
Chimeric Antigen Receptor T Cell Therapy Targeting Epithelial Cell Adhesion Molecule in Gastric Cancer: Mechanisms of Tumor Resistance
 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Lentiviral Vector Construction
2.3. Transduction of Human Primary T Cells
2.4. Flow Cytometry
2.5. Mouse Experiments
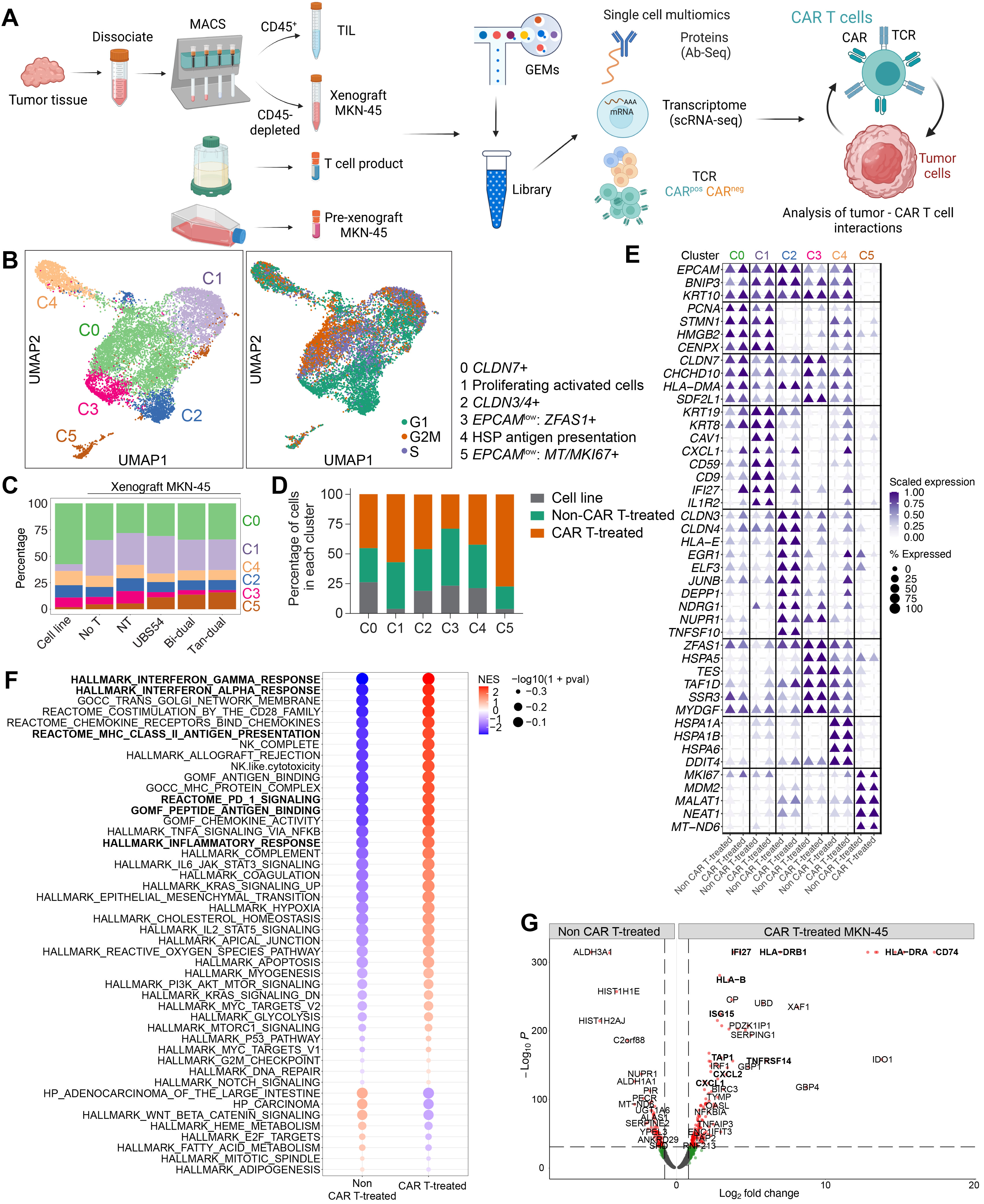
2.6. Tumor Dissociation and Single Cell Isolation
2.7. Single-Cell Sequencing Procedure
2.8. Pre-Processing of Single-Cell RNA-Seq, Protein, and TCR Data
2.9. Integration, Clustering, and Identification of Subsets
2.10. Differential Expression Analysis
2.11. Gene Set Analysis
2.12. Statistical Analysis
3. Results
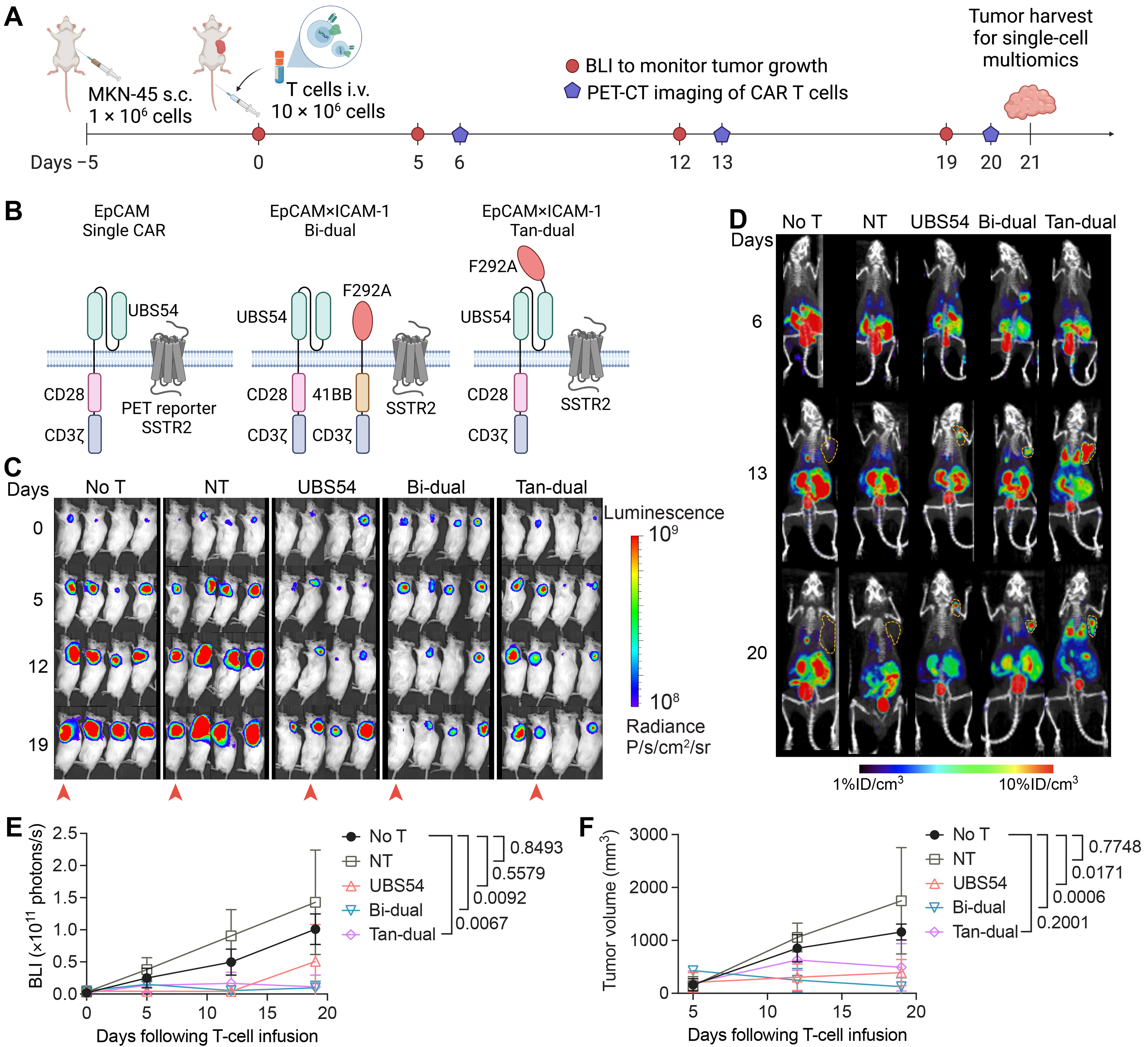
3.1. CAR T Cell Imaging Reveals Penetration and Expansion of CAR T Cells in Tumor
3.2. Single-Cell Analysis of Tumor Cells Revealed a Remodeling towards External Stimuli following CAR T Cell Infusion
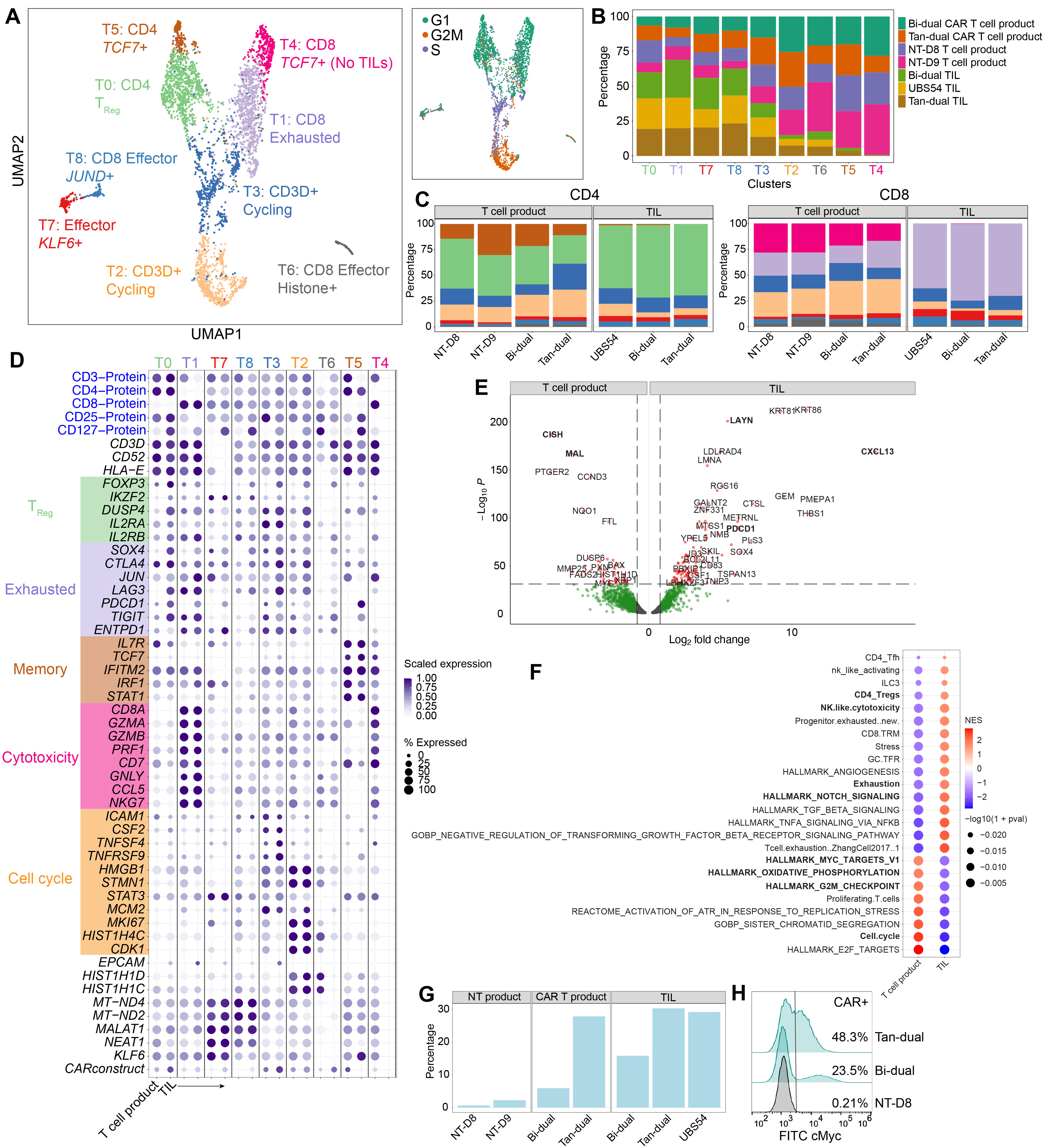
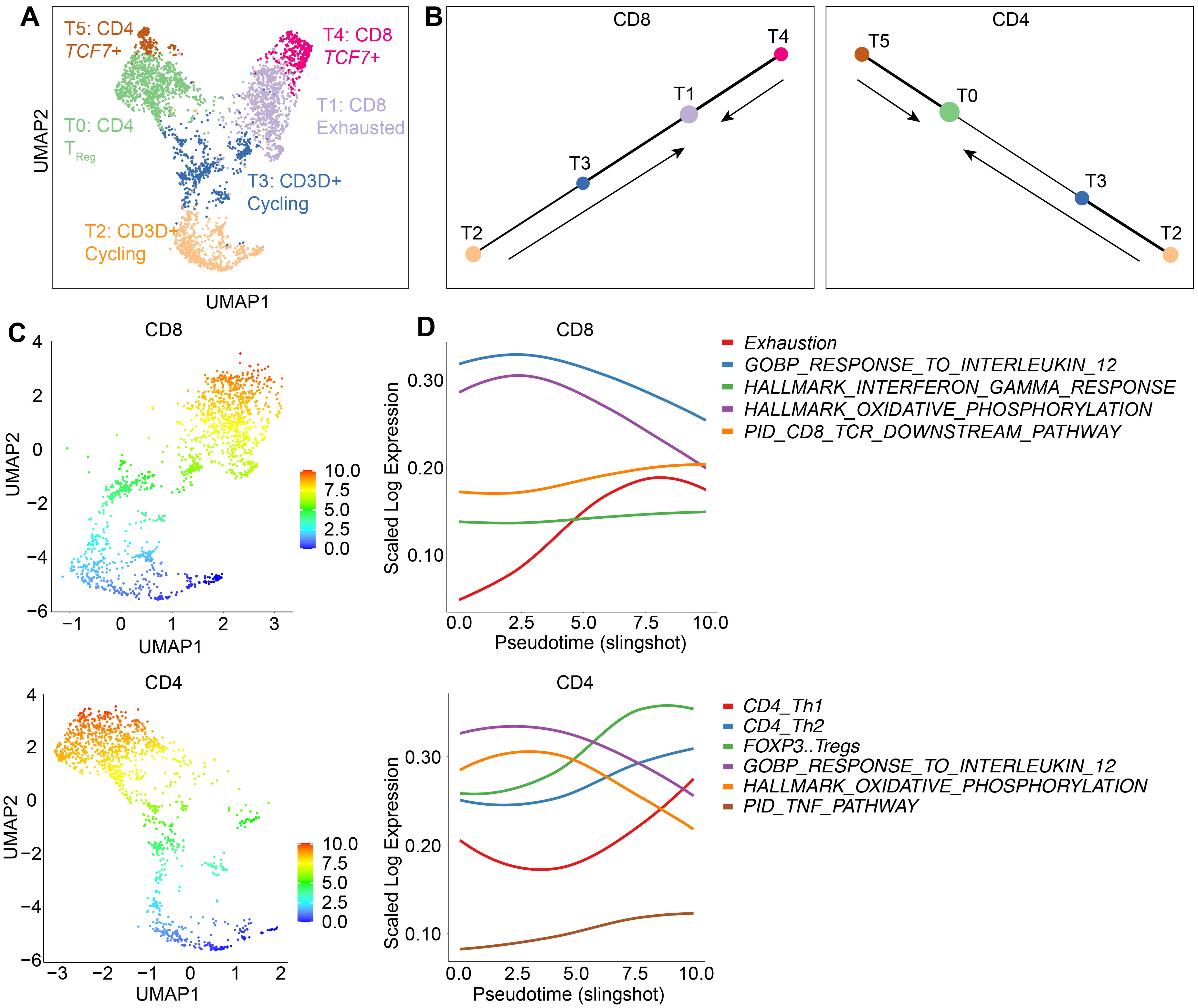
3.3. T Cell Analysis Revealed a Rapid Differentiation Process That Resulted in CD8 T Cell Exhaustion and CD4 T Cells Adopting a Regulatory Phenotype
3.4. Detection of CAR T Cell Transcript
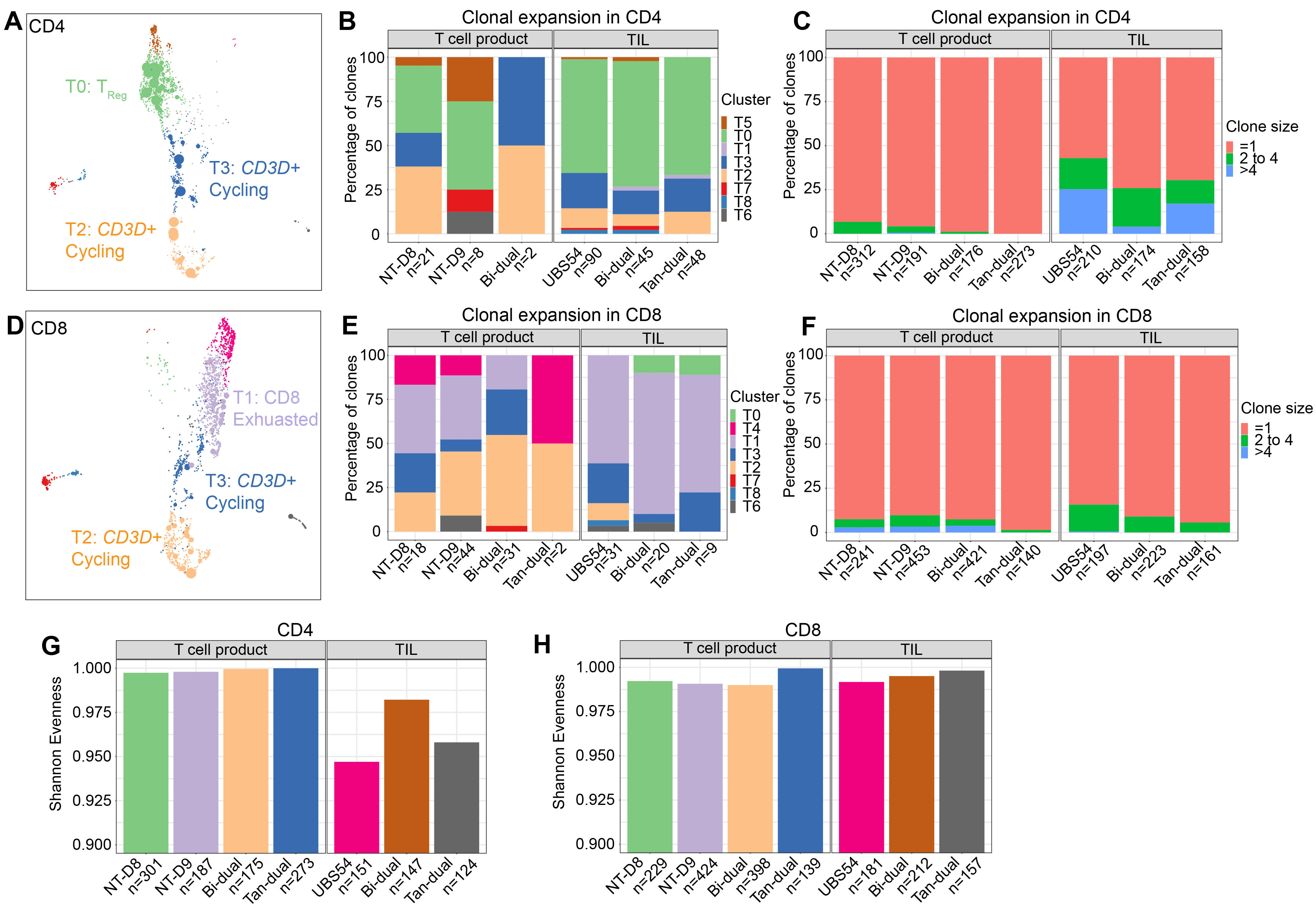
3.5. Analysis of TCR Sequences Revealed Clonally Expanded CAR T Cells in the Tumor
3.6. Trajectory Analysis Demonstrates a Differentiation of CAR T Cells towards Exhausted and TReg Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Pauken, K.E.; Wherry, E.J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015, 36, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Sanmamed, M.F.; Nie, X.; Desai, S.S.; Villaroel-Espindola, F.; Badri, T.; Zhao, D.; Kim, A.W.; Ji, L.; Zhang, T.; Quinlan, E.; et al. A Burned-Out CD8+ T-cell Subset Expands in the Tumor Microenvironment and Curbs Cancer Immunotherapy. Cancer Discov. 