
Therapeutic Targeting of Glioblastoma and the Interactions with Its Microenvironment
 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Molecular Subtype and GBM Stem Cells
3. Blood–Brain Barrier Disruption in GBM
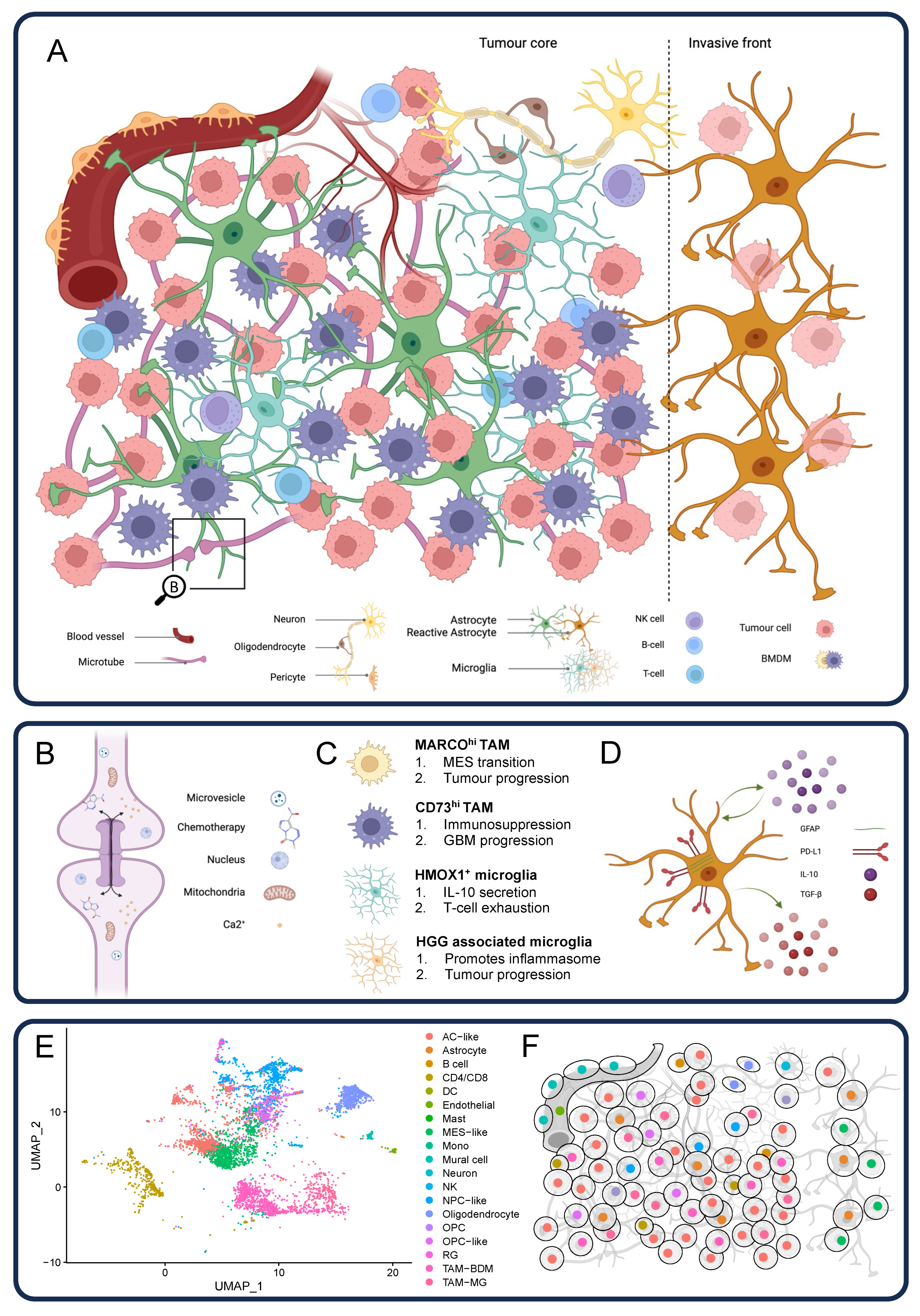
4. The Complex Glioblastoma Microenvironment
4.1. Astrocytes
4.2. Neurons
4.3. Glioblastoma Connectome
4.4. Myeloid Cells
4.5. Dendritic Cells
4.6. Lymphoid Cells
5. Therapeutic Perspectives

{kind=link}
| Cell Targeted | Mechanism Targeted | Potential Strategy | Potential Limitations |
|---|---|---|---|
| GBM | |||
| Tumour cells | Enhancing immunogenicity | Increasing MHC-I expression Increasing TMB and, therefore, neoantigen presentation with RT/TMZ treatment | Dampened NK cell response Sub-clonal TMB is associated with poor response to ICIs Effect may be restricted to MGMT-methylated GBM |
| Tumour cells, immune cells | Blocking negative regulators of antitumour immune response | ICIs | Lack of T cell infiltration, highly immunosuppressive TME |
| GSCs | Targeting specific markers Promoting GSC differentiation | Inhibition of CD133/ GPD1/ L1CAM Graphene oxide, Sulindac | Lack of truly specific targets, intrinsic treatment resistance |
| Tumour cells | Limiting the impact of hypoxia | HIF1⍺ inhibition | Limited efficacy thus far |
| TME—normal brain | |||
| Astrocytes | Limiting astrogliosis to suppress reactive astrocyte formation | JAK/STAT inhibition | Limited data available |
| Neurons | Targeting AMPAR signal | Perempanel treatment | Limited data available |
| Connectome | Targeting gap junctions | Connexin 43 targeting | Limited data available |
| TME—immune component | |||
| TAMs | Limiting infiltration | CCL2 inhibition | Limited data available |
| Limiting M2 polarization | CSF-1R inhibition | Induced resistance | |
| Enhancing phagocytosis | CD47 inhibition | Haematological side effects | |
| Depleting specific populations | CD73+ or MARCOhigh depletion | Limited data available | |
| DCs | Enhancing immune activation | Therapeutic vaccines | Efficacy dependent on T cell homing to the tumour |
| Neutrophils | Limiting infiltration | Blocking chemokines | |
| Tregs | Depleting cells | IL-25 depleting Ab | Limited data available |
| T cells | Enhancing targeting and activation | CAR-T cell therapy | Antigen loss, on-target off-tumour effect |
| NK cells | Enhancing activation | Activating cytokines | Limited data available |
| CNS—integrity | |||
| BBB | Increasing permeability | Focused ultrasound | Transient effect |

6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Brown, N.F.; Ottaviani, D.; Tazare, J.; Gregson, J.; Kitchen, N.; Brandner, S.; Fersht, N.; Mulholland, P. Survival Outcomes and Prognostic Factors in Glioblastoma. Cancers 2022, 14, 3161. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma. JAMA 2017, 318, 2306. [Google Scholar] [CrossRef] [PubMed]
- Liau, L.M.; Ashkan, K.; Brem, S.; Campian, J.L.; Trusheim, J.E.; Iwamoto, F.M.; Tran, D.D.; Ansstas, G.; Cobbs, C.S.; Heth, J.A.; et al. Association of Autologous Tumor Lysate-Loaded Dendritic Cell Vaccination With Extension of Survival Among Patients With Newly Diagnosed and Recurrent Glioblastoma: A Phase 3 Prospective Externally Controlled Cohort Trial. JAMA Oncol. 2023, 9, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bahr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef]
- Mulholland, P.J.; Brown, N.F.; McBain, C.; Brazil, L.; Peoples, S.; Jefferies, S.; Harris, F.; Plaha, P.; Vinayan, A.; Brooks, C.; et al. A randomised phase II multicentre study of ipilimumab with temozolomide vs temozolomide alone after surgery and chemoradiotherapy in patients with recently diagnosed glioblastoma: Ipi-Glio. J. Clin. Oncol. 2023, 41, LBA2023. [Google Scholar] [CrossRef]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; Lopez-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inoges, S.; de Andrea, C.; Lopez-Diaz de Cerio, A.; Tejada, S.; et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Migliorini, D.; Dietrich, P.Y.; Stupp, R.; Linette, G.P.; Posey, A.D., Jr.; June, C.H. CAR T-Cell Therapies in Glioblastoma: A First Look. Clin. Cancer Res. 2018, 24, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pages, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Berghoff, A.S.; Kiesel, B.; Widhalm, G.; Rajky, O.; Ricken, G.; Wohrer, A.; Dieckmann, K.; Filipits, M.; Brandstetter, A.; Weller, M.; et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol. 2015, 17, 1064–1075. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Wiesner, S.; Xiao, J.; Ericson, K.; Chen, W.; Hall, W.A.; Low, W.C.; Ohlfest, J.R. Expression of MHC I and NK ligands on human CD133+ glioma cells: Possible targets of immunotherapy. J. Neurooncol. 2007, 83, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Venkataramani, V.; Yang, Y.; Schubert, M.C.; Reyhan, E.; Tetzlaff, S.K.; Wissmann, N.; Botz, M.; Soyka, S.J.; Beretta, C.A.; Pramatarov, R.L.; et al. Glioblastoma hijacks neuronal mechanisms for brain invasion. Cell 2022, 185, 2899–2917.e2831. [Google Scholar] [CrossRef] [PubMed]
