integrating tissue and circulating tumor DNA (ctDNA) analysis in the era of immuno-oncology-blood TMB is not the same as tissue TMB. J. Immunother. Cancer 2021, 9, e002551. [Google Scholar] [CrossRef]
- Wu, H.-X.; Wang, Z.-X.; Zhao, Q.; Chen, D.-L.; He, M.-M.; Yang, L.-P.; Wang, Y.-N.; Jin, Y.; Ren, C.; Luo, H.-Y. Tumor mutational and indel burden: A systematic pan-cancer evaluation as prognostic biomarkers. Ann. Transl. Med. 2019, 7, 640. [Google Scholar] [CrossRef] [PubMed]
- Riviere, P.; Goodman, A.M.; Okamura, R.; Barkauskas, D.A.; Whitchurch, T.J.; Lee, S.; Khalid, N.; Collier, R.; Mareboina, M.; Frampton, G.M. High tumor mutational burden correlates with longer survival in immunotherapy-naïve patients with diverse cancers. Mol. Cancer Ther. 2020, 19, 2139–2145. [Google Scholar] [CrossRef]
- Forde, P.M.; Spicer, J.; Lu, S.; Provencio, M.; Mitsudomi, T.; Awad, M.M.; Felip, E.; Broderick, S.R.; Brahmer, J.R.; Swanson, S.J. Neoadjuvant nivolumab plus chemotherapy in resectable lung cancer. N. Engl. J. Med. 2022, 386, 1973–1985. [Google Scholar] [CrossRef]
- Sorokin, M.; Gorelyshev, A.; Efimov, V.; Zotova, E.; Zolotovskaia, M.; Rabushko, E.; Kuzmin, D.; Seryakov, A.; Kamashev, D.; Li, X. RNA sequencing data for FFPE tumor blocks can be used for robust estimation of tumor mutation burden in individual biosamples. Front. Oncol. 2021, 11, 732644. [Google Scholar] [CrossRef] [PubMed]
- Jessen, E.; Liu, Y.; Davila, J.; Kocher, J.-P.; Wang, C. Determining mutational burden and signature using RNA-seq from tumor-only samples. BMC Med. Genom. 2021, 14, 65. [Google Scholar] [CrossRef]
- Katzir, R.; Rudberg, N.; Yizhak, K. Estimating tumor mutational burden from RNA-sequencing without a matched-normal sample. Nat. Commun. 2022, 13, 3092. [Google Scholar] [CrossRef] [PubMed]
- Valero, C.; Lee, M.; Hoen, D.; Zehir, A.; Berger, M.F.; Seshan, V.E.; Chan, T.A.; Morris, L.G. Response rates to anti–PD-1 immunotherapy in microsatellite-stable solid tumors with 10 or more mutations per megabase. JAMA Oncol. 2021, 7, 739–743. [Google Scholar] [CrossRef]
- Mo, S.-F.; Cai, Z.-Z.; Kuai, W.-H.; Li, X.; Chen, Y.-T. Universal cutoff for tumor mutational burden in predicting the efficacy of anti-PD-(L) 1 therapy for advanced cancers. Front. Cell Dev. Biol. 2023, 11, 1209243. [Google Scholar] [CrossRef]
- Vokes, N.I.; Liu, D.; Ricciuti, B.; Jimenez-Aguilar, E.; Rizvi, H.; Dietlein, F.; He, M.X.; Margolis, C.A.; Elmarakeby, H.A.; Girshman, J. Harmonization of tumor mutational burden quantification and association with response to immune checkpoint blockade in non–small-cell lung cancer. JCO Precis. Oncol. 2019, 3, 1–12. [Google Scholar] [CrossRef]
- Ricciuti, B.; Wang, X.; Alessi, J.V.; Rizvi, H.; Mahadevan, N.R.; Li, Y.Y.; Polio, A.; Lindsay, J.; Umeton, R.; Sinha, R. Association of high tumor mutation burden in non–small cell lung cancers with increased immune infiltration and improved clinical outcomes of PD-L1 blockade across PD-L1 expression levels. JAMA Oncol. 2022, 8, 1160–1168. [Google Scholar] [CrossRef]
- Wood, M.A.; Weeder, B.R.; David, J.K.; Nellore, A.; Thompson, R.F. Burden of tumor mutations, neoepitopes, and other variants are weak predictors of cancer immunotherapy response and overall survival. Genome Med. 2020, 12, 33. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Ganguly, A.; Chatterjee, K.; Spada, S.; Mukherjee, S. Targets of Immune Escape Mechanisms in Cancer: Basis for Development and Evolution of Cancer Immune Checkpoint Inhibitors. Biology 2023, 12, 218. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo Gutiérrez, E.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Hong, Z.; Zhang, C.; Wang, L.; Han, Z.; Ma, D. Immune checkpoint therapy for solid tumours: Clinical dilemmas and future trends. Signal Transduct. Target. Ther. 2023, 8, 320. [Google Scholar] [CrossRef] [PubMed]
