
Epigenetic and Genetic Keys to Fight HPV-Related Cancers
 , , , , , , ,
, , , , , , ,
Abstract
:Simple Summary
Abstract
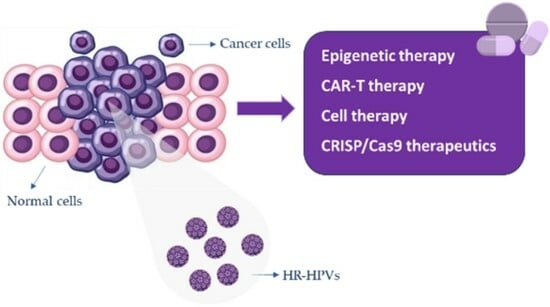
1. Introduction
2. Human HPV Features
2.1. HPV Structures
2.2. HPV Life Cycle and Treatment Strategies
2.3. HPV Carcinogenesis and Vaccine/Treatment Strategies
3. HPV: Epigenetic Modulators
3.1. HPV Chromatin Structure and Epigenetic Regulation of Transcription
3.2. HPV-Induced Host Epigenetic Changes
3.3. Epigenetic Modulators for the Treatment of HPV-Induced Infections and Tumors
3.3.1. HPV and HDAC Inhibitor
3.3.2. HPV and DNMT Modulators
3.3.3. Considerations: Epigenetic Drugs
4. HPV: Genetic and Immunotherapy
4.1. Engineered T-Cell Therapy
Considerations: Immunotherapy
4.2. CRISPR-CAS9
Considerations: CRISPR-CAS9
5. Conclusions and Future Directions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Munger, K. Viruses Associated with Human Cancer. Biochim. Biophys. Acta 2008, 1782, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Soheili, M.; Keyvani, H.; Soheili, M.; Nasseri, S. Human Papilloma Virus: A Review Study of Epidemiology, Carcinogenesis, Diagnostic Methods, and Treatment of All HPV-Related Cancers. Med. J. Islam. Repub. Iran 2021, 35, 65. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, X. N6-Methyladenosine and Its Implications in Viruses. Genom. Proteom. Bioinform. 2022. [Google Scholar] [CrossRef] [PubMed]
- Dom-Chima, N.; Ajang, Y.A.; Dom-Chima, C.I.; Biswas-Fiss, E.; Aminu, M.; Biswas, S.B. Human Papillomavirus Spectrum of HPV-Infected Women in Nigeria: An Analysis by next-Generation Sequencing and Type-Specific PCR. Virol. J. 2023, 20, 144. [Google Scholar] [CrossRef] [PubMed]
- Gusho, E.; Laimins, L. Human Papillomaviruses Target the DNA Damage Repair and Innate Immune Response Pathways to Allow for Persistent Infection. Viruses 2021, 13, 1390. [Google Scholar] [CrossRef] [PubMed]
- Hofstetter, A.M.; Rosenthal, S.L. Factors Impacting HPV Vaccination: Lessons for Health Care Professionals. Expert Rev. Vaccines 2014, 13, 1013–1026. [Google Scholar] [CrossRef]
- Graham, S.V. The Human Papillomavirus Replication Cycle, and Its Links to Cancer Progression: A Comprehensive Review. Clin. Sci. 2017, 131, 2201–2221. [Google Scholar] [CrossRef]
- Mac, M.; Moody, C.A. Epigenetic Regulation of the Human Papillomavirus Life Cycle. Pathogens 2020, 9, 483. [Google Scholar] [CrossRef]
- Soto, D.; Song, C.; McLaughlin-Drubin, M.E. Epigenetic Alterations in Human Papillomavirus-Associated Cancers. Viruses 2017, 9, 248. [Google Scholar] [CrossRef]
- Durzynska, J.; Lesniewicz, K.; Poreba, E. Human Papillomaviruses in Epigenetic Regulations. Mutat. Res. Rev. Mutat. Res. 2017, 772, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Amaro-Filho, S.M.; Pereira Chaves, C.B.; Felix, S.P.; Basto, D.L.; de Almeida, L.M.; Moreira, M.A.M. HPV DNA Methylation at the Early Promoter and E1/E2 Integrity: A Comparison between HPV16, HPV18 and HPV45 in Cervical Cancer. Papillomavirus Res. 2018, 5, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Hewavisenti, R.V.; Arena, J.; Ahlenstiel, C.L.; Sasson, S.C. Human Papillomavirus in the Setting of Immunodeficiency: Pathogenesis and the Emergence of next-Generation Therapies to Reduce the High Associated Cancer Risk. Front. Immunol. 2023, 14, 1112513. [Google Scholar] [CrossRef] [PubMed]
- Antra, N.; Parashar, P.; Hungyo, H.; Jain, A.; Ahmad, S.; Tandon, V. Unraveling Molecular Mechanisms of Head and Neck Cancer. Crit. Rev. Oncol. Hematol. 2022, 178, 103778. [Google Scholar] [CrossRef]
- Gheit, T. Mucosal and Cutaneous Human Papillomavirus Infections and Cancer Biology. Front. Oncol. 2019, 9, 355. [Google Scholar] [CrossRef]
- Keiffer, T.R.; Soorya, S.; Sapp, M.J. Recent Advances in Our Understanding of the Infectious Entry Pathway of Human Papillomavirus Type 16. Microorganisms 2021, 9, 2076. [Google Scholar] [CrossRef]
- Cerqueira, C.; Schiller, J.T. Papillomavirus Assembly: An Overview and Perspectives. Virus Res. 2017, 231, 103–107. [Google Scholar] [CrossRef]
- DiGiuseppe, S.; Luszczek, W.; Keiffer, T.R.; Bienkowska-Haba, M.; Guion, L.G.M.; Sapp, M.J. Incoming Human Papillomavirus Type 16 Genome Resides in a Vesicular Compartment throughout Mitosis. Proc. Natl. Acad. Sci. USA 2016, 113, 6289–6294. [Google Scholar] [CrossRef]
- Pešut, E.; Đukić, A.; Lulić, L.; Skelin, J.; Šimić, I.; Milutin Gašperov, N.; Tomaić, V.; Sabol, I.; Grce, M. Human Papillomaviruses-Associated Cancers: An Update of Current Knowledge. Viruses 2021, 13, 2234. [Google Scholar] [CrossRef]
- Haręża, D.A.; Wilczyński, J.R.; Paradowska, E. Human Papillomaviruses as Infectious Agents in Gynecological Cancers. Oncogenic Properties of Viral Proteins. Int. J. Mol. Sci. 2022, 23, 1818. [Google Scholar] [CrossRef]
- Yu, L.; Majerciak, V.; Zheng, Z.-M. HPV16 and HPV18 Genome Structure, Expression, and Post-Transcriptional Regulation. Int. J. Mol. Sci. 2022, 23, 4943. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; Liu, M.; Li, W.; Sun, B.; Liu, D.; Wang, G.; Shi, J.; Li, L. Protein Post-Translational Modification in SARS-CoV-2 and Host Interaction. Front. Immunol. 2022, 13, 1068449. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, A.; Félez-Sánchez, M.; Bravo, I.G. Genome Plasticity in Papillomaviruses and De Novo Emergence of E5 Oncogenes. Genome Biol. Evol. 2019, 11, 1602–1617. [Google Scholar] [CrossRef] [PubMed]
- Olmedo-Nieva, L.; Muñoz-Bello, J.O.; Contreras-Paredes, A.; Lizano, M. The Role of E6 Spliced Isoforms (E6*) in Human Papillomavirus-Induced Carcinogenesis. Viruses 2018, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Coursey, T.L.; Van Doorslaer, K.; McBride, A.A. Regulation of Human Papillomavirus 18 Genome Replication, Establishment, and Persistence by Sequences in the Viral Upstream Regulatory Region. J. Virol. 2021, 95, e0068621. [Google Scholar] [CrossRef] [PubMed]
- Ajiro, M.; Tang, S.; Doorbar, J.; Zheng, Z.-M. Serine/Arginine-Rich Splicing Factor 3 and Heterogeneous Nuclear Ribonucleoprotein A1 Regulate Alternative RNA Splicing and Gene Expression of Human Papillomavirus 18 through Two Functionally Distinguishable Cis Elements. J. Virol. 2016, 90, 9138–9152. [Google Scholar] [CrossRef]
- Yilmaz, G.; Biswas-Fiss, E.E.; Biswas, S.B. Sequence-Dependent Interaction of the Human Papillomavirus E2 Protein with the DNA Elements on Its DNA Replication Origin. Int. J. Mol. Sci. 2023, 24, 6555. [Google Scholar] [CrossRef]
- Castro-Muñoz, L.J.; Manzo-Merino, J.; Muñoz-Bello, J.O.; Olmedo-Nieva, L.; Cedro-Tanda, A.; Alfaro-Ruiz, L.A.; Hidalgo-Miranda, A.; Madrid-Marina, V.; Lizano, M. The Human Papillomavirus (HPV) E1 Protein Regulates the Expression of Cellular Genes Involved in Immune Response. Sci. Rep. 2019, 9, 13620. [Google Scholar] [CrossRef]
- Chojnacki, M.; Melendy, T. The HPV E2 Transcriptional Transactivation Protein Stimulates Cellular DNA Polymerase Epsilon. Viruses 2018, 10, 321. [Google Scholar] [CrossRef]