- DeCordova, S.; Shastri, A.; Tsolaki, A.G.; Yasmin, H.; Klein, L.; Singh, S.K.; Kishore, U. Molecular Heterogeneity and Immunosuppressive Microenvironment in Glioblastoma. Front. Immunol. 2020, 11, 1402. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e821. [Google Scholar] [CrossRef]
- Alcantara Llaguno, S.R.; Wang, Z.; Sun, D.; Chen, J.; Xu, J.; Kim, E.; Hatanpaa, K.J.; Raisanen, J.M.; Burns, D.K.; Johnson, J.E.; et al. Adult Lineage-Restricted CNS Progenitors Specify Distinct Glioblastoma Subtypes. Cancer Cell 2015, 28, 429–440. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.Y.; Kim, W.K.; Lee, J.K.; Park, J.; et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 2018, 560, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Eramo, A.; Ricci-Vitiani, L.; Zeuner, A.; Pallini, R.; Lotti, F.; Sette, G.; Pilozzi, E.; Larocca, L.M.; Peschle, C.; De Maria, R. Chemotherapy resistance of glioblastoma stem cells. Cell. Death Differ. 2006, 13, 1238–1241. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, B.; Spencer, D.; Pytel, P.; Ahmed, A.U.; Lesniak, M.S. The role of glioma stem cells in chemotherapy resistance and glioblastoma multiforme recurrence. Expert Rev. Neurother 2015, 15, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Prager, B.C.; Bhargava, S.; Mahadev, V.; Hubert, C.G.; Rich, J.N. Glioblastoma Stem Cells: Driving Resilience through Chaos. Trends Cancer 2020, 6, 223–235. [Google Scholar] [CrossRef]
- Zheng, Y.; Carrillo-Perez, F.; Pizurica, M.; Heiland, D.H.; Gevaert, O. Spatial cellular architecture predicts prognosis in glioblastoma. Nat. Commun. 2023, 14, 4122. [Google Scholar] [CrossRef]
- Turnquist, C.; Harris, B.T.; Harris, C.C. Radiation-induced brain injury: Current concepts and therapeutic strategies targeting neuroinflammation. Neuro-Oncol. Adv. 2020, 2, vdaa057. [Google Scholar] [CrossRef] [PubMed]
- Richards, L.M.; Whitley, O.K.N.; MacLeod, G.; Cavalli, F.M.G.; Coutinho, F.J.; Jaramillo, J.E.; Svergun, N.; Riverin, M.; Croucher, D.C.; Kushida, M.; et al. Gradient of Developmental and Injury Response transcriptional states defines functional vulnerabilities underpinning glioblastoma heterogeneity. Nat. Cancer 2021, 2, 157–173. [Google Scholar] [CrossRef]
- Bien-Moller, S.; Balz, E.; Herzog, S.; Plantera, L.; Vogelgesang, S.; Weitmann, K.; Seifert, C.; Fink, M.A.; Marx, S.; Bialke, A.; et al. Association of Glioblastoma Multiforme Stem Cell Characteristics, Differentiation, and Microglia Marker Genes with Patient Survival. Stem Cells Int. 2018, 2018, 9628289. [Google Scholar] [CrossRef]
- Torrisi, F.; Alberghina, C.; D’Aprile, S.; Pavone, A.M.; Longhitano, L.; Giallongo, S.; Tibullo, D.; Di Rosa, M.; Zappalà, A.; Cammarata, F.P. The hallmarks of glioblastoma: Heterogeneity, intercellular crosstalk and molecular signature of invasiveness and progression. Biomedicines 2022, 10, 806. [Google Scholar] [CrossRef]
- Couturier, C.P.; Ayyadhury, S.; Le, P.U.; Nadaf, J.; Monlong, J.; Riva, G.; Allache, R.; Baig, S.; Yan, X.; Bourgey, M.; et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat. Commun. 2020, 11, 3406. [Google Scholar] [CrossRef]
- Hara, T.; Chanoch-Myers, R.; Mathewson, N.D.; Myskiw, C.; Atta, L.; Bussema, L.; Eichhorn, S.W.; Greenwald, A.C.; Kinker, G.S.; Rodman, C. Interactions between cancer cells and immune cells drive transitions to mesenchymal-like states in glioblastoma. Cancer Cell 2021, 39, 779–792.e711. [Google Scholar] [CrossRef] [PubMed]
- Brooks, L.J.; Simpson Ragdale, H.; Hill, C.S.; Clements, M.; Parrinello, S. Injury programs shape glioblastoma. Trends Neurosci. 2022, 45, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef]
- Gangoso, E.; Southgate, B.; Bradley, L.; Rus, S.; Galvez-Cancino, F.; McGivern, N.; Guc, E.; Kapourani, C.A.; Byron, A.; Ferguson, K.M.; et al. Glioblastomas acquire myeloid-affiliated transcriptional programs via epigenetic immunoediting to elicit immune evasion. Cell 2021, 184, 2454–2470. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, C.; Wang, L.; Chen, Y. A comprehensive review in improving delivery of small-molecule chemotherapeutic agents overcoming the blood-brain/brain tumor barriers for glioblastoma treatment. Drug Deliv. 2019, 26, 551–565. [Google Scholar] [CrossRef]
- Wolburg, H.; Noell, S.; Fallier-Becker, P.; Mack, A.F.; Wolburg-Buchholz, K. The disturbed blood-brain barrier in human glioblastoma. Mol. Aspects Med. 2012, 33, 579–589. [Google Scholar] [CrossRef]