- Klein, O.; Kee, D.; Markman, B.; Carlino, M.S.; Underhill, C.; Palmer, J.; Power, D.; Cebon, J.; Behren, A. Evaluation of TMB as a predictive biomarker in patients with solid cancers treated with anti-PD-1/CTLA-4 combination immunotherapy. Cancer Cell 2021, 39, 592–593. [Google Scholar] [CrossRef]
- Adashek, J.J.; Kato, S.; Pabla, S.; Nesline, M.; Conroy, J.M.; Subbiah, V.; DePietro, P.; Kurzrock, R. LAG3 transcriptomic expression correlates with high levels of PD-1, PD-L1, PD-L2, and CTLA-4 checkpoints and with high tumor mutational burden across cancers. J. Clin. Oncol. 2022, 40, 2561. [Google Scholar] [CrossRef]
- Mezheyeuski, A.; Backman, M.; Mattsson, J.; Martín-Bernabé, A.; Larsson, C.; Hrynchyk, I.; Hammarström, K.; Ström, S.; Ekström, J.; Mauchanski, S. An immune score reflecting pro-and anti-tumoural balance of tumour microenvironment has major prognostic impact and predicts immunotherapy response in solid cancers. EBioMedicine 2023, 88, 104452. [Google Scholar] [CrossRef]
- Forde, P.M.; Chaft, J.E.; Smith, K.N.; Anagnostou, V.; Cottrell, T.R.; Hellmann, M.D.; Zahurak, M.; Yang, S.C.; Jones, D.R.; Broderick, S. Neoadjuvant PD-1 blockade in resectable lung cancer. N. Engl. J. Med. 2018, 378, 1976–1986. [Google Scholar] [CrossRef]
- Wang, H.-C.; Moi, S.-H.; Chan, L.-P.; Wu, C.-C.; Du, J.-S.; Liu, P.-L.; Chou, M.-C.; Wu, C.-W.; Huang, C.-J.; Hsiao, H.-H. The role of the genomic mutation signature and tumor mutation burden on relapse risk prediction in head and neck squamous cell carcinoma after concurrent chemoradiotherapy. Exp. Mol. Med. 2023, 55, 926–938. [Google Scholar] [CrossRef]
- Kwiatkowski, D.J.; Rusch, V.W.; Chaft, J.E.; Johnson, B.E.; Nicholas, A.; Wistuba, I.I.; Merritt, R.; Lee, J.M.; Bunn, P.A.; Tang, Y. Neoadjuvant atezolizumab in resectable non-small cell lung cancer (NSCLC): Interim analysis and biomarker data from a multicenter study (LCMC3). J. Clin. Oncol. 2019, 37, 8503. [Google Scholar] [CrossRef]
- Niknafs, N.; Balan, A.; Cherry, C.; Hummelink, K.; Monkhorst, K.; Shao, X.M.; Belcaid, Z.; Marrone, K.A.; Murray, J.; Smith, K.N. Persistent mutation burden drives sustained anti-tumor immune responses. Nat. Med. 2023, 29, 440–449. [Google Scholar] [CrossRef]
- Wang, J.; Chen, P.; Su, M.; Zhong, G.; Zhang, S.; Gou, D. Integrative modeling of multiomics data for predicting tumor mutation burden in patients with lung cancer. BioMed Res. Int. 2022, 2022, 2698190. [Google Scholar] [CrossRef]
- Jain, M.S.; Massoud, T.F. Predicting tumour mutational burden from histopathological images using multiscale deep learning. Nat. Mach. Intell. 2020, 2, 356–362. [Google Scholar] [CrossRef]
- Lam, L.H.T.; Chu, N.T.; Tran, T.-O.; Do, D.T.; Le, N.Q.K. A radiomics-based machine learning model for prediction of tumor mutational burden in lower-grade gliomas. Cancers 2022, 14, 3492. [Google Scholar] [CrossRef]
- Brawley, O.W.; Luhn, P.; Reese-White, D.; Ogbu, U.C.; Madhavan, S.; Wilson, G.; Cox, M.; Ewing, A.; Hammer, C.; Richie, N. Disparities in Tumor Mutational Burden, Immunotherapy Use, and Outcomes Based on Genomic Ancestry in Non–Small-Cell Lung Cancer. JCO Glob. Oncol. 2021, 7, 1537–1546. [Google Scholar] [CrossRef] [PubMed]
- Asmann, Y.W.; Parikh, K.; Bergsagel, P.L.; Dong, H.; Adjei, A.A.; Borad, M.J.; Mansfield, A.S. Inflation of tumor mutation burden by tumor-only sequencing in under-represented groups. NPJ Precis. Oncol. 2021, 5, 22. [Google Scholar] [CrossRef]