- Mu, Y.; Tews, B.A.; Luttermann, C.; Meyers, G. Interaction of Pestiviral E1 and E2 Sequences in Dimer Formation and Intracellular Retention. Int. J. Mol. Sci. 2021, 22, 7285. [Google Scholar] [CrossRef]
- Gammoh, N.; Grm, H.S.; Massimi, P.; Banks, L. Regulation of Human Papillomavirus Type 16 E7 Activity through Direct Protein Interaction with the E2 Transcriptional Activator. J. Virol. 2006, 80, 1787–1797. [Google Scholar] [CrossRef]
- Bellanger, S.; Blachon, S.; Mechali, F.; Bonne-Andrea, C.; Thierry, F. High-Risk but Not Low-Risk HPV E2 Proteins Bind to the APC Activators Cdh1 and Cdc20 and Cause Genomic Instability. Cell Cycle 2005, 4, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A.; McPhillips, M.G.; Oliveira, J.G. Brd4: Tethering, Segregation and Beyond. Trends Microbiol. 2004, 12, 527–529. [Google Scholar] [CrossRef]
- Parish, J.L.; Kowalczyk, A.; Chen, H.-T.; Roeder, G.E.; Sessions, R.; Buckle, M.; Gaston, K. E2 Proteins from High- and Low-Risk Human Papillomavirus Types Differ in Their Ability to Bind P53 and Induce Apoptotic Cell Death. J. Virol. 2006, 80, 4580–4590. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.-M.; Baker, C.C. Papillomavirus Genome Structure, Expression, and Post-Transcriptional Regulation. Front. Biosci. 2006, 11, 2286–2302. [Google Scholar] [CrossRef] [PubMed]
- Prati, B.; Marangoni, B.; Boccardo, E. Human Papillomavirus and Genome Instability: From Productive Infection to Cancer. Clinics 2018, 73, e539s. [Google Scholar] [CrossRef] [PubMed]
- Yajid, A.I.; Zakariah, M.A.; Mat Zin, A.A.; Othman, N.H. Potential Role of E4 Protein in Human Papillomavirus Screening: A Review. Asian Pac. J. Cancer Prev. 2017, 18, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Davy, C.; Doorbar, J. G2/M Cell Cycle Arrest in the Life Cycle of Viruses. Virology 2007, 368, 219–226. [Google Scholar] [CrossRef]
- Ren, S.; Gaykalova, D.A.; Guo, T.; Favorov, A.V.; Fertig, E.J.; Tamayo, P.; Callejas-Valera, J.L.; Allevato, M.; Gilardi, M.; Santos, J.; et al. HPV E2, E4, E5 Drive Alternative Carcinogenic Pathways in HPV Positive Cancers. Oncogene 2020, 39, 6327–6339. [Google Scholar] [CrossRef]
- Przybylski, M.; Pruski, D.; Millert-Kalińska, S.; Krzyżaniak, M.; de Mezer, M.; Frydrychowicz, M.; Jach, R.; Żurawski, J. Expression of E4 Protein and HPV Major Capsid Protein (L1) as A Novel Combination in Squamous Intraepithelial Lesions. Biomedicines 2023, 11, 225. [Google Scholar] [CrossRef]
- Venuti, A.; Paolini, F.; Nasir, L.; Corteggio, A.; Roperto, S.; Campo, M.S.; Borzacchiello, G. Papillomavirus E5: The Smallest Oncoprotein with Many Functions. Mol. Cancer 2011, 10, 140. [Google Scholar] [CrossRef]
- Ilahi, N.E.; Bhatti, A. Impact of HPV E5 on Viral Life Cycle via EGFR Signaling. Microb. Pathog. 2020, 139, 103923. [Google Scholar] [CrossRef] [PubMed]
- de Freitas, A.C.; de Oliveira, T.H.A.; Barros, M.R.; Venuti, A. HrHPV E5 Oncoprotein: Immune Evasion and Related Immunotherapies. J. Exp. Clin. Cancer Res. 2017, 36, 71. [Google Scholar] [CrossRef] [PubMed]
- Tomaić, V. Functional Roles of E6 and E7 Oncoproteins in HPV-Induced Malignancies at Diverse Anatomical Sites. Cancers 2016, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Shimada, M.; Yamashita, A.; Saito, M.; Ichino, M.; Kinjo, T.; Mizuki, N.; Klinman, D.M.; Okuda, K. The Human Papillomavirus E6 Protein Targets Apoptosis-Inducing Factor (AIF) for Degradation. Sci. Rep. 2020, 10, 14195. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hong, X.; Wei, Z.; Xie, M.; Li, W.; Liu, G.; Guo, H.; Yang, J.; Wei, W.; Zhang, S. Ubiquitination of the HPV Oncoprotein E6 Is Critical for E6/E6AP-Mediated P53 Degradation. Front. Microbiol. 2019, 10, 2483. [Google Scholar] [CrossRef] [PubMed]
- Scarth, J.A.; Patterson, M.R.; Morgan, E.L.; Macdonald, A. The Human Papillomavirus Oncoproteins: A Review of the Host Pathways Targeted on the Road to Transformation. J. Gen. Virol. 2021, 102, 001540. [Google Scholar] [CrossRef] [PubMed]
- Roman, A.; Munger, K. The Papillomavirus E7 Proteins. Virology 2013, 445, 138–168. [Google Scholar] [CrossRef]
- Dick, F.A.; Rubin, S.M. Molecular Mechanisms Underlying RB Protein Function. Nat. Rev. Mol. Cell Biol. 2013, 14, 297–306. [Google Scholar] [CrossRef]
- Nor Rashid, N.; Yusof, R.; Watson, R.J. Disruption of Repressive P130-DREAM Complexes by Human Papillomavirus 16 E6/E7 Oncoproteins Is Required for Cell-Cycle Progression in Cervical Cancer Cells. J. Gen. Virol. 2011, 92, 2620–2627. [Google Scholar] [CrossRef]
- Nor Rashid, N.; Yong, Z.L.; Yusof, R.; Watson, R.J. HPV 16E7 and 48E7 Proteins Use Different Mechanisms to Target P130 to Overcome Cell Cycle Block. Virol. J. 2016, 13, 2. [Google Scholar] [CrossRef]
- James, C.D.; Saini, S.; Sesay, F.; Ko, K.; Felthousen-Rusbasan, J.; Iness, A.N.; Nulton, T.; Windle, B.; Dozmorov, M.G.; Morgan, I.M.; et al. Restoring the DREAM Complex Inhibits the Proliferation of High-Risk HPV Positive Human Cells. Cancers 2021, 13, 489. [Google Scholar] [CrossRef] [PubMed]
- Emanuele, M.J.; Enrico, T.P.; Mouery, R.D.; Wasserman, D.; Nachum, S.; Tzur, A. Complex Cartography: Regulation of E2F Transcription Factors by Cyclin F and Ubiquitin. Trends Cell Biol. 2020, 30, 640–652. [Google Scholar] [CrossRef] [PubMed]
- Guiley, K.Z.; Liban, T.J.; Felthousen, J.G.; Ramanan, P.; Litovchick, L.; Rubin, S.M. Structural Mechanisms of DREAM Complex Assembly and Regulation. Genes Dev. 2015, 29, 961–974. [Google Scholar] [CrossRef] [PubMed]
- Engeland, K. Cell Cycle Regulation: P53-P21-RB Signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef] [PubMed]
- Jansma, A.L.; Martinez-Yamout, M.A.; Liao, R.; Sun, P.; Dyson, H.J.; Wright, P.E. The High-Risk HPV16 E7 Oncoprotein Mediates Interaction between the Transcriptional Coactivator CBP and the Retinoblastoma Protein PRb. J. Mol. Biol. 2014, 426, 4030–4048. [Google Scholar] [CrossRef]
- Basukala, O.; Banks, L. The Not-So-Good, the Bad and the Ugly: HPV E5, E6 and E7 Oncoproteins in the Orchestration of Carcinogenesis. Viruses 2021, 13, 1892. [Google Scholar] [CrossRef] [PubMed]
- Tsakogiannis, D.; Nikolaidis, M.; Zagouri, F.; Zografos, E.; Kottaridi, C.; Kyriakopoulou, Z.; Tzioga, L.; Markoulatos, P.; Amoutzias, G.D.; Bletsa, G. Mutation Profile of HPV16 L1 and L2 Genes in Different Geographic Areas. Viruses 2022, 15, 141. [Google Scholar] [CrossRef]
- Joyce, J.G.; Tung, J.S.; Przysiecki, C.T.; Cook, J.C.; Lehman, E.D.; Sands, J.A.; Jansen, K.U.; Keller, P.M. The L1 Major Capsid Protein of Human Papillomavirus Type 11 Recombinant Virus-like Particles Interacts with Heparin and Cell-Surface Glycosaminoglycans on Human Keratinocytes. J. Biol. Chem. 1999, 274, 5810–5822. [Google Scholar] [CrossRef]
- Wang, J.W.; Roden, R.B.S. L2, the Minor Capsid Protein of Papillomavirus. Virology 2013, 445, 175–186. [Google Scholar] [CrossRef]