- Dubois, L.G.; Campanati, L.; Righy, C.; D’Andrea-Meira, I.; Spohr, T.C.; Porto-Carreiro, I.; Pereira, C.M.; Balca-Silva, J.; Kahn, S.A.; DosSantos, M.F.; et al. Gliomas and the vascular fragility of the blood brain barrier. Front. Cell Neurosci. 2014, 8, 418. [Google Scholar] [CrossRef]
- Yang, J.; Yan, J.; Liu, B. Targeting VEGF/VEGFR to Modulate Antitumor Immunity. Front. Immunol. 2018, 9, 978. [Google Scholar] [CrossRef]
- Liebelt, B.D.; Shingu, T.; Zhou, X.; Ren, J.; Shin, S.A.; Hu, J. Glioma Stem Cells: Signaling, Microenvironment, and Therapy. Stem Cells Int. 2016, 2016, 7849890. [Google Scholar] [CrossRef]
- Hardee, M.E.; Zagzag, D. Mechanisms of glioma-associated neovascularization. Am. J. Pathol. 2012, 181, 1126–1141. [Google Scholar] [CrossRef]
- Jhaveri, N.; Chen, T.C.; Hofman, F.M. Tumor vasculature and glioma stem cells: Contributions to glioma progression. Cancer Lett. 2016, 380, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Uribe, D.; Torres, A.; Rocha, J.D.; Niechi, I.; Oyarzun, C.; Sobrevia, L.; San Martin, R.; Quezada, C. Multidrug resistance in glioblastoma stem-like cells: Role of the hypoxic microenvironment and adenosine signaling. Mol. Aspects Med. 2017, 55, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.Y.; Ho, N.P.; Lee, T.K. Cancer Stem Cells and Their Microenvironment: Biology and Therapeutic Implications. Stem Cells Int. 2017, 2017, 3714190. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010, 29, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Nardinocchi, L.; Pantisano, V.; Puca, R.; Porru, M.; Aiello, A.; Grasselli, A.; Leonetti, C.; Safran, M.; Rechavi, G.; Givol, D.; et al. Zinc downregulates HIF-1alpha and inhibits its activity in tumor cells in vitro and in vivo. PLoS ONE 2010, 5, e15048. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, E.M.; Bandopadhyay, G.; Coyle, B.; Grabowska, A. A HIF-independent, CD133-mediated mechanism of cisplatin resistance in glioblastoma cells. Cell Oncol. 2018, 41, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Dalerba, P.; Cho, R.W.; Clarke, M.F. Cancer stem cells: Models and concepts. Annu. Rev. Med. 2007, 58, 267–284. [Google Scholar] [CrossRef]
- Guo, X.; Xue, H.; Shao, Q.; Wang, J.; Guo, X.; Chen, X.; Zhang, J.; Xu, S.; Li, T.; Zhang, P.; et al. Hypoxia promotes glioma-associated macrophage infiltration via periostin and subsequent M2 polarization by upregulating TGF-beta and M-CSFR. Oncotarget 2016, 7, 80521–80542. [Google Scholar] [CrossRef]
- Matyash, V.; Kettenmann, H. Heterogeneity in astrocyte morphology and physiology. Brain Res. Rev. 2010, 63, 2–10. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef]
- Henrik Heiland, D.; Ravi, V.M.; Behringer, S.P.; Frenking, J.H.; Wurm, J.; Joseph, K.; Garrelfs, N.W.C.; Strahle, J.; Heynckes, S.; Grauvogel, J.; et al. Tumor-associated reactive astrocytes aid the evolution of immunosuppressive environment in glioblastoma. Nat. Commun. 2019, 10, 2541. [Google Scholar] [CrossRef]
- Pittet, C.L.; Newcombe, J.; Antel, J.P.; Arbour, N. The majority of infiltrating CD8 T lymphocytes in multiple sclerosis lesions is insensitive to enhanced PD-L1 levels on CNS cells. Glia 2011, 59, 841–856. [Google Scholar] [CrossRef]
- He, F.; Ge, W.; Martinowich, K.; Becker-Catania, S.; Coskun, V.; Zhu, W.; Wu, H.; Castro, D.; Guillemot, F.; Fan, G.; et al. A positive autoregulatory loop of Jak-STAT signaling controls the onset of astrogliogenesis. Nat. Neurosci. 2005, 8, 616–625. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef]
- Krishna, S.; Choudhury, A.; Keough, M.B.; Seo, K.; Ni, L.; Kakaizada, S.; Lee, A.; Aabedi, A.; Popova, G.; Lipkin, B.; et al. Glioblastoma remodelling of human neural circuits decreases survival. Nature 2023, 617, 599–607. [Google Scholar] [CrossRef]
- Radin, D.P.; Tsirka, S.E. Interactions between Tumor Cells, Neurons, and Microglia in the Glioma Microenvironment. Int. J. Mol. Sci. 2020, 21, 8476. [Google Scholar] [CrossRef]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Korber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, H.S.; Tam, L.T.; Woo, P.J.; Lennon, J.; Nagaraja, S.; Gillespie, S.M.; Ni, J.; Duveau, D.Y.; Morris, P.J.; Zhao, J.J.; et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature 2017, 549, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.W.; Polepalli, J.; Mitra, S.S.; et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 2015, 161, 803–816. [Google Scholar] [CrossRef]
- Buckingham, S.C.; Campbell, S.L.; Haas, B.R.; Montana, V.; Robel, S.; Ogunrinu, T.; Sontheimer, H. Glutamate release by primary brain tumors induces epileptic activity. Nat. Med. 2011, 17, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.L.; Buckingham, S.C.; Sontheimer, H. Human glioma cells induce hyperexcitability in cortical networks. Epilepsia 2012, 53, 1360–1370. [Google Scholar] [CrossRef]