2021, 11, 1700–1715. [Google Scholar] [CrossRef]
- Li, H.; van der Leun, A.M.; Yofe, I.; Lubling, Y.; Gelbard-Solodkin, D.; van Akkooi, A.C.; Braber, M.v.D.; Rozeman, E.A.; Haanen, J.B.; Blank, C.U.; et al. Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell 2019, 176, 775–789.e18. [Google Scholar] [CrossRef]
- Davidson, S.; Efremova, M.; Riedel, A.; Mahata, B.; Pramanik, J.; Huuhtanen, J.; Kar, G.; Vento-Tormo, R.; Hagai, T.; Chen, X.; et al. Single-Cell RNA Sequencing Reveals a Dynamic Stromal Niche That Supports Tumor Growth. Cell Rep. 2020, 31, 107628. [Google Scholar] [CrossRef]
- Moon, E.K.; Wang, L.C.; Dolfi, D.V.; Wilson, C.B.; Ranganathan, R.; Sun, J.; Kapoor, V.; Scholler, J.; Puré, E.; Milone, M.C.; et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin. Cancer Res. 2014, 20, 4262–4273. [Google Scholar] [CrossRef]
- Haradhvala, N.J.; Leick, M.B.; Maurer, K.; Gohil, S.H.; Larson, R.C.; Yao, N.; Gallagher, K.M.E.; Katsis, K.; Frigault, M.J.; Southard, J.; et al. Distinct cellular dynamics associated with response to CAR-T therapy for refractory B cell lymphoma. Nat. Med. 2022, 28, 1848–1859. [Google Scholar] [CrossRef]
- Good, Z.; Spiegel, J.Y.; Sahaf, B.; Malipatlolla, M.B.; Ehlinger, Z.J.; Kurra, S.; Desai, M.H.; Reynolds, W.D.; Lin, A.W.; Vandris, P.; et al. Post-infusion CAR T(Reg) cells identify patients resistant to CD19-CAR therapy. Nat. Med. 2022, 28, 1860–1871. [Google Scholar] [CrossRef]