- Darshan, M.S.; Lucchi, J.; Harding, E.; Moroianu, J. The L2 Minor Capsid Protein of Human Papillomavirus Type 16 Interacts with a Network of Nuclear Import Receptors. J. Virol. 2004, 78, 12179–12188. [Google Scholar] [CrossRef]
- Yarbrough, M.L.; Mata, M.A.; Sakthivel, R.; Fontoura, B.M.A. Viral Subversion of Nucleocytoplasmic Trafficking. Traffic 2014, 15, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Nelson, L.M.; Rose, R.C.; Moroianu, J. Nuclear Import Strategies of High Risk HPV16 L1 Major Capsid Protein. J. Biol. Chem. 2002, 277, 23958–23964. [Google Scholar] [CrossRef]
- Morante, A.V.; Baboolal, D.D.; Simon, X.; Pan, E.C.-Y.; Meneses, P.I. Human Papillomavirus Minor Capsid Protein L2 Mediates Intracellular Trafficking into and Passage beyond the Endoplasmic Reticulum. Microbiol. Spectr. 2022, 10, e0150522. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, X.; Jiao, Y.; Wu, C. High-Risk Human Papillomavirus Oncogenic E6/E7 MRNAs Splicing Regulation. Front. Cell Infect. Microbiol. 2022, 12, 929666. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J. The E4 Protein; Structure, Function and Patterns of Expression. Virology 2013, 445, 80–98. [Google Scholar] [CrossRef] [PubMed]
- Boxman, I.L.; Hogewoning, A.; Mulder, L.H.; Bouwes Bavinck, J.N.; ter Schegget, J. Detection of Human Papillomavirus Types 6 and 11 in Pubic and Perianal Hair from Patients with Genital Warts. J. Clin. Microbiol. 1999, 37, 2270–2273. [Google Scholar] [CrossRef]
- Egawa, N.; Egawa, K.; Griffin, H.; Doorbar, J. Human Papillomaviruses; Epithelial Tropisms, and the Development of Neoplasia. Viruses 2015, 7, 3863–3890. [Google Scholar] [CrossRef]
- Budhwani, M.; Lukowski, S.W.; Porceddu, S.V.; Frazer, I.H.; Chandra, J. Dysregulation of Stemness Pathways in HPV Mediated Cervical Malignant Transformation Identifies Potential Oncotherapy Targets. Front. Cell Infect. Microbiol. 2020, 10, 307. [Google Scholar] [CrossRef]
- Richards, R.M.; Lowy, D.R.; Schiller, J.T.; Day, P.M. Cleavage of the Papillomavirus Minor Capsid Protein, L2, at a Furin Consensus Site Is Necessary for Infection. Proc. Natl. Acad. Sci. USA 2006, 103, 1522–1527. [Google Scholar] [CrossRef]
- Surviladze, Z.; Dziduszko, A.; Ozbun, M.A. Essential Roles for Soluble Virion-Associated Heparan Sulfonated Proteoglycans and Growth Factors in Human Papillomavirus Infections. PLoS Pathog. 2012, 8, e1002519. [Google Scholar] [CrossRef]
- Abban, C.Y.; Meneses, P.I. Usage of Heparan Sulfate, Integrins, and FAK in HPV16 Infection. Virology 2010, 403, 1–16. [Google Scholar] [CrossRef]
- Evander, M.; Frazer, I.H.; Payne, E.; Qi, Y.M.; Hengst, K.; McMillan, N.A. Identification of the Alpha6 Integrin as a Candidate Receptor for Papillomaviruses. J. Virol. 1997, 71, 2449–2456. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, K.D.; Gawlitza, A.; Spoden, G.A.; Zhang, X.A.; Lambert, C.; Berditchevski, F.; Florin, L. Tetraspanin CD151 Mediates Papillomavirus Type 16 Endocytosis. J. Virol. 2013, 87, 3435–3446. [Google Scholar] [CrossRef] [PubMed]
- Culp, T.D.; Budgeon, L.R.; Marinkovich, M.P.; Meneguzzi, G.; Christensen, N.D. Keratinocyte-Secreted Laminin 5 Can Function as a Transient Receptor for Human Papillomaviruses by Binding Virions and Transferring Them to Adjacent Cells. J. Virol. 2006, 80, 8940–8950. [Google Scholar] [CrossRef]
- Shafti-Keramat, S.; Handisurya, A.; Kriehuber, E.; Meneguzzi, G.; Slupetzky, K.; Kirnbauer, R. Different Heparan Sulfate Proteoglycans Serve as Cellular Receptors for Human Papillomaviruses. J. Virol. 2003, 77, 13125–13135. [Google Scholar] [CrossRef] [PubMed]
- Woodham, A.W.; Da Silva, D.M.; Skeate, J.G.; Raff, A.B.; Ambroso, M.R.; Brand, H.E.; Isas, J.M.; Langen, R.; Kast, W.M. The S100A10 Subunit of the Annexin A2 Heterotetramer Facilitates L2-Mediated Human Papillomavirus Infection. PLoS ONE 2012, 7, e43519. [Google Scholar] [CrossRef] [PubMed]
- Dziduszko, A.; Ozbun, M.A. Annexin A2 and S100A10 Regulate Human Papillomavirus Type 16 Entry and Intracellular Trafficking in Human Keratinocytes. J. Virol. 2013, 87, 7502–7515. [Google Scholar] [CrossRef] [PubMed]
- Võsa, L.; Sudakov, A.; Remm, M.; Ustav, M.; Kurg, R. Identification and Analysis of Papillomavirus E2 Protein Binding Sites in the Human Genome. J. Virol. 2012, 86, 348–357. [Google Scholar] [CrossRef]
- Pal, A.; Kundu, R. Human Papillomavirus E6 and E7: The Cervical Cancer Hallmarks and Targets for Therapy. Front. Microbiol. 2019, 10, 3116. [Google Scholar] [CrossRef]
- Vega, T. Outcome Standards for Public Cancer Education: The Foundation for Community Education Programs. Oncol. Nurs. Forum 1985, 12, 66–67. [Google Scholar]
- White, E.A. Manipulation of Epithelial Differentiation by HPV Oncoproteins. Viruses 2019, 11, 369. [Google Scholar] [CrossRef]
- McLachlin, C.M.; Tate, J.E.; Zitz, J.C.; Sheets, E.E.; Crum, C.P. Human Papillomavirus Type 18 and Intraepithelial Lesions of the Cervix. Am. J. Pathol. 1994, 144, 141–147. [Google Scholar] [PubMed]
- Stoler, M.H.; Rhodes, C.R.; Whitbeck, A.; Wolinsky, S.M.; Chow, L.T.; Broker, T.R. Human Papillomavirus Type 16 and 18 Gene Expression in Cervical Neoplasias. Hum. Pathol. 1992, 23, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Middleton, K.; Peh, W.; Southern, S.; Griffin, H.; Sotlar, K.; Nakahara, T.; El-Sherif, A.; Morris, L.; Seth, R.; Hibma, M.; et al. Organization of Human Papillomavirus Productive Cycle during Neoplastic Progression Provides a Basis for Selection of Diagnostic Markers. J. Virol. 2003, 77, 10186–10201. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Xicotencatl, L.; Pedroza-Saavedra, A.; Chihu-Amparan, L.; Salazar-Piña, A.; Maldonado-Gama, M.; Esquivel-Guadarrama, F. Cellular Functions of HPV16 E5 Oncoprotein during Oncogenic Transformation. Mol. Cancer Res. 2021, 19, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Gunasekharan, V.; Laimins, L.A. Human Papillomaviruses Modulate MicroRNA 145 Expression to Directly Control Genome Amplification. J. Virol. 2013, 87, 6037–6043. [Google Scholar] [CrossRef]
- Graham, S.V.; Faizo, A.A.A. Control of Human Papillomavirus Gene Expression by Alternative Splicing. Virus Res. 2017, 231, 83–95. [Google Scholar] [CrossRef]
- Johansson, C.; Schwartz, S. Regulation of Human Papillomavirus Gene Expression by Splicing and Polyadenylation. Nat. Rev. Microbiol. 2013, 11, 239–251. [Google Scholar] [CrossRef]
- Guion, L.; Bienkowska-Haba, M.; DiGiuseppe, S.; Florin, L.; Sapp, M. PML Nuclear Body-Residing Proteins Sequentially Associate with HPV Genome after Infectious Nuclear Delivery. PLoS Pathog. 2019, 15, e1007590. [Google Scholar] [CrossRef]
- Sofiani, V.H.; Veisi, P.; Rukerd, M.R.Z.; Ghazi, R.; Nakhaie, M. The complexity of human papilloma virus in cancers: A narrative review. Infect. Agent Cancer 2023, 18, 13. [Google Scholar] [CrossRef]