- Campbell, S.L.; Robel, S.; Cuddapah, V.A.; Robert, S.; Buckingham, S.C.; Kahle, K.T.; Sontheimer, H. GABAergic disinhibition and impaired KCC2 cotransporter activity underlie tumor-associated epilepsy. Glia 2015, 63, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Hua, T.; Shi, H.; Zhu, M.; Chen, C.; Su, Y.; Wen, S.; Zhang, X.; Chen, J.; Huang, Q.; Wang, H. Glioma-neuronal interactions in tumor progression: Mechanism, therapeutic strategies and perspectives (Review). Int. J. Oncol. 2022, 61, 104. [Google Scholar] [CrossRef] [PubMed]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Weil, S.; Osswald, M.; Solecki, G.; Grosch, J.; Jung, E.; Lemke, D.; Ratliff, M.; Hanggi, D.; Wick, W.; Winkler, F. Tumor microtubes convey resistance to surgical lesions and chemotherapy in gliomas. Neuro Oncol. 2017, 19, 1316–1326. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Locker, J.; Sahai, E.; Segall, J.E. Classifying collective cancer cell invasion. Nat. Cell Biol. 2012, 14, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Gritsenko, P.G.; Atlasy, N.; Dieteren, C.E.J.; Navis, A.C.; Venhuizen, J.H.; Veelken, C.; Schubert, D.; Acker-Palmer, A.; Westerman, B.A.; Wurdinger, T.; et al. p120-catenin-dependent collective brain infiltration by glioma cell networks. Nat. Cell Biol. 2020, 22, 97–107. [Google Scholar] [CrossRef]
- Cuddapah, V.A.; Robel, S.; Watkins, S.; Sontheimer, H. A neurocentric perspective on glioma invasion. Nat. Rev. Neurosci. 2014, 15, 455–465. [Google Scholar] [CrossRef]
- Garofano, L.; Migliozzi, S.; Oh, Y.T.; D’Angelo, F.; Najac, R.D.; Ko, A.; Frangaj, B.; Caruso, F.P.; Yu, K.; Yuan, J.; et al. Pathway-based classification of glioblastoma uncovers a mitochondrial subtype with therapeutic vulnerabilities. Nat. Cancer 2021, 2, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Varn, F.S.; Johnson, K.C.; Martinek, J.; Huse, J.T.; Nasrallah, M.P.; Wesseling, P.; Cooper, L.A.D.; Malta, T.M.; Wade, T.E.; Sabedot, T.S.; et al. Glioma progression is shaped by genetic evolution and microenvironment interactions. Cell 2022, 185, 2184–2199.e2116. [Google Scholar] [CrossRef] [PubMed]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., 3rd; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef]
- Hong, S.; Dissing-Olesen, L.; Stevens, B. New insights on the role of microglia in synaptic pruning in health and disease. Curr. Opin. Neurobiol. 2016, 36, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Gielen, P.R.; Schulte, B.M.; Kers-Rebel, E.D.; Verrijp, K.; Bossman, S.A.; Ter Laan, M.; Wesseling, P.; Adema, G.J. Elevated levels of polymorphonuclear myeloid-derived suppressor cells in patients with glioblastoma highly express S100A8/9 and arginase and suppress T cell function. Neuro Oncol. 2016, 18, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Richard, S.A. Explicating the Pivotal Pathogenic, Diagnostic, and Therapeutic Biomarker Potentials of Myeloid-Derived Suppressor Cells in Glioblastoma. Dis. Markers 2020, 2020, 8844313. [Google Scholar] [CrossRef]
- Alban, T.J.; Alvarado, A.G.; Sorensen, M.D.; Bayik, D.; Volovetz, J.; Serbinowski, E.; Mulkearns-Hubert, E.E.; Sinyuk, M.; Hale, J.S.; Onzi, G.R.; et al. Global immune fingerprinting in glioblastoma patient peripheral blood reveals immune-suppression signatures associated with prognosis. JCI Insight 2018, 3, e122264. [Google Scholar] [CrossRef]
- Jurga, A.M.; Paleczna, M.; Kuter, K.Z. Overview of General and Discriminating Markers of Differential Microglia Phenotypes. Front. Cell Neurosci. 2020, 14, 198. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.; Brandenburg, S.; Turkowski, K.; Muller, S.; Vajkoczy, P. Resident microglia, and not peripheral macrophages, are the main source of brain tumor mononuclear cells. Int. J. Cancer 2015, 137, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Landry, A.P.; Balas, M.; Alli, S.; Spears, J.; Zador, Z. Distinct regional ontogeny and activation of tumor associated macrophages in human glioblastoma. Sci. Rep. 2020, 10, 19542. [Google Scholar] [CrossRef] [PubMed]
- Ochocka, N.; Segit, P.; Walentynowicz, K.A.; Wojnicki, K.; Cyranowski, S.; Swatler, J.; Mieczkowski, J.; Kaminska, B. Single-cell RNA sequencing reveals functional heterogeneity of glioma-associated brain macrophages. Nat. Commun. 2021, 12, 1151. [Google Scholar] [CrossRef] [PubMed]
- Pombo Antunes, A.R.; Scheyltjens, I.; Lodi, F.; Messiaen, J.; Antoranz, A.; Duerinck, J.; Kancheva, D.; Martens, L.; De Vlaminck, K.; Van Hove, H.; et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat. Neurosci. 2021, 24, 595–610. [Google Scholar] [CrossRef]