- Walker, A.J.; Majzner, R.G.; Zhang, L.; Wanhainen, K.; Long, A.H.; Nguyen, S.M.; Lopomo, P.; Vigny, M.; Fry, T.J.; Orentas, R.J.; et al. Tumor Antigen and Receptor Densities Regulate Efficacy of a Chimeric Antigen Receptor Targeting Anaplastic Lymphoma Kinase. Mol. Ther. 2017, 25, 2189–2201. [Google Scholar] [CrossRef]
- Chinnasamy, D.; Yu, Z.; Kerkar, S.P.; Zhang, L.; Morgan, R.A.; Restifo, N.P.; Rosenberg, S.A. Local Delivery of lnterleukin-12 Using T Cells Targeting VEGF Receptor-2 Eradicates Multiple Vascularized Tumors in Mice. Clin. Cancer Res. 2012, 18, 1672–1683. [Google Scholar] [CrossRef] [PubMed]
- Gargett, T.; Ebert, L.M.; Truong, N.T.; Kollis, P.M.; Sedivakova, K.; Yu, W.; Yeo, E.C.; Wittwer, N.L.; Gliddon, B.L.; Tea, M.N.; et al. GD2-targeting CAR-T cells enhanced by transgenic IL-15 expression are an effective and clinically feasible therapy for glioblastoma. J. Immunother. Cancer 2022, 10, e005187. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, H.; Alcaina, Y.; Puc, J.; Birt, A.; Vedvyas, Y.; Gallagher, M.; Alla, S.; Riascos, M.C.; McCloskey, J.E.; et al. Inducible expression of interleukin-12 augments the efficacy of affinity-tuned chimeric antigen receptors in murine solid tumor models. Nat. Commun. 2023, 14, 2068. [Google Scholar] [CrossRef] [PubMed]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, R.; Sadelain, M.; Adusumilli, P.S.; Human, P.S. CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef] [PubMed]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation and Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef]