- Calton, C.M.; Bronnimann, M.P.; Manson, A.R.; Li, S.; Chapman, J.A.; Suarez-Berumen, M.; Williamson, T.R.; Molugu, S.K.; Bernal, R.A.; Campos, S.K. Translocation of the Papillomavirus L2/VDNA Complex across the Limiting Membrane Requires the Onset of Mitosis. PLoS Pathog. 2017, 13, e1006200. [Google Scholar] [CrossRef] [PubMed]
- Archambault, J.; Melendy, T. Targeting Human Papillomavirus Genome Replication for Antiviral Drug Discovery. Antivir. Ther. 2013, 18, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.G.; Vidmar, T.J.; Koeller, K.; Bashkin, J.K.; Fisher, C. DNA Damage Repair Genes Controlling Human Papillomavirus (HPV) Episome Levels under Conditions of Stability and Extreme Instability. PLoS ONE 2013, 8, e75406. [Google Scholar] [CrossRef] [PubMed]
- Okunade, K.S. Human Papillomavirus and Cervical Cancer. J. Obs. Gynaecol. 2020, 40, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, A.R.; Nyitray, A.G.; Kreimer, A.R.; Pierce Campbell, C.M.; Goodman, M.T.; Sudenga, S.L.; Monsonego, J.; Franceschi, S. EUROGIN 2014 Roadmap: Differences in Human Papillomavirus Infection Natural History, Transmission and Human Papillomavirus-Related Cancer Incidence by Gender and Anatomic Site of Infection. Int. J. Cancer 2015, 136, 2752–2760. [Google Scholar] [CrossRef]
- Koneva, L.A.; Zhang, Y.; Virani, S.; Hall, P.B.; McHugh, J.B.; Chepeha, D.B.; Wolf, G.T.; Carey, T.E.; Rozek, L.S.; Sartor, M.A. HPV Integration in HNSCC Correlates with Survival Outcomes, Immune Response Signatures, and Candidate Drivers. Mol. Cancer Res. 2018, 16, 90–102. [Google Scholar] [CrossRef]
- Schrank, T.P.; Kim, S.; Rehmani, H.; Kothari, A.; Wu, D.; Yarbrough, W.G.; Issaeva, N. Direct Comparison of HPV16 Viral Genomic Integration, Copy Loss, and Structural Variants in Oropharyngeal and Uterine Cervical Cancers Reveal Distinct Relationships to E2 Disruption and Somatic Alteration. Cancers 2022, 14, 4488. [Google Scholar] [CrossRef]
- Valle, G.; Crisma, M.; Toniolo, C.; Holt, E.M.; Tamura, M.; Bland, J.; Stammer, C.H. Crystallographic Characterization of Conformation of 1-Aminocyclopropane-1-Carboxylic Acid Residue (Ac3c) in Simple Derivatives and Peptides. Int. J. Pept. Protein Res. 1989, 34, 56–65. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA Damage, Repair, and Mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef]
- Yeo-Teh, N.S.L.; Ito, Y.; Jha, S. High-Risk Human Papillomaviral Oncogenes E6 and E7 Target Key Cellular Pathways to Achieve Oncogenesis. Int. J. Mol. Sci. 2018, 19, 1706. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human Papillomavirus Molecular Biology and Disease Association. Rev. Med. Virol. 2015, 25 (Suppl. S1), 2–23. [Google Scholar] [CrossRef]
- Leeman, J.E.; Li, Y.; Bell, A.; Hussain, S.S.; Majumdar, R.; Rong-Mullins, X.; Blecua, P.; Damerla, R.; Narang, H.; Ravindran, P.T.; et al. Human Papillomavirus 16 Promotes Microhomology-Mediated End-Joining. Proc. Natl. Acad. Sci. USA 2019, 116, 21573–21579. [Google Scholar] [CrossRef] [PubMed]
- Desfarges, S.; Ciuffi, A. Viral Integration and Consequences on Host Gene Expression. In Viruses: Essential Agents of Life; Witzany, G., Ed.; Springer: Dordrecht, The Netherlands, 2012; pp. 147–175. ISBN 978-94-007-4898-9. [Google Scholar]
- Berman, T.A.; Schiller, J.T. Human Papillomavirus in Cervical Cancer and Oropharyngeal Cancer: One Cause, Two Diseases. Cancer 2017, 123, 2219–2229. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, C.T.; Scholtz, C.H. A Review on the Effect of Macrocyclic Lactones on Dung-Dwelling Insects: Toxicity of Macrocyclic Lactones to Dung Beetles. Onderstepoort J. Vet. Res. 2015, 82, 858. [Google Scholar] [CrossRef]
- Garbuglia, A.R.; Lapa, D.; Sias, C.; Capobianchi, M.R.; Del Porto, P. The Use of Both Therapeutic and Prophylactic Vaccines in the Therapy of Papillomavirus Disease. Front. Immunol. 2020, 11, 188. [Google Scholar] [CrossRef] [PubMed]
- Leite, K.R.M.; Pimenta, R.; Canavez, J.; Canavez, F.; de Souza, F.R.; Vara, L.; Estivallet, C.; Camara-Lopes, L.H. HPV Genotype Prevalence and Success of Vaccination to Prevent Cervical Cancer. Acta Cytol. 2020, 64, 420–424. [Google Scholar] [CrossRef]
- Chan, C.K.; Aimagambetova, G.; Ukybassova, T.; Kongrtay, K.; Azizan, A. Human Papillomavirus Infection and Cervical Cancer: Epidemiology, Screening, and Vaccination-Review of Current Perspectives. J. Oncol. 2019, 2019, 3257939. [Google Scholar] [CrossRef]
- Stanley, M.; Pinto, L.A.; Trimble, C. Human Papillomavirus Vaccines--Immune Responses. Vaccine 2012, 30 (Suppl. S5), F83–F87. [Google Scholar] [CrossRef]
- Kirnbauer, R.; Booy, F.; Cheng, N.; Lowy, D.R.; Schiller, J.T. Papillomavirus L1 Major Capsid Protein Self-Assembles into Virus-like Particles That Are Highly Immunogenic. Proc. Natl. Acad. Sci. USA 1992, 89, 12180–12184. [Google Scholar] [CrossRef]
- Arbyn, M.; Xu, L. Efficacy and Safety of Prophylactic HPV Vaccines. A Cochrane Review of Randomized Trials. Expert Rev. Vaccines 2018, 17, 1085–1091. [Google Scholar] [CrossRef]
- Chabeda, A.; Yanez, R.J.R.; Lamprecht, R.; Meyers, A.E.; Rybicki, E.P.; Hitzeroth, I.I. Therapeutic Vaccines for High-Risk HPV-Associated Diseases. Papillomavirus Res. 2018, 5, 46–58. [Google Scholar] [CrossRef]
- Meyer, T.; Stockfleth, E. Clinical Investigations of Toll-like Receptor Agonists. Expert Opin. Investig. Drugs 2008, 17, 1051–1065. [Google Scholar] [CrossRef]
- Hemmi, H.; Kaisho, T.; Takeuchi, O.; Sato, S.; Sanjo, H.; Hoshino, K.; Horiuchi, T.; Tomizawa, H.; Takeda, K.; Akira, S. Small Anti-Viral Compounds Activate Immune Cells via the TLR7 MyD88-Dependent Signaling Pathway. Nat. Immunol. 2002, 3, 196–200. [Google Scholar] [CrossRef]
- Tran, H.; Moreno, G.; Shumack, S. Imiquimod as a Dermatological Therapy. Expert. Opin. Pharmacother. 2004, 5, 427–438. [Google Scholar] [CrossRef]
- Bilu, D.; Sauder, D.N. Imiquimod: Modes of Action. Br. J. Dermatol. 2003, 149 (Suppl. S66), 5–8. [Google Scholar] [CrossRef]
- Andrei, G.; De Clercq, E.; Snoeck, R. Drug Targets in Cytomegalovirus Infection. Infect. Disord. Drug Targets 2009, 9, 201–222. [Google Scholar] [CrossRef] [PubMed]
- Amine, A.; Rivera, S.; Opolon, P.; Dekkal, M.; Biard, D.S.F.; Bouamar, H.; Louache, F.; McKay, M.J.; Bourhis, J.; Deutsch, E.; et al. Novel Anti-Metastatic Action of Cidofovir Mediated by Inhibition of E6/E7, CXCR4 and Rho/ROCK Signaling in HPV Tumor Cells. PLoS ONE 2009, 4, e5018. [Google Scholar] [CrossRef]
- Donne, A.J.; Hampson, L.; He, X.T.; Day, P.J.R.; Salway, F.; Rothera, M.P.; Homer, J.J.; Hampson, I.N. Potential Risk Factors Associated with the Use of Cidofovir to Treat Benign Human Papillomavirus-Related Disease. Antivir. Ther. 2009, 14, 939–952. [Google Scholar] [CrossRef] [PubMed]
- Snoeck, R.; Noel, J.C.; Muller, C.; De Clercq, E.; Bossens, M. Cidofovir, a New Approach for the Treatment of Cervix Intraepithelial Neoplasia Grade III (CIN III). J. Med. Virol. 2000, 60, 205–209. [Google Scholar] [CrossRef]