- Chen, P.; Zhao, D.; Li, J.; Liang, X.; Li, J.; Chang, A.; Henry, V.K.; Lan, Z.; Spring, D.J.; Rao, G.; et al. Symbiotic Macrophage-Glioma Cell Interactions Reveal Synthetic Lethality in PTEN-Null Glioma. Cancer Cell 2019, 35, 868–884.e866. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- Xuan, W.; Lesniak, M.S.; James, C.D.; Heimberger, A.B.; Chen, P. Context-Dependent Glioblastoma-Macrophage/Microglia Symbiosis and Associated Mechanisms. Trends Immunol. 2021, 42, 280–292. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Abdelfattah, N.; Kumar, P.; Wang, C.; Leu, J.S.; Flynn, W.F.; Gao, R.; Baskin, D.S.; Pichumani, K.; Ijare, O.B.; Wood, S.L.; et al. Single-cell analysis of human glioma and immune cells identifies S100A4 as an immunotherapy target. Nat. Commun. 2022, 13, 767. [Google Scholar] [CrossRef]
- Muller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017, 18, 234. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lage, M.; Lynch, T.M.; Bi, Y.; Cocito, C.; Way, G.P.; Pal, S.; Haller, J.; Yan, R.E.; Ziober, A.; Nguyen, A.; et al. Immune landscapes associated with different glioblastoma molecular subtypes. Acta Neuropathol. Commun. 2019, 7, 203. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Ishikawa, E.; Matsuda, M.; Sugii, N.; Kohzuki, H.; Akutsu, H.; Sakamoto, N.; Takano, S.; Matsumura, A. Infiltration of CD163-positive macrophages in glioma tissues after treatment with anti-PD-L1 antibody and role of PI3Kgamma inhibitor as a combination therapy with anti-PD-L1 antibody in in vivo model using temozolomide-resistant murine glioma-initiating cells. Brain Tumor Pathol. 2020, 37, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Ravi, V.M.; Will, P.; Kueckelhaus, J.; Sun, N.; Joseph, K.; Salie, H.; Vollmer, L.; Kuliesiute, U.; von Ehr, J.; Benotmane, J.K.; et al. Spatially resolved multi-omics deciphers bidirectional tumor-host interdependence in glioblastoma. Cancer Cell 2022, 40, 639–655.e613. [Google Scholar] [CrossRef] [PubMed]
- Sa, J.K.; Chang, N.; Lee, H.W.; Cho, H.J.; Ceccarelli, M.; Cerulo, L.; Yin, J.; Kim, S.S.; Caruso, F.P.; Lee, M.; et al. Transcriptional regulatory networks of tumor-associated macrophages that drive malignancy in mesenchymal glioblastoma. Genome Biol. 2020, 21, 216. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Sun, Y.; Zhang, Q.; Jin, W.; Gordon, R.E.; Zhang, Y.; Wang, J.; Sun, C.; Wang, Z.J.; Qi, X.; et al. Pro-inflammatory and proliferative microglia drive progression of glioblastoma. Cell Rep. 2021, 36, 109718. [Google Scholar] [CrossRef]
- Goswami, S.; Walle, T.; Cornish, A.E.; Basu, S.; Anandhan, S.; Fernandez, I.; Vence, L.; Blando, J.; Zhao, H.; Yadav, S.S.; et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat. Med. 2020, 26, 39–46. [Google Scholar] [CrossRef]
- Mi, Y.; Guo, N.; Luan, J.; Cheng, J.; Hu, Z.; Jiang, P.; Jin, W.; Gao, X. The Emerging Role of Myeloid-Derived Suppressor Cells in the Glioma Immune Suppressive Microenvironment. Front. Immunol. 2020, 11, 737. [Google Scholar] [CrossRef]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef]
- Massara, M.; Persico, P.; Bonavita, O.; Mollica Poeta, V.; Locati, M.; Simonelli, M.; Bonecchi, R. Neutrophils in Gliomas. Front. Immunol. 2017, 8, 1349. [Google Scholar] [CrossRef]
- Maas, R.R.; Soukup, K.; Fournier, N.; Massara, M.; Galland, S.; Kornete, M.; Wischnewski, V.; Lourenco, J.; Croci, D.; Alvarez-Prado, A.F.; et al. The local microenvironment drives activation of neutrophils in human brain tumors. Cell 2023, 186, 4546–4566. [Google Scholar] [CrossRef]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, X.H. Tumor-Associated Neutrophils and Macrophages-Heterogenous but Not Chaotic. Front. Immunol. 2020, 11, 553967. [Google Scholar] [CrossRef]
- Khan, S.; Mittal, S.; McGee, K.; Alfaro-Munoz, K.D.; Majd, N.; Balasubramaniyan, V.; de Groot, J.F. Role of Neutrophils and Myeloid-Derived Suppressor Cells in Glioma Progression and Treatment Resistance. Int. J. Mol. Sci. 2020, 21, 1954. [Google Scholar] [CrossRef]
- Mildner, A.; Jung, S. Development and function of dendritic cell subsets. Immunity 2014, 40, 642–656. [Google Scholar] [CrossRef] [PubMed]
- Bottcher, J.P.; Reis e Sousa, C. The Role of Type 1 Conventional Dendritic Cells in Cancer Immunity. Trends Cancer 2018, 4, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.J.; Ornelles, D.A.; Mitchell, L.M.; Brzoza-Lewis, K.L.; Hiltbold, E.M. IL-12 produced by dendritic cells augments CD8+ T cell activation through the production of the chemokines CCL1 and CCL17. J. Immunol. 2008, 181, 8576–8584. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Jackson, C.; Kim, T.; Choi, J.; Lim, M. A Characterization of Dendritic Cells and Their Role in Immunotherapy in Glioblastoma: From Preclinical Studies to Clinical Trials. Cancers 2019, 11, 537. [Google Scholar] [CrossRef]