- Narayan, V.; Barber-Rotenberg, J.S.; Jung, I.-Y.; Lacey, S.F.; Rech, A.J.; Davis, M.M.; Hwang, W.-T.; Lal, P.; Carpenter, E.L.; Maude, S.L.; et al. PSMA-targeting TGFβ-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: A phase 1 trial. Nat. Med. 2022, 28, 724–734. [Google Scholar] [CrossRef]
- Li, W.; Qiu, S.; Chen, J.; Jiang, S.; Chen, W.; Jiang, J.; Wang, F.; Si, W.; Shu, Y.; Wei, P.; et al. Chimeric Antigen Receptor Designed to Prevent Ubiquitination and Downregulation Showed Durable Antitumor Efficacy. Immunity 2020, 53, 456–470.e456. [Google Scholar] [CrossRef]
- Weber, E.W.; Parker, K.R.; Sotillo, E.; Lynn, R.C.; Anbunathan, H.; Lattin, J.; Good, Z.; Belk, J.A.; Daniel, B.; Klysz, D.; et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science 2021, 372, eaba1786. [Google Scholar] [CrossRef]
- Wang, D.; Prager, B.C.; Gimple, R.C.; Aguilar, B.; Alizadeh, D.; Tang, H.; Lv, D.; Starr, R.; Brito, A.; Wu, Q.; et al. CRISPR Screening of CAR T Cells and Cancer Stem Cells Reveals Critical Dependencies for Cell-Based Therapies. Cancer Discov. 2021, 11, 1192–1211. [Google Scholar] [CrossRef]
- Singh, N.; Lee, Y.G.; Shestova, O.; Ravikumar, P.; Hayer, K.E.; Hong, S.J.; Lu, X.M.; Pajarillo, R.; Agarwal, S.; Kuramitsu, S.; et al. Impaired Death Receptor Signaling in Leukemia Causes Antigen-Independent Resistance by Inducing CAR T-cell Dysfunction. Cancer Discov. 2020, 10, 552–567. [Google Scholar] [CrossRef]