- Da Silva, M.L.R.; De Albuquerque, B.H.D.R.; Allyrio, T.A.D.M.F.; De Almeida, V.D.; Cobucci, R.N.D.O.; Bezerra, F.L.; Andrade, V.S.; Lanza, D.C.F.; De Azevedo, J.C.V.; De Araújo, J.M.G.; et al. The Role of HPV-Induced Epigenetic Changes in Cervical Carcinogenesis (Review). Biomed. Rep. 2021, 15, 60. [Google Scholar] [CrossRef] [PubMed]
- Tronick, E.; Hunter, R.G. Waddington, Dynamic Systems, and Epigenetics. Front. Behav. Neurosci. 2016, 10, 107. [Google Scholar] [CrossRef] [PubMed]
- Porter, S.S.; Liddle, J.C.; Browne, K.; Pastrana, D.V.; Garcia, B.A.; Buck, C.B.; Weitzman, M.D.; McBride, A.A. Histone Modifications in Papillomavirus Virion Minichromosomes. mBio 2021, 12, e03274-20. [Google Scholar] [CrossRef]
- Favre, M.; Breitburd, F.; Croissant, O.; Orth, G. Chromatin-like Structures Obtained after Alkaline Disruption of Bovine and Human Papillomaviruses. J. Virol. 1977, 21, 1205–1209. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.L.; Caodaglio, A.S.; Sichero, L. Regulation of HPV Transcription. Clinics 2018, 73, e486s. [Google Scholar] [CrossRef]
- Murakami, I.; Iwata, T.; Morisada, T.; Tanaka, K.; Aoki, D. Nucleosome Positioning on Episomal Human Papillomavirus DNA in Cultured Cells. Pathogens 2021, 10, 772. [Google Scholar] [CrossRef]
- Azmi, I.F.; Watanabe, S.; Maloney, M.F.; Kang, S.; Belsky, J.A.; MacAlpine, D.M.; Peterson, C.L.; Bell, S.P. Nucleosomes Influence Multiple Steps during Replication Initiation. Elife 2017, 6, e22512. [Google Scholar] [CrossRef]
- Songock, W.K.; Kim, S.-M.; Bodily, J.M. The Human Papillomavirus E7 Oncoprotein as a Regulator of Transcription. Virus Res. 2017, 231, 56–75. [Google Scholar] [CrossRef]
- Jacquin, E.; Baraquin, A.; Ramanah, R.; Carcopino, X.; Morel, A.; Valmary-Degano, S.; Bravo, I.G.; de Sanjosé, S.; Riethmuller, D.; Mougin, C.; et al. Methylation of Human Papillomavirus Type 16 CpG Sites at E2-Binding Site 1 (E2BS1), E2BS2, and the Sp1-Binding Site in Cervical Cancer Samples as Determined by High-Resolution Melting Analysis-PCR. J. Clin. Microbiol. 2013, 51, 3207–3215. [Google Scholar] [CrossRef]
- Lee, H.-T.; Oh, S.; Ro, D.H.; Yoo, H.; Kwon, Y.-W. The Key Role of DNA Methylation and Histone Acetylation in Epigenetics of Atherosclerosis. J. Lipid Atheroscler. 2020, 9, 419–434. [Google Scholar] [CrossRef]
- Liu, S.; Chang, W.; Jin, Y.; Feng, C.; Wu, S.; He, J.; Xu, T. The Function of Histone Acetylation in Cervical Cancer Development. Biosci. Rep. 2019, 39, BSR20190527. [Google Scholar] [CrossRef]
- Imhof, A.; Yang, X.J.; Ogryzko, V.V.; Nakatani, Y.; Wolffe, A.P.; Ge, H. Acetylation of General Transcription Factors by Histone Acetyltransferases. Curr. Biol. 1997, 7, 689–692. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. The CBP Co-Activator Is a Histone Acetyltransferase. Nature 1996, 384, 641–643. [Google Scholar] [CrossRef] [PubMed]
- Bernat, A.; Avvakumov, N.; Mymryk, J.S.; Banks, L. Interaction between the HPV E7 Oncoprotein and the Transcriptional Coactivator P300. Oncogene 2003, 22, 7871–7881. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; He, D.; Cheng, L.; Huang, C.; Zhang, Y.; Rao, X.; Kong, Y.; Li, C.; Zhang, Z.; Liu, J.; et al. P300/CBP Inhibition Enhances the Efficacy of Programmed Death-Ligand 1 Blockade Treatment in Prostate Cancer. Oncogene 2020, 39, 3939–3951. [Google Scholar] [CrossRef] [PubMed]
- Langsfeld, E.S.; Bodily, J.M.; Laimins, L.A. The Deacetylase Sirtuin 1 Regulates Human Papillomavirus Replication by Modulating Histone Acetylation and Recruitment of DNA Damage Factors NBS1 and Rad51 to Viral Genomes. PLoS Pathog. 2015, 11, e1005181. [Google Scholar] [CrossRef]
- Das, D.; Bristol, M.L.; Smith, N.W.; James, C.D.; Wang, X.; Pichierri, P.; Morgan, I.M. Werner Helicase Control of Human Papillomavirus 16 E1-E2 DNA Replication Is Regulated by SIRT1 Deacetylation. mBio 2019, 10, e00263-19. [Google Scholar] [CrossRef]
- Groves, I.J.; Knight, E.L.A.; Ang, Q.Y.; Scarpini, C.G.; Coleman, N. HPV16 Oncogene Expression Levels during Early Cervical Carcinogenesis Are Determined by the Balance of Epigenetic Chromatin Modifications at the Integrated Virus Genome. Oncogene 2016, 35, 4773–4786. [Google Scholar] [CrossRef]
- Gautam, D.; Johnson, B.A.; Mac, M.; Moody, C.A. SETD2-Dependent H3K36me3 Plays a Critical Role in Epigenetic Regulation of the HPV31 Life Cycle. PLoS Pathog. 2018, 14, e1007367. [Google Scholar] [CrossRef]
- Thain, A.; Webster, K.; Emery, D.; Clarke, A.R.; Gaston, K. DNA Binding and Bending by the Human Papillomavirus Type 16 E2 Protein. Recognition of an Extended Binding Site. J. Biol. Chem. 1997, 272, 8236–8242. [Google Scholar] [CrossRef]
- Kim, K.; Garner-Hamrick, P.A.; Fisher, C.; Lee, D.; Lambert, P.F. Methylation Patterns of Papillomavirus DNA, Its Influence on E2 Function, and Implications in Viral Infection. J. Virol. 2003, 77, 12450–12459. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Peng, Q.; Jiang, X.; Tan, S.; Yang, Y.; Yang, W.; Han, Y.; Chen, Y.; Oyang, L.; Lin, J.; et al. Metabolic Reprogramming and Epigenetic Modifications in Cancer: From the Impacts and Mechanisms to the Treatment Potential. Exp. Mol. Med. 2023, 55, 1357–1370. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Gonzalez, E.A.; Rameshwar, P.; Etchegaray, J.-P. Non-Coding RNAs as Mediators of Epigenetic Changes in Malignancies. Cancers 2020, 12, 3657. [Google Scholar] [CrossRef]
- Lakshminarasimhan, R.; Liang, G. The Role of DNA Methylation in Cancer. Adv. Exp. Med. Biol. 2016, 945, 151–172. [Google Scholar] [CrossRef] [PubMed]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Ganguly, P.; Ganguly, N. Modulation of DNA Methylation by Human Papillomavirus E6 and E7 Oncoproteins in Cervical Cancer. Oncol. Lett. 2018, 15, 11–22. [Google Scholar] [CrossRef]
- Wang, Z.; Sturgis, E.M.; Zhang, F.; Lei, D.; Liu, Z.; Xu, L.; Song, X.; Wei, Q.; Li, G. Genetic Variants of P27 and P21 as Predictors for Risk of Second Primary Malignancy in Patients with Index Squamous Cell Carcinoma of Head and Neck. Mol. Cancer 2012, 11, 17. [Google Scholar] [CrossRef]
- Qi, Q.; Ling, Y.; Zhu, M.; Zhou, L.; Wan, M.; Bao, Y.; Liu, Y. Promoter Region Methylation and Loss of Protein Expression of PTEN and Significance in Cervical Cancer. Biomed. Rep. 2014, 2, 653–658. [Google Scholar] [CrossRef]
- Rudnizky, S.; Malik, O.; Bavly, A.; Pnueli, L.; Melamed, P.; Kaplan, A. Nucleosome Mobility and the Regulation of Gene Expression: Insights from Single-Molecule Studies. Protein Sci. 2017, 26, 1266–1277. [Google Scholar] [CrossRef]
- Boscolo-Rizzo, P.; Furlan, C.; Lupato, V.; Polesel, J.; Fratta, E. Novel Insights into Epigenetic Drivers of Oropharyngeal Squamous Cell Carcinoma: Role of HPV and Lifestyle Factors. Clin. Epigenet. 2017, 9, 124. [Google Scholar] [CrossRef]