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.; Han, S.J.; Sughrue, M.E.; Tihan, T.; Parsa, A.T. Immune cell infiltrate differences in pilocytic astrocytoma and glioblastoma: Evidence of distinct immunological microenvironments that reflect tumor biology. J. Neurosurg. 2011, 115, 505–511. [Google Scholar] [CrossRef]
- Fadul, C.E.; Fisher, J.L.; Gui, J.; Hampton, T.H.; Cote, A.L.; Ernstoff, M.S. Immune modulation effects of concomitant temozolomide and radiation therapy on peripheral blood mononuclear cells in patients with glioblastoma multiforme. Neuro Oncol. 2011, 13, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Facoetti, A.; Nano, R.; Zelini, P.; Morbini, P.; Benericetti, E.; Ceroni, M.; Campoli, M.; Ferrone, S. Human leukocyte antigen and antigen processing machinery component defects in astrocytic tumors. Clin. Cancer Res. 2005, 11, 8304–8311. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Wu, W.; Zhang, H.; Zhang, X.; Zhang, D.; Wang, Q.; Huang, L.; Wang, Y.; Hang, C. High expression of B7-H6 in human glioma tissues promotes tumor progression. Oncotarget 2017, 8, 37435–37447. [Google Scholar] [CrossRef] [PubMed]
- Speranza, M.C.; Passaro, C.; Ricklefs, F.; Kasai, K.; Klein, S.R.; Nakashima, H.; Kaufmann, J.K.; Ahmed, A.K.; Nowicki, M.O.; Obi, P.; et al. Preclinical investigation of combined gene-mediated cytotoxic immunotherapy and immune checkpoint blockade in glioblastoma. Neuro Oncol. 2018, 20, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Tomaszowski, K.H.; Marisetty, A.; Kong, L.Y.; Wei, J.; Duna, M.; Blumberg, K.; Ji, X.; Jacobs, C.; Fuller, G.N.; et al. Profiling of patients with glioma reveals the dominant immunosuppressive axis is refractory to immune function restoration. JCI Insight 2020, 5, e134386. [Google Scholar] [CrossRef]
- Sayour, E.J.; McLendon, P.; McLendon, R.; De Leon, G.; Reynolds, R.; Kresak, J.; Sampson, J.H.; Mitchell, D.A. Increased proportion of FoxP3+ regulatory T cells in tumor infiltrating lymphocytes is associated with tumor recurrence and reduced survival in patients with glioblastoma. Cancer Immunol. Immunother 2015, 64, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Heimberger, A.B.; Abou-Ghazal, M.; Reina-Ortiz, C.; Yang, D.S.; Sun, W.; Qiao, W.; Hiraoka, N.; Fuller, G.N. Incidence and prognostic impact of FoxP3+ regulatory T cells in human gliomas. Clin. Cancer Res. 2008, 14, 5166–5172. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Barbi, J.; Pan, F. The regulation of immune tolerance by FOXP3. Nat. Rev. Immunol. 2017, 17, 703–717. [Google Scholar] [CrossRef]
- Vilgelm, A.E.; Richmond, A. Chemokines Modulate Immune Surveillance in Tumorigenesis, Metastasis, and Response to Immunotherapy. Front. Immunol. 2019, 10, 333. [Google Scholar] [CrossRef]
- Jarnicki, A.G.; Lysaght, J.; Todryk, S.; Mills, K.H. Suppression of antitumor immunity by IL-10 and TGF-beta-producing T cells infiltrating the growing tumor: Influence of tumor environment on the induction of CD4+ and CD8+ regulatory T cells. J. Immunol. 2006, 177, 896–904. [Google Scholar] [CrossRef]
- Li, C.; Jiang, P.; Wei, S.; Xu, X.; Wang, J. Regulatory T cells in tumor microenvironment: New mechanisms, potential therapeutic strategies and future prospects. Mol. Cancer 2020, 19, 116. [Google Scholar] [CrossRef]
- Johnson, L.A.; Scholler, J.; Ohkuri, T.; Kosaka, A.; Patel, P.R.; McGettigan, S.E.; Nace, A.K.; Dentchev, T.; Thekkat, P.; Loew, A.; et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci. Transl. Med. 2015, 7, 275ra222. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Thaci, B.; Crawford, A.C.; Sampath, P. Interleukin-13 receptor alpha 2-targeted glioblastoma immunotherapy. Biomed Res. Int. 2014, 2014, 952128. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-B.; Karpova, A.; Gritsenko, M.A.; Kyle, J.E.; Cao, S.; Li, Y.; Rykunov, D.; Colaprico, A.; Rothstein, J.H.; Hong, R. Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell 2021, 39, 509–528.e520. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Stuart, T.; Kowalski, M.H.; Choudhary, S.; Hoffman, P.; Hartman, A.; Srivastava, A.; Molla, G.; Madad, S.; Fernandez-Granda, C.; et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 2023, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Moreno, C.; Salas, S.M.; Samuelsson, E.; Brandner, S.; Kranendonk, M.E.; Nilsson, M.; Stunnenberg, H.G. Harmonized single-cell landscape, intercellular crosstalk and tumor architecture of glioblastoma. BioRxiv 2022. [Google Scholar]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e3529. [Google Scholar] [CrossRef]