- Boroughs, A.C.; Larson, R.C.; Marjanovic, N.D.; Gosik, K.; Castano, A.P.; Porter, C.B.; Lorrey, S.J.; Ashenberg, O.; Jerby, L.; Hofree, M.; et al. A Distinct Transcriptional Program in Human CAR T Cells Bearing the 4-1BB Signaling Domain Revealed by scRNA-Seq. Mol. Ther. 2020, 28, 2577–2592. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Han, G.; Puebla-Osorio, N.; Ma, M.C.J.; Strati, P.; Chasen, B.; Dai, E.; Dang, M.; Jain, N.; Yang, H.; et al. Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat. Med. 2020, 26, 1878–1887. [Google Scholar] [CrossRef]
- Melenhorst, J.J.; Chen, G.M.; Wang, M.; Porter, D.L.; Chen, C.; Collins, M.A.; Gao, P.; Bandyopadhyay, S.; Sun, H.; Zhao, Z.; et al. Decade-long leukaemia remissions with persistence of CD4+ CAR T cells. Nature 2022, 602, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Good, C.R.; Aznar, M.A.; Kuramitsu, S.; Samareh, P.; Agarwal, S.; Donahue, G.; Ishiyama, K.; Wellhausen, N.; Rennels, A.K.; Ma, Y.; et al. An NK-like CAR T cell transition in CAR T cell dysfunction. Cell 2021, 184, 6081–6100.e26. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, D.; Wong, R.A.; Gholamin, S.; Maker, M.; Aftabizadeh, M.; Yang, X.; Pecoraro, J.R.; Jeppson, J.D.; Wang, D.; Aguilar, B.; et al. IFNγ Is Critical for CAR T Cell–Mediated Myeloid Activation and Induction of Endogenous Immunity. Cancer Discov. 2021, 11, 2248–2265. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Palmer, D.C.; Robeson, A.C.; Shou, P.; Bommiasamy, H.; Laurie, S.J.; Willis, C.; Dotti, G.; Vincent, B.G.; Restifo, N.P.; et al. STING agonist promotes CAR T cell trafficking and persistence in breast cancer. J. Exp. Med. 2021, 218, e20200844. [Google Scholar] [CrossRef]