- Hsiehchen, D.; Espinoza, M.; Valero, C.; Ahn, C.; Morris, L.G. Impact of tumor mutational burden on checkpoint inhibitor drug eligibility and outcomes across racial groups. J. Immunother. Cancer 2021, 9, e003683. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, N.J.; Eghtesad, M.; Kadri, S.; Cursio, J.; Ritterhouse, L.; Segal, J.; Husain, A.; Patel, J.D. Fewer actionable mutations but higher tumor mutational burden characterizes NSCLC in black patients at an urban academic medical center. Oncotarget 2019, 10, 5817. [Google Scholar] [CrossRef]
- Nassar, A.H.; Adib, E.; Abou Alaiwi, S.; El Zarif, T.; Groha, S.; Akl, E.W.; Nuzzo, P.V.; Mouhieddine, T.H.; Perea-Chamblee, T.; Taraszka, K. Ancestry-driven recalibration of tumor mutational burden and disparate clinical outcomes in response to immune checkpoint inhibitors. Cancer Cell 2022, 40, 1161–1172. [Google Scholar] [CrossRef]
- Don, J.; Byron, S.A.; Zhang, G.; Izatt, T.; Zhang, J.; Davis, B.; Turner, B.; Keats, J.J.; Trent, J.M.; Rodriguez-Rodriguez, L. Abstract A006: Increased germline mutational burden in individuals of African ancestry: Implications for interpretation of tumor mutation burden. Cancer Epidemiol. Biomark. Prev. 2023, 32, A006. [Google Scholar] [CrossRef]
- Florez, M.A.; Kemnade, J.O.; Chen, N.; Du, W.; Sabichi, A.L.; Wang, D.Y.; Huang, Q.; Miller-Chism, C.N.; Jotwani, A.; Chen, A.C. Persistent Ethnicity-Associated Disparity in Antitumor Effectiveness of Immune Checkpoint Inhibitors Despite Equal Access. Cancer Res. Commun. 2022, 2, 806–813. [Google Scholar] [CrossRef]
- Morad, G.; Helmink, B.A.; Sharma, P.; Wargo, J.A. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell 2021, 184, 5309–5337. [Google Scholar] [CrossRef] [PubMed]
- Siu, L.L.; Ivy, S.P.; Dixon, E.L.; Gravell, A.E.; Reeves, S.A.; Rosner, G.L. Challenges and opportunities in adapting clinical trial design for immunotherapies. Clin. Cancer Res. 2017, 23, 4950–4958. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| (A) Challenges associated with TMB assessment | |
| Tumor/Biopsy Specific | Cold vs. hot, tumor purity, intra- and inter-tumor heterogeneity, type of biopsy and tumor tissue (fresh versus archival, FFPE, ctDNA), tumor cell quantification, tumor enrichment |
| Assay Specific | Composition, platform, coverage, depth, mutation calling, Input, Indexes, Sensitivity, Specificity, sample and library preparation and conversion, sequencing, exon coverage, data analysis |
| Bioinformatics | Variation in bioinformatic platforms and TMB (germline variants, deduction of driver mutations, CH, resistance genes), VAF, variant calling, Cutoff validation, QC |
| Interpretation of results | Variations in TMB thresholds, variation in patient populations including race/assays used/cancer treatments in different clinical trials, turnaround time, scoring failure rates, intra-patient factors |
| (B) Potential solutions | |
| Factors affecting TMB measurement | Potential Solution |
| Assay Specific | |
| Panel Size | Minimum of 1 Mb of coding sequence is required for reliable measurement |
| Mutations to be counted for measurement of TMB | Identification of true somatic mutations |
| Robust identification of mutations through NGS assay (specimen quality, sequencing depth) | |
| Minimum tumor tissue content | Tumor tissue enrichment (FoundationOne®CDx optimum 30% tumor content) |
| Bioinformatics Algorithm | |
| Somatic mutation identification | Removal of germline mutation using large population genome sequencing databases |
| Using algorithms to further identity somatic mutations | |
| Removal of pathogenic mutations | |
| Removal of germline variants from rare populations | Calibrating the cut-off values for germline identification from population variant databases. |
| Interpretation of Results | |
| Histology and clinical-context-specific interpretation of TMB values | Using disease-specific stratification of TMB-high patients |
| Different cut-offs for different treatments and histologies | Universal/histology-specific cut-offs |