- Parbin, S.; Kar, S.; Shilpi, A.; Sengupta, D.; Deb, M.; Rath, S.K.; Patra, S.K. Histone Deacetylases: A Saga of Perturbed Acetylation Homeostasis in Cancer. J. Histochem. Cytochem. 2014, 62, 11–33. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dakic, A.; Chen, R.; Dai, Y.; Schlegel, R.; Liu, X. Direct HPV E6/Myc interactions induce histone modifications, Pol II phosphorylation, and hTERT promoter activation. Oncotarget 2017, 8, 96323–96339. [Google Scholar] [CrossRef] [PubMed]
- Pańczyszyn, A.; Boniewska-Bernacka, E.; Głąb, G. Telomeres and Telomerase During Human Papillomavirus-Induced Carcinogenesis. Mol. Diagn. Ther. 2018, 22, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Morel, A.; Baguet, A.; Perrard, J.; Demeret, C.; Jacquin, E.; Guenat, D.; Mougin, C.; Prétet, J.-L. 5azadC Treatment Upregulates MiR-375 Level and Represses HPV16 E6 Expression. Oncotarget 2017, 8, 46163–46176. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulou, A.F.; Velentzas, A.D.; Konstantakou, E.G.; Avgeris, M.; Katarachia, S.A.; Papandreou, N.C.; Kalavros, N.I.; Mpakou, V.E.; Iconomidou, V.; Anastasiadou, E.; et al. Revisiting Histone Deacetylases in Human Tumorigenesis: The Paradigm of Urothelial Bladder Cancer. Int. J. Mol. Sci. 2019, 20, 1291. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Galloway, D.A. Novel Functions of the Human Papillomavirus E6 Oncoproteins. Annu. Rev. Virol. 2015, 2, 403–423. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Loo, J.X.; Shi, X.; Xiong, W.; Guo, Y.; Ke, H.; Yang, M.; Jiang, Y.; Xia, S.; Zhao, M.; et al. E6 Protein Expressed by High-Risk HPV Activates Super-Enhancers of the EGFR and c-MET Oncogenes by Destabilizing the Histone Demethylase KDM5C. Cancer Res. 2018, 78, 1418–1430. [Google Scholar] [CrossRef]
- Rasi Bonab, F.; Baghbanzadeh, A.; Ghaseminia, M.; Bolandi, N.; Mokhtarzadeh, A.; Amini, M.; Dadashzadeh, K.; Hajiasgharzadeh, K.; Baradaran, B.; Bannazadeh Baghi, H. Molecular Pathways in the Development of HPV-Induced Cervical Cancer. EXCLI J. 2021, 20, 320–337. [Google Scholar] [CrossRef]
- Annese, T.; Tamma, R.; De Giorgis, M.; Ribatti, D. MicroRNAs Biogenesis, Functions and Role in Tumor Angiogenesis. Front. Oncol. 2020, 10, 581007. [Google Scholar] [CrossRef]
- Yang, M.; Zhai, X.; Xia, B.; Wang, Y.; Lou, G. Long Noncoding RNA CCHE1 Promotes Cervical Cancer Cell Proliferation via Upregulating PCNA. Tumour Biol. 2015, 36, 7615–7622. [Google Scholar] [CrossRef]
- Teng, J.; Guo, X.; Wang, H. CCEPR Is a Novel Clinical Biomarker for Prognosis and Regulates Cell Proliferation through PCNA in Osteosarcoma. J. Cell Biochem. 2019, 120, 12796–12802. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Fan, H. Long Noncoding RNA CCHE1 Indicates a Poor Prognosis of Hepatocellular Carcinoma and Promotes Carcinogenesis via Activation of the ERK/MAPK Pathway. Biomed. Pharmacother. 2016, 83, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.-C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long Noncoding RNA as Modular Scaffold of Histone Modification Complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Mandal, P.; Sadhukhan, T.; Roy Chowdhury, R.; Ranjan Mondal, N.; Chakravarty, B.; Chatterjee, T.; Roy, S.; Sengupta, S. Bridging Links between Long Noncoding RNA HOTAIR and HPV Oncoprotein E7 in Cervical Cancer Pathogenesis. Sci. Rep. 2015, 5, 11724. [Google Scholar] [CrossRef] [PubMed]
- Carse, S.; Bergant, M.; Schäfer, G. Advances in Targeting HPV Infection as Potential Alternative Prophylactic Means. Int. J. Mol. Sci. 2021, 22, 2201. [Google Scholar] [CrossRef] [PubMed]
- Rajan, P.K.; Udoh, U.-A.; Sanabria, J.D.; Banerjee, M.; Smith, G.; Schade, M.S.; Sanabria, J.; Sodhi, K.; Pierre, S.; Xie, Z.; et al. The Role of Histone Acetylation-/Methylation-Mediated Apoptotic Gene Regulation in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 8894. [Google Scholar] [CrossRef] [PubMed]
- Haery, L.; Thompson, R.C.; Gilmore, T.D. Histone Acetyltransferases and Histone Deacetylases in B- and T-Cell Development, Physiology and Malignancy. Genes Cancer 2015, 6, 184–213. [Google Scholar] [CrossRef]
- Nitsch, S.; Zorro Shahidian, L.; Schneider, R. Histone Acylations and Chromatin Dynamics: Concepts, Challenges, and Links to Metabolism. EMBO Rep. 2021, 22, e52774. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Mottamal, M.; Zheng, S.; Huang, T.L.; Wang, G. Histone Deacetylase Inhibitors in Clinical Studies as Templates for New Anticancer Agents. Molecules 2015, 20, 3898–3941. [Google Scholar] [CrossRef]
- Manta, A.; Kazanas, S.; Karamaroudis, S.; Gogas, H.; Ziogas, D.C. Histone Deacetylase Inhibitors as a Novel Therapeutic Approach for Pheochromocytomas and Paragangliomas. Oncol. Res. 2022, 30, 211–219. [Google Scholar] [CrossRef]
- Sangwan, R.; Rajan, R.; Mandal, P.K. HDAC as Onco Target: Reviewing the Synthetic Approaches with SAR Study of Their Inhibitors. Eur. J. Med. Chem. 2018, 158, 620–706. [Google Scholar] [CrossRef]
- Banerjee, S.; Adhikari, N.; Amin, S.A.; Jha, T. Histone Deacetylase 8 (HDAC8) and Its Inhibitors with Selectivity to Other Isoforms: An Overview. Eur. J. Med. Chem. 2019, 164, 214–240. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Wang, F.; Elhassan, R.M.; Cheng, Y.; Tang, X.; Chen, W.; Fang, H.; Hou, X. Targeting Histone Deacetylases for Cancer Therapy: Trends and Challenges. Acta Pharm. Sin. B 2023, 13, 2425–2463. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Zhou, J.; Fang, M.; Wang, G.; Fu, S.; Sun, B.; Lv, J. The Anti-Inflammatory Mechanism of SAHA in Acute Pancreatitis through HDAC5/SLIT2/Akt/β-Catenin Axis. Hum. Mol. Genet. 2022, 31, 2023–2034. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.; Gupta, S. The Role of Histone Deacetylases in Prostate Cancer. Epigenetics 2008, 3, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Bazzaro, M.; Wang, M.-C.; Chan, K.C.; Peng, S.; Roden, R.B.S. Combination of Proteasome and HDAC Inhibitors for Uterine Cervical Cancer Treatment. Clin. Cancer Res. 2009, 15, 570–577. [Google Scholar] [CrossRef]
- Banerjee, N.S.; Moore, D.W.; Broker, T.R.; Chow, L.T. Vorinostat, a Pan-HDAC Inhibitor, Abrogates Productive HPV-18 DNA Amplification. Proc. Natl. Acad. Sci. USA 2018, 115, E11138–E11147. [Google Scholar] [CrossRef]
- Li, J.; Li, X.; Wang, X.; Hou, J.; Zang, J.; Gao, S.; Xu, W.; Zhang, Y. PXD101 Analogs with L-Phenylglycine-Containing Branched Cap as Histone Deacetylase Inhibitors. Chem. Biol. Drug Des. 2016, 88, 574–584. [Google Scholar] [CrossRef]
- Wahaib, K.; Beggs, A.E.; Campbell, H.; Kodali, L.; Ford, P.D. Panobinostat: A Histone Deacetylase Inhibitor for the Treatment of Relapsed or Refractory Multiple Myeloma. Am. J. Health Syst. Pharm. 2016, 73, 441–450. [Google Scholar] [CrossRef]
- Wasim, L.; Chopra, M. Panobinostat Induces Apoptosis via Production of Reactive Oxygen Species and Synergizes with Topoisomerase Inhibitors in Cervical Cancer Cells. Biomed. Pharmacother. 2016, 84, 1393–1405. [Google Scholar] [CrossRef] [PubMed]