- Srivastava, S.; Furlan, S.N.; Jaeger-Ruckstuhl, C.A.; Sarvothama, M.; Berger, C.; Smythe, K.S.; Garrison, S.M.; Specht, J.M.; Lee, S.M.; Amezquita, R.A.; et al. Immunogenic Chemotherapy Enhances Recruitment of CAR-T Cells to Lung Tumors and Improves Antitumor Efficacy when Combined with Checkpoint Blockade. Cancer Cell 2021, 39, 193–208.e10. [Google Scholar] [CrossRef]
- Yang, Y.; McCloskey, J.E.; Yang, H.; Puc, J.; Alcaina, Y.; Vedvyas, Y.; Gallegos, A.A.G.; Ortiz-Sánchez, E.; de Stanchina, E.; Min, I.M.; et al. Bispecific CAR T Cells against EpCAM and Inducible ICAM-1 Overcome Antigen Heterogeneity and Generate Superior Antitumor Responses. Cancer Immunol. Res. 2021, 9, 1158–1174. [Google Scholar] [CrossRef]
- Huls, G.A.; Heijnen, I.A.; Cuomo, M.E.; Koningsberger, J.C.; Wiegman, L.; Boel, E.; van der Vuurst de Vries, A.-R.; Loyson, S.A.; Helfrich, W.; van Berge Henegouwen, G.P.; et al. A recombinant, fully human monoclonal antibody with antitumor activity constructed from phage-displayed antibody fragments. Nat. Biotechnol. 1999, 17, 276–281. [Google Scholar] [CrossRef]
- Jin, M.; Song, G.; Carman, C.V.; Kim, Y.S.; Astrof, N.S.; Shimaoka, M.; Wittrup, D.K.; Springer, T.A. Directed evolution to probe protein allostery and integrin I domains of 200,000-fold higher affinity. Proc. Natl. Acad. Sci. USA 2006, 103, 5758–5763. [Google Scholar] [CrossRef]
- Park, S.; Shevlin, E.; Vedvyas, Y.; Zaman, M.; Park, S.; Hsu, Y.-M.S.; Min, I.M.; Jin, M.M. Micromolar affinity CAR T cells to ICAM-1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci. Rep. 2017, 7, 14366. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef] [PubMed]
- Finak, G.; McDavid, A.; Yajima, M.; Deng, J.; Gersuk, V.; Shalek, A.K.; Slichter, C.K.; Miller, H.W.; McElrath, M.J.; Prlic, M.; et al. MAST: A flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015, 16, 278. [Google Scholar] [CrossRef] [PubMed]