| References: [4,12,25,26,27,28,29,30] | |
| Clinical Trial | Treatment | Phase/Design | Stage of Cancer | TMB Cut-off (mut/Mb) | TMB Assay | Results | Additional Points |
|---|---|---|---|---|---|---|---|
| POPLAR trial NCT01903993 [122,123] | Atezolizumab vs. Docetaxel | Phase 2 randomized controlled trial | Previously treated, Stage IIIB or IVNSCLC | ≥10 to ≥20 | bTMB assay compared to tTMB using Foundation-ACT (FACT) (targets 1.1 Mb coding region) | PFS HR 0.68, 0.57, 0.58 at bTMB cut-offs ≥10, ≥16 and ≥20; OS HR 0.59, 0.56, 0.51 at bTMB cut-offs ≥10, ≥16 and ≥20. mOS 12.6 vs. 9.7 mo in Atezolizumab vs. decetaxel (HR 0.76) At bTMB cut-off of ≥16, mPFS 4.2 vs. 2.9 mo and mOS 13 vs. 7.4 mo in atezolizumab vs. docetaxel | Training set; PPA 64%, NPA 88%. Spearman rho 0.64 |
| OAK Trial NCT02008227 [124,125] | Atezolizumab vs. Docetaxel | Phase 3 randomized controlled trial | Previously treated, Stage IIIB or IV NSCLC | ≥10 to ≥20 | bTMB assay compared to FoundationACT (FACT) (targets 1.1 Mb coding region) | mOS 13.3 vs. 9.8 mo HR = 0.78. | Validation set; significant PFS benefit for bTMB ≥16 (HR 0.65, p = 0.013) |
| BFAST Cohort C NCT03178552 [126] | Atezolizumab vs. Platinum Chemotherapy | Phase 3 | First line Stage IIIB–IV NSCLC | ≥16 | Foundation Medicine bTMB Clinical Trial Assay (targets 1.1 Mb coding region) * | mFPS 4.5 vs. 4.3 mo, mOS 13.3 vs. 10.3 mo in Atezolizumab vs. chemotherapy (not significant) | Concordance: bTMB CTA ≥ 16 equivalent to a F1 CDx value of 13.6 mut/Mb; 10 mutations were equivalent to 8.3 Mut/Mb, PPA of 82.9%, NPA of 91.5% |
| B-FIRST NCT02848651 [127] | Atezolizumab | Phase 2 | First line Stage IIIB–IVB NSCLC | ≥16 | FoundationOne bTMB Assay (targets 1.1 Mb coding region) | ORR 17% in ITT, mPFS 5 vs. 3.5 mo in bTMB ≥ 16 vs. <16 (HR 0.8, p = 0.35), mOS 23.9 vs. 13.4 mo in the bTMB ≥ 16 vs. <16 group (HR = 0.6, p = 0.18) not significant | Exploratory analysis showed patients with MSAF < 1% had significantly longer PFS (6.8 vs. 3.6 mo, p = 0.047) |
| MYSTIC NCT02453282 [128,129] | Durvalumab +/− Tremelimumab vs chemotherapy | Phase 3, Randomized controlled trial | First Line metastatic NSCLC | ≥10 to ≥20 | bTMB GuardantOMNI ctDNA platform; tTMB FoundationOne® CDx Assay (1.1 Mb coding region) | bTMB ≥ 20 mut/Mb associated with improved OS and FPS. mOS 12.6 mo (HR 0.72 for durvalumab vs chemo); HR 0.74 for Durvalumab + trememelimumab vs. durvalumab | Concordance Spearman rho 0.6 |
| NEPTUNE NCT02542293 | Durvalumab + Temelimumab | Phase 3, Randomized controlled trial | First line metastatic NSCLC | ≥20 Mut/Mb | GuardantOMNI plasma next-generation sequencing platform | For bTMB ≥ 20 mOS 11.7 vs. 9.1 in durvalumab/tremelimumab vs chemo, HR 0.71; mPFS 4.2 vs. 5.1 mo, HR 0.77. Not significant | No correlation between bTMB and PD-L1 status (Spearman’s rho 0.018) |
| BUDDY trial NCT04059887 [119] | Atezolizumab | Prospective | Relapsed or metastatic NSCLC | CT-ULTRA | No difference in ORR between bTMB low and high divided by median bTMB of 11.5 mut/Mb | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, J.; Das, B.; Shin, S.; Chen, A. Challenges and Future Directions in the Management of Tumor Mutational Burden-High (TMB-H) Advanced Solid Malignancies. Cancers 2023, 15, 5841. https://doi.org/10.3390/cancers15245841
Ahmed J, Das B, Shin S, Chen A. Challenges and Future Directions in the Management of Tumor Mutational Burden-High (TMB-H) Advanced Solid Malignancies. Cancers. 2023; 15(24):5841. https://doi.org/10.3390/cancers15245841
Chicago/Turabian StyleAhmed, Jibran, Biswajit Das, Sarah Shin, and Alice Chen. 2023. "Challenges and Future Directions in the Management of Tumor Mutational Burden-High (TMB-H) Advanced Solid Malignancies" Cancers 15, no. 24: 5841. https://doi.org/10.3390/cancers15245841