- Faghihloo, E.; Akbari, A.; Adjaminezhad-Fard, F.; Mokhtari-Azad, T. Transcriptional Regulation of E-Cadherin and Oncoprotein E7 by Valproic Acid in HPV Positive Cell Lines. Iran. J. Basic. Med. Sci. 2016, 19, 601–607. [Google Scholar] [PubMed]
- Coronel, J.; Cetina, L.; Pacheco, I.; Trejo-Becerril, C.; González-Fierro, A.; de la Cruz-Hernandez, E.; Perez-Cardenas, E.; Taja-Chayeb, L.; Arias-Bofill, D.; Candelaria, M.; et al. A Double-Blind, Placebo-Controlled, Randomized Phase III Trial of Chemotherapy plus Epigenetic Therapy with Hydralazine Valproate for Advanced Cervical Cancer. Preliminary Results. Med. Oncol. 2011, 28 (Suppl. S1), S540–S546. [Google Scholar] [CrossRef]
- Marques, A.E.M.; do Nascimento Filho, C.H.V.; Marinho Bezerra, T.M.; Guerra, E.N.S.; Castilho, R.M.; Squarize, C.H. Entinostat Is a Novel Therapeutic Agent to Treat Oral Squamous Cell Carcinoma. J. Oral. Pathol. Med. 2020, 49, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.L. DNA Methylation. The Effect of Minor Bases on DNA-Protein Interactions. Biochem. J. 1990, 265, 309–320. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The Epigenomics of Cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef]
- Cui, D.; Xu, X. DNA Methyltransferases, DNA Methylation, and Age-Associated Cognitive Function. Int. J. Mol. Sci. 2018, 19, 1315. [Google Scholar] [CrossRef]
- Leonhardt, H.; Page, A.W.; Weier, H.U.; Bestor, T.H. A Targeting Sequence Directs DNA Methyltransferase to Sites of DNA Replication in Mammalian Nuclei. Cell 1992, 71, 865–873. [Google Scholar] [CrossRef]
- Liu, Y.; Oakeley, E.J.; Sun, L.; Jost, J.P. Multiple Domains Are Involved in the Targeting of the Mouse DNA Methyltransferase to the DNA Replication Foci. Nucleic Acids Res. 1998, 26, 1038–1045. [Google Scholar] [CrossRef]
- Yoder, J.A.; Soman, N.S.; Verdine, G.L.; Bestor, T.H. DNA (Cytosine-5)-Methyltransferases in Mouse Cells and Tissues. Studies with a Mechanism-Based Probe. J. Mol. Biol. 1997, 270, 385–395. [Google Scholar] [CrossRef]
- Suetake, I.; Shinozaki, F.; Miyagawa, J.; Takeshima, H.; Tajima, S. DNMT3L Stimulates the DNA Methylation Activity of Dnmt3a and Dnmt3b through a Direct Interaction. J. Biol. Chem. 2004, 279, 27816–27823. [Google Scholar] [CrossRef] [PubMed]
- Kulis, M.; Esteller, M. DNA Methylation and Cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M. DNA Hypermethylation in Disease: Mechanisms and Clinical Relevance. Epigenetics 2019, 14, 1141–1163. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, Z.; Ding, Y.; Wang, L.; Wang, S.; Wang, H.; Qin, Y. DNA Methylation: From Cancer Biology to Clinical Perspectives. Front. Biosci. 2022, 27, 326. [Google Scholar] [CrossRef] [PubMed]
- Biktasova, A.; Hajek, M.; Sewell, A.; Gary, C.; Bellinger, G.; Deshpande, H.A.; Bhatia, A.; Burtness, B.; Judson, B.; Mehra, S.; et al. Demethylation Therapy as a Targeted Treatment for Human Papillomavirus-Associated Head and Neck Cancer. Clin. Cancer Res. 2017, 23, 7276–7287. [Google Scholar] [CrossRef] [PubMed]
- Castro-Oropeza, R.; Piña-Sánchez, P. Epigenetic and Transcriptomic Regulation Landscape in HPV+ Cancers: Biological and Clinical Implications. Front. Genet. 2022, 13, 886613. [Google Scholar] [CrossRef]
- Kumar, A.R.; Devan, A.R.; Nair, B.; Vinod, B.S.; Nath, L.R. Harnessing the Immune System against Cancer: Current Immunotherapy Approaches and Therapeutic Targets. Mol. Biol. Rep. 2021, 48, 8075–8095. [Google Scholar] [CrossRef]
- Maffuid, K.; Cao, Y. Decoding the Complexity of Immune-Cancer Cell Interactions: Empowering the Future of Cancer Immunotherapy. Cancers 2023, 15, 4188. [Google Scholar] [CrossRef]
- Alnefaie, A.; Albogami, S.; Asiri, Y.; Ahmad, T.; Alotaibi, S.S.; Al-Sanea, M.M.; Althobaiti, H. Chimeric Antigen Receptor T-Cells: An Overview of Concepts, Applications, Limitations, and Proposed Solutions. Front. Bioeng. Biotechnol. 2022, 10, 797440. [Google Scholar] [CrossRef]
- Moreno, C.; Haynie, C.; Cheever, A.; Weber, K.S. Alternative CAR Therapies: Recent Approaches in Engineering Chimeric Antigen Receptor Immune Cells to Combat Cancer. Biomedicines 2022, 10, 1493. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Zhong, J.F.; Zhang, X. Engineering CAR-T Cells. Biomark. Res. 2017, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- De Marco, R.C.; Monzo, H.J.; Ojala, P.M. CAR T Cell Therapy: A Versatile Living Drug. Int. J. Mol. Sci. 2023, 24, 6300. [Google Scholar] [CrossRef] [PubMed]
- Roselli, E.; Boucher, J.C.; Li, G.; Kotani, H.; Spitler, K.; Reid, K.; Cervantes, E.V.; Bulliard, Y.; Tu, N.; Lee, S.B.; et al. 4-1BB and Optimized CD28 Co-Stimulation Enhances Function of Human Mono-Specific and Bi-Specific Third-Generation CAR T Cells. J. Immunother. Cancer 2021, 9, e003354. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Hu, Y.; Xiao, W.; Tian, Z. Chimeric Antigen Receptor- and Natural Killer Cell Receptor-Engineered Innate Killer Cells in Cancer Immunotherapy. Cell Mol. Immunol. 2021, 18, 2083–2100. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, D.; Wong, R.A.; Gholamin, S.; Maker, M.; Aftabizadeh, M.; Yang, X.; Pecoraro, J.R.; Jeppson, J.D.; Wang, D.; Aguilar, B.; et al. IFNγ Is Critical for CAR T Cell-Mediated Myeloid Activation and Induction of Endogenous Immunity. Cancer Discov. 2021, 11, 2248–2265. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Huang, X.; Huang, J. CAR-T Cell Therapy for Hematological Malignancies: Limitations and Optimization Strategies. Front. Immunol. 2022, 13, 1019115. [Google Scholar] [CrossRef] [PubMed]
- Guzman, G.; Reed, M.R.; Bielamowicz, K.; Koss, B.; Rodriguez, A. CAR-T Therapies in Solid Tumors: Opportunities and Challenges. Curr. Oncol. Rep. 2023, 25, 479–489. [Google Scholar] [CrossRef]
- Yu, L.; Lanqing, G.; Huang, Z.; Xin, X.; Minglin, L.; Fa-Hui, L.; Zou, H.; Min, J. T Cell Immunotherapy for Cervical Cancer: Challenges and Opportunities. Front. Immunol. 2023, 14, 1105265. [Google Scholar] [CrossRef]
- Chen, L.; Xie, T.; Wei, B.; Di, D.-L. Current Progress in CAR-T Cell Therapy for Tumor Treatment. Oncol. Lett. 2022, 24, 358. [Google Scholar] [CrossRef]
- Chandran, S.S.; Klebanoff, C.A. T Cell Receptor-Based Cancer Immunotherapy: Emerging Efficacy and Pathways of Resistance. Immunol. Rev. 2019, 290, 127–147. [Google Scholar] [CrossRef]
- Menon, A.P.; Moreno, B.; Meraviglia-Crivelli, D.; Nonatelli, F.; Villanueva, H.; Barainka, M.; Zheleva, A.; van Santen, H.M.; Pastor, F. Modulating T Cell Responses by Targeting CD3. Cancers 2023, 15, 1189. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Cheng, X.-X.; Xue, J.Z.; Xue, S.-A. Emerging Strategies in TCR-Engineered T Cells. Front. Immunol. 2022, 13, 850358. [Google Scholar] [CrossRef] [PubMed]
- Rs, J. The Immune Microenvironment in Human Papilloma Virus-Induced Cervical Lesions-Evidence for Estrogen as an Immunomodulator. Front. Cell Infect. Microbiol. 2021, 11, 649815. [Google Scholar] [CrossRef]