- Squair, J.W.; Gautier, M.; Kathe, C.; Anderson, M.A.; James, N.D.; Hutson, T.H.; Hudelle, R.; Qaiser, T.; Matson, K.J.E.; Barraud, Q.; et al. Confronting false discoveries in single-cell differential expression. Nat. Commun. 2021, 12, 5692. [Google Scholar] [CrossRef]
- Wolf, F.A.; Hamey, F.K.; Plass, M.; Solana, J.; Dahlin, J.S.; Gottgens, B.; Rajewsky, N.; Simon, L.; Theis, F.J. PAGA: Graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 2019, 20, 59. [Google Scholar] [CrossRef]
- Street, K.; Risso, D.; Fletcher, R.B.; Das, D.; Ngai, J.; Yosef, N.; Purdom, E.; Dudoit, S. Slingshot: Cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 2018, 19, 477. [Google Scholar] [CrossRef]
- Alexey, A.S. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv 2016, 060012. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Vedvyas, Y.; Shevlin, E.; Zaman, M.; Min, I.M.; Amor-Coarasa, A.; Park, S.; Park, S.; Kwon, K.-W.; Smith, T.; Luo, Y.; et al. Longitudinal PET imaging demonstrates biphasic CAR T cell responses in survivors. JCI Insight 2016, 1, e90064. [Google Scholar] [CrossRef]
- Wu, Z.; Shi, J.; Song, Y.; Zhao, J.; Sun, J.; Chen, X.; Gao, P.; Wang, Z. Claudin-7 (CLDN7) is overexpressed in gastric cancer and promotes gastric cancer cell proliferation, invasion and maintains mesenchymal state. Neoplasma 2018, 65, 349–359. [Google Scholar] [CrossRef]
- Supino, D.; Minute, L.; Mariancini, A.; Riva, F.; Magrini, E.; Garlanda, C. Negative Regulation of the IL-1 System by IL-1R2 and IL-1R8: Relevance in Pathophysiology and Disease. Front. Immunol. 2022, 13, 804641. [Google Scholar] [CrossRef]