- Khan, F.; Pang, L.; Dunterman, M.; Lesniak, M.S.; Heimberger, A.B.; Chen, P. Macrophages and microglia in glioblastoma: Heterogeneity, plasticity, and therapy. J. Clin. Invest. 2023, 133, e163446. [Google Scholar] [CrossRef]
- Seano, G. Targeting the perivascular niche in brain tumors. Curr. Opin. Oncol. 2018, 30, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Persano, L.; Rampazzo, E.; Basso, G.; Viola, G. Glioblastoma cancer stem cells: Role of the microenvironment and therapeutic targeting. Biochem. Pharmacol. 2013, 85, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Beier, D.; Rohrl, S.; Pillai, D.R.; Schwarz, S.; Kunz-Schughart, L.A.; Leukel, P.; Proescholdt, M.; Brawanski, A.; Bogdahn, U.; Trampe-Kieslich, A.; et al. Temozolomide preferentially depletes cancer stem cells in glioblastoma. Cancer Res. 2008, 68, 5706–5715. [Google Scholar] [CrossRef] [PubMed]
- Garnier, D.; Meehan, B.; Kislinger, T.; Daniel, P.; Sinha, A.; Abdulkarim, B.; Nakano, I.; Rak, J. Divergent evolution of temozolomide resistance in glioblastoma stem cells is reflected in extracellular vesicles and coupled with radiosensitization. Neuro Oncol. 2018, 20, 236–248. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, S.X.; Ma, J.W.; Li, H.Y.; Ye, J.C.; Xie, S.M.; Du, B.; Zhong, X.Y. EGCG inhibits properties of glioma stem-like cells and synergizes with temozolomide through downregulation of P-glycoprotein inhibition. J. Neurooncol. 2015, 121, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Xiong, X.; Sun, Y. Functional characterization of SOX2 as an anticancer target. Signal Transduct. Target Ther. 2020, 5, 135. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, G.; Bozek, D.A.; Rajakulendran, N.; Monteiro, V.; Ahmadi, M.; Steinhart, Z.; Kushida, M.M.; Yu, H.; Coutinho, F.J.; Cavalli, F.M.G.; et al. Genome-Wide CRISPR-Cas9 Screens Expose Genetic Vulnerabilities and Mechanisms of Temozolomide Sensitivity in Glioblastoma Stem Cells. Cell Rep. 2019, 27, 971–986.e979. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Akkari, L.; Bowman, R.L.; Tessier, J.; Klemm, F.; Handgraaf, S.M.; de Groot, M.; Quail, D.F.; Tillard, L.; Gadiot, J.; Huse, J.T.; et al. Dynamic changes in glioma macrophage populations after radiotherapy reveal CSF-1R inhibition as a strategy to overcome resistance. Sci. Transl Med. 2020, 12, eaaw7843. [Google Scholar] [CrossRef]
- Wang, L.; Jung, J.; Babikir, H.; Shamardani, K.; Jain, S.; Feng, X.; Gupta, N.; Rosi, S.; Chang, S.; Raleigh, D.; et al. A single-cell atlas of glioblastoma evolution under therapy reveals cell-intrinsic and cell-extrinsic therapeutic targets. Nat. Cancer 2022, 3, 1534–1552. [Google Scholar] [CrossRef]
- Song, J.H.; Bellail, A.; Tse, M.C.; Yong, V.W.; Hao, C. Human astrocytes are resistant to Fas ligand and tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. J. Neurosci. 2006, 26, 3299–3308. [Google Scholar] [CrossRef] [PubMed]
- Okolie, O.; Bago, J.R.; Schmid, R.S.; Irvin, D.M.; Bash, R.E.; Miller, C.R.; Hingtgen, S.D. Reactive astrocytes potentiate tumor aggressiveness in a murine glioma resection and recurrence model. Neuro Oncol. 2016, 18, 1622–1633. [Google Scholar] [CrossRef]
- Schneider, M.; Vollmer, L.; Potthoff, A.L.; Ravi, V.M.; Evert, B.O.; Rahman, M.A.; Sarowar, S.; Kueckelhaus, J.; Will, P.; Zurhorst, D.; et al. Meclofenamate causes loss of cellular tethering and decoupling of functional networks in glioblastoma. Neuro Oncol. 2021, 23, 1885–1897. [Google Scholar] [CrossRef] [PubMed]
- Salmaggi, A.; Corno, C.; Maschio, M.; Donzelli, S.; D’Urso, A.; Perego, P.; Ciusani, E. Synergistic Effect of Perampanel and Temozolomide in Human Glioma Cell Lines. J. Pers. Med. 2021, 11, 390. [Google Scholar] [CrossRef] [PubMed]
- Flores-Toro, J.A.; Luo, D.; Gopinath, A.; Sarkisian, M.R.; Campbell, J.J.; Charo, I.F.; Singh, R.; Schall, T.J.; Datta, M.; Jain, R.K.; et al. CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. Proc. Natl. Acad. Sci. USA 2020, 117, 1129–1138. [Google Scholar] [CrossRef]
- Shao, Z.; Tan, Y.; Shen, Q.; Hou, L.; Yao, B.; Qin, J.; Xu, P.; Mao, C.; Chen, L.N.; Zhang, H.; et al. Molecular insights into ligand recognition and activation of chemokine receptors CCR2 and CCR3. Cell Discov. 2022, 8, 44. [Google Scholar] [CrossRef]
- Quail, D.F.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 2016, 352, aad3018. [Google Scholar] [CrossRef]
- Cui, X.; Ma, C.; Vasudevaraja, V.; Serrano, J.; Tong, J.; Peng, Y.; Delorenzo, M.; Shen, G.; Frenster, J.; Morales, R.T.; et al. Dissecting the immunosuppressive tumor microenvironments in Glioblastoma-on-a-Chip for optimized PD-1 immunotherapy. Elife 2020, 9, e52253. [Google Scholar] [CrossRef]
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro Oncol. 2016, 18, 557–564. [Google Scholar] [CrossRef]