- Almeida, A.M.; Queiroz, J.A.; Sousa, F.; Sousa, Â. Cervical Cancer and HPV Infection: Ongoing Therapeutic Research to Counteract the Action of E6 and E7 Oncoproteins. Drug Discov. Today 2019, 24, 2044–2057. [Google Scholar] [CrossRef]
- Jin, B.Y.; Campbell, T.E.; Draper, L.M.; Stevanović, S.; Weissbrich, B.; Yu, Z.; Restifo, N.P.; Rosenberg, S.A.; Trimble, C.L.; Hinrichs, C.S. Engineered T Cells Targeting E7 Mediate Regression of Human Papillomavirus Cancers in a Murine Model. JCI Insight 2018, 3, e99488. [Google Scholar] [CrossRef]
- Poorebrahim, M.; Quiros-Fernandez, I.; Marmé, F.; Burdach, S.E.; Cid-Arregui, A. A Costimulatory Chimeric Antigen Receptor Targeting TROP2 Enhances the Cytotoxicity of NK Cells Expressing a T Cell Receptor Reactive to Human Papillomavirus Type 16 E7. Cancer Lett. 2023, 566, 216242. [Google Scholar] [CrossRef]
- Zhu, Y. Advances in CRISPR/Cas9. Biomed. Res. Int. 2022, 2022, 9978571. [Google Scholar] [CrossRef]
- Najafi, S.; Tan, S.C.; Aghamiri, S.; Raee, P.; Ebrahimi, Z.; Jahromi, Z.K.; Rahmati, Y.; Sadri Nahand, J.; Piroozmand, A.; Jajarmi, V.; et al. Therapeutic Potentials of CRISPR-Cas Genome Editing Technology in Human Viral Infections. Biomed. Pharmacother. 2022, 148, 112743. [Google Scholar] [CrossRef]
- Zhen, S.; Hua, L.; Takahashi, Y.; Narita, S.; Liu, Y.-H.; Li, Y. In Vitro and in Vivo Growth Suppression of Human Papillomavirus 16-Positive Cervical Cancer Cells by CRISPR/Cas9. Biochem. Biophys. Res. Commun. 2014, 450, 1422–1426. [Google Scholar] [CrossRef]
- Ehrke-Schulz, E.; Schiwon, M.; Leitner, T.; Dávid, S.; Bergmann, T.; Liu, J.; Ehrhardt, A. CRISPR/Cas9 Delivery with One Single Adenoviral Vector Devoid of All Viral Genes. Sci. Rep. 2017, 7, 17113. [Google Scholar] [CrossRef] [PubMed]
- Jubair, L.; Fallaha, S.; McMillan, N.A.J. Systemic Delivery of CRISPR/Cas9 Targeting HPV Oncogenes Is Effective at Eliminating Established Tumors. Mol. Ther. 2019, 27, 2091–2099. [Google Scholar] [CrossRef] [PubMed]
- Ehrke-Schulz, E.; Heinemann, S.; Schulte, L.; Schiwon, M.; Ehrhardt, A. Adenoviral Vectors Armed with PAPILLOMAVIRUs Oncogene Specific CRISPR/Cas9 Kill Human-Papillomavirus-Induced Cervical Cancer Cells. Cancers 2020, 12, 1934. [Google Scholar] [CrossRef] [PubMed]
- Tomar, S.; Graves, C.A.; Altomare, D.; Kowli, S.; Kassler, S.; Sutkowski, N.; Gillespie, M.B.; Creek, K.E.; Pirisi, L. Human Papillomavirus Status and Gene Expression Profiles of Oropharyngeal and Oral Cancers from European American and African American Patients. Head Neck 2016, 38, E694–E704. [Google Scholar] [CrossRef]
- Abboodi, F.; Buckhaults, P.; Altomare, D.; Liu, C.; Hosseinipour, M.; Banister, C.E.; Creek, K.E.; Pirisi, L. HPV-Inactive Cell Populations Arise from HPV16-Transformed Human Keratinocytes after P53 Knockout. Virology 2021, 554, 9–16. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Function | Source |
|---|---|---|
| E1 |
| [28] |
| E2 |
| [30,31,32,34,65] |
| E4 |
| [38,39,66] |
| E5 |
| [42,43] |
| E6 |
| [43,47] |
| E7 |
| [56,57] |
| L1 |
| [58] |
| L2 |
| [64] |
| Epigenetic Drug | FDA Approved | Clinical Trial Phase | NCT Number | Deseas |
|---|---|---|---|---|
| SAHA | ✓ | I, II | NCT00948688 NCT00857324 NCT00574587 | Pancreatic cancer Multiple myeloma Breast cancer |
| TSA | ✓ | I | NCT03828926 | Relapsed or refractory Hematologic malignancies |
| VPA | ✓ | II, I, II | NCT01900730 NCT00374075 NCT00326170 | Breast cancer Spinal muscular atrophy Myelodysplastic syndrome Acute myelogenous leukemia |
| PXD101 | ✓ | I | NCT01583777 NCT00421889 | Advanced cancer Ovarian cancer |
| Panobinostat | ✓ | I, II, III | NCT00840346 NCT01023308 | Acute myeloblastic leukemia Multiple myeloma |
| MS-275 | ✓ | I, II, II | NCT04708470 NCT00828854 NCT04708470 | Cancer, solid tumor, microsatellite stable colon Cancer (MSS); ER + breast cancer Metastatic checkpoint Refractory HPV associated malignancies |
| Epigenetic Drug | FDA Approved | Clinical Trial Phase | NCT Number | Deseas |
|---|---|---|---|---|
| 5-azacytidine | ✓ | I, I, II | NCT02940483 NCT00886457 NCT00744757 | Brain tumor recurrent cancer Myelodysplastic syndrome |
| decitabine | ✓ | I | NCT04252248 | Head and neck cancer Anogenital cancer |
| Trial N. | Target | Condition | Phase | Patients N. | Start | Status |
|---|---|---|---|---|---|---|
| NCT01583686 | Mesothelin | Metastatic/unresectable Mesothelin-positive cancer | I, II | 15 | 2012 | Term. (2018) |
| NCT03356795 | GD2, PSMA, MUC1, Mesothelin | Patients with stage III, IV, or relapsed cervical cancer | I, II | 20 | 2017 | Pending |
| NCT04556669 | CD22 + PD-L1 | Advanced malignant solid tumors | I | 30 | 2020 | Pending Est. compl.: 2025 |
| NCT05518253 | CD70 | CD70-positive advanced/metastatic solid tumors | I | 36 | 2022 | Pending Est. compl.: 2025 |
| NCT05468190 | CD70 | Advanced/metastatic CD70-positive solid tumors | I | 48 | 2022 | Pending Est. compl.: 2025 |
| Trial N. | Target | Condition | Phase | Patients N. | Start | Status |
|---|---|---|---|---|---|---|
| NCT02280811 | E6 | HLA-A*02:01-positive HPV-16-associated metastatic/recurrent cancer | I, II | 12 | 2014 | Compl. (2016) |
| NCT02153905 | MAGE-A3 | HLA-A 01-positive metastatic/recurrent cancer expressing MAGE-A3 | I, II | 3 | 2014 | Compl. (2018) |
| NCT02111850 | MAGE-A3 | (HLA)-DP0401/0402-positive metastatic/recurrent cancer expressing MAGE-A3-DP4 | I, II | 21 | 2014 | Compl. (2021) |
| NCT02858310 | E7 | HLA-A*02:01-positive HPV-16-associated metastatic/recurrent cancer | I, II | 180 | 2017 | Pending Est. compl.: 2026 |
| NCT03578406 | E6 + PD-1 | Metastatic/recurrent HPV-16-positive cancers | I | 20 | 2018 | Pending |
| NCT05357027 | E6 | HLA-A*02- and HPV16-positive metastatic/recurrent positive cervical cancer | I, II | 18 | 2022 | Pending Est. compl.: 2024 |
| NCT05122221 | E7 | HLA-A*02- and HPV16-positive cancer | I | 12 | 2022 | Pending Est. compl.: 2024 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Folliero, V.; Dell’Annunziata, F.; Chianese, A.; Morone, M.V.; Mensitieri, F.; Di Spirito, F.; Mollo, A.; Amato, M.; Galdiero, M.; Dal Piaz, F.; et al. Epigenetic and Genetic Keys to Fight HPV-Related Cancers. Cancers 2023, 15, 5583. https://doi.org/10.3390/cancers15235583
Folliero V, Dell’Annunziata F, Chianese A, Morone MV, Mensitieri F, Di Spirito F, Mollo A, Amato M, Galdiero M, Dal Piaz F, et al. Epigenetic and Genetic Keys to Fight HPV-Related Cancers. Cancers. 2023; 15(23):5583. https://doi.org/10.3390/cancers15235583
Chicago/Turabian StyleFolliero, Veronica, Federica Dell’Annunziata, Annalisa Chianese, Maria Vittoria Morone, Francesca Mensitieri, Federica Di Spirito, Antonio Mollo, Massimo Amato, Massimiliano Galdiero, Fabrizio Dal Piaz, and et al. 2023. "Epigenetic and Genetic Keys to Fight HPV-Related Cancers" Cancers 15, no. 23: 5583. https://doi.org/10.3390/cancers15235583