- Chen, P.; Hsu, W.H.; Han, J.; Xia, Y.; DePinho, R.A. Cancer Stemness Meets Immunity: From Mechanism to Therapy. Cell Rep. 2021, 34, 108597. [Google Scholar] [CrossRef]
- Ma, D.; Liu, S.; Lal, B.; Wei, S.; Wang, S.; Zhan, D.; Zhang, H.; Lee, R.S.; Gao, P.; Lopez-Bertoni, H.; et al. Extracellular Matrix Protein Tenascin C Increases Phagocytosis Mediated by CD47 Loss of Function in Glioblastoma. Cancer Res. 2019, 79, 2697–2708. [Google Scholar] [CrossRef]
- Guo, M.; Abd-Rabbo, D.; Bertol, B.C.; Carew, M.; Lukhele, S.; Snell, L.M.; Xu, W.; Boukhaled, G.M.; Elsaesser, H.; Halaby, M.J.; et al. Molecular, metabolic, and functional CD4 T cell paralysis in the lymph node impedes tumor control. Cell Rep. 2023, 42, 113047. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Kienzler, J.C.; Reynoso, J.G.; Lee, A.; Shiuan, E.; Li, S.; Kim, J.; Ding, L.; Monteleone, A.J.; Owens, G.C.; et al. Immune checkpoint blockade induces distinct alterations in the microenvironments of primary and metastatic brain tumors. J. Clin. Invest. 2023, 133, e169314. [Google Scholar] [CrossRef] [PubMed]
- Simonds, E.F.; Lu, E.D.; Badillo, O.; Karimi, S.; Liu, E.V.; Tamaki, W.; Rancan, C.; Downey, K.M.; Stultz, J.; Sinha, M.; et al. Deep immune profiling reveals targetable mechanisms of immune evasion in immune checkpoint inhibitor-refractory glioblastoma. J. Immunother Cancer 2021, 9, e002181. [Google Scholar] [CrossRef]
- Bielamowicz, K.; Fousek, K.; Byrd, T.T.; Samaha, H.; Mukherjee, M.; Aware, N.; Wu, M.F.; Orange, J.S.; Sumazin, P.; Man, T.K.; et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol. 2018, 20, 506–518. [Google Scholar] [CrossRef] [PubMed]
- Solomon, I.; Amann, M.; Goubier, A.; Arce Vargas, F.; Zervas, D.; Qing, C.; Henry, J.Y.; Ghorani, E.; Akarca, A.U.; Marafioti, T.; et al. CD25-T(reg)-depleting antibodies preserving IL-2 signaling on effector T cells enhance effector activation and antitumor immunity. Nat. Cancer 2020, 1, 1153–1166. [Google Scholar] [CrossRef]
- Kong, D.S.; Nam, D.H.; Kang, S.H.; Lee, J.W.; Chang, J.H.; Kim, J.H.; Lim, Y.J.; Koh, Y.C.; Chung, Y.G.; Kim, J.M.; et al. Phase III randomized trial of autologous cytokine-induced killer cell immunotherapy for newly diagnosed glioblastoma in Korea. Oncotarget 2017, 8, 7003–7013. [Google Scholar] [CrossRef]
- Sanders, S.; Debinski, W. Challenges to Successful Implementation of the Immune Checkpoint Inhibitors for Treatment of Glioblastoma. Int. J. Mol. Sci. 2020, 21, 2759. [Google Scholar] [CrossRef]
- Papademetriou, I.T.; Porter, T. Promising approaches to circumvent the blood-brain barrier: Progress, pitfalls and clinical prospects in brain cancer. Ther. Deliv. 2015, 6, 989–1016. [Google Scholar] [CrossRef]
- Sonabend, A.M.; Gould, A.; Amidei, C.; Ward, R.; Schmidt, K.A.; Zhang, D.Y.; Gomez, C.; Bebawy, J.F.; Liu, B.P.; Bouchoux, G.; et al. Repeated blood-brain barrier opening with an implantable ultrasound device for delivery of albumin-bound paclitaxel in patients with recurrent glioblastoma: A phase 1 trial. Lancet Oncol. 2023, 24, 509–522. [Google Scholar] [CrossRef]
- Liu, L.; Huh, J.R.; Shah, K. Microbiota and the gut-brain-axis: Implications for new therapeutic design in the CNS. EBioMedicine 2022, 77, 103908. [Google Scholar] [CrossRef]
- Patrizz, A.; Dono, A.; Zorofchian, S.; Hines, G.; Takayasu, T.; Husein, N.; Otani, Y.; Arevalo, O.; Choi, H.A.; Savarraj, J.; et al. Glioma and temozolomide induced alterations in gut microbiome. Sci. Rep. 2020, 10, 21002. [Google Scholar] [CrossRef]
- D’Alessandro, G.; Antonangeli, F.; Marrocco, F.; Porzia, A.; Lauro, C.; Santoni, A.; Limatola, C. Gut microbiota alterations affect glioma growth and innate immune cells involved in tumor immunosurveillance in mice. Eur. J. Immunol. 2020, 50, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Naghavian, R.; Faigle, W.; Oldrati, P.; Wang, J.; Toussaint, N.C.; Qiu, Y.; Medici, G.; Wacker, M.; Freudenmann, L.K.; Bonte, P.E.; et al. Microbial peptides activate tumour-infiltrating lymphocytes in glioblastoma. Nature 2023, 617, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Genoud, V.; Kinnersley, B.; Brown, N.F.; Ottaviani, D.; Mulholland, P. Therapeutic Targeting of Glioblastoma and the Interactions with Its Microenvironment. Cancers 2023, 15, 5790. https://doi.org/10.3390/cancers15245790
Genoud V, Kinnersley B, Brown NF, Ottaviani D, Mulholland P. Therapeutic Targeting of Glioblastoma and the Interactions with Its Microenvironment. Cancers. 2023; 15(24):5790. https://doi.org/10.3390/cancers15245790
Chicago/Turabian StyleGenoud, Vassilis, Ben Kinnersley, Nicholas F. Brown, Diego Ottaviani, and Paul Mulholland. 2023. "Therapeutic Targeting of Glioblastoma and the Interactions with Its Microenvironment" Cancers 15, no. 24: 5790. https://doi.org/10.3390/cancers15245790