- Maehara, Y.; Oki, E.; Abe, T.; Tokunaga, E.; Shibahara, K.; Kakeji, Y.; Sugimachi, K. Overexpression of the heat shock protein HSP70 family and p53 protein and prognosis for patients with gastric cancer. Oncology 2000, 58, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xie, Y.; Zhang, W.; Gao, J.; Wang, M.; Zheng, G.; Yin, X.; Xia, H.; Tao, X. Interferon alpha-inducible protein 27 promotes epithelial-mesenchymal transition and induces ovarian tumorigenicity and stemness. J. Surg. Res. 2015, 193, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Zhao, J.; Song, J.; Li, Y.; Zuo, R.; Sa, Y.; Ma, Z.; Ouyang, H. Interferon alpha-inducible protein 27 (IFI27) is a prognostic marker for pancreatic cancer based on comprehensive bioinformatics analysis. Bioengineered 2021, 12, 8515–8528. [Google Scholar] [CrossRef] [PubMed]
- Workel, H.H.; Lubbers, J.M.; Arnold, R.; Prins, T.M.; van der Vlies, P.; de Lange, K.; Bosse, T.; van Gool, I.C.; Eggink, F.A.; Wouters, M.C.; et al. A Transcriptionally Distinct CXCL13+CD103+CD8+ T-cell Population Is Associated with B-cell Recruitment and Neoantigen Load in Human Cancer. Cancer Immunol. Res. 2019, 7, 784–796. [Google Scholar] [CrossRef]
- Palmer, D.C.; Guittard, G.C.; Franco, Z.; Crompton, J.G.; Eil, R.L.; Patel, S.J.; Ji, Y.; Van Panhuys, N.; Klebanoff, C.A.; Sukumar, M.; et al. Cish actively silences TCR signaling in CD8+ T cells to maintain tumor tolerance. J. Exp. Med. 2015, 212, 2095–2113. [Google Scholar] [CrossRef]
- Anton, O.; Batista, A.; Millan, J.; Andres-Delgado, L.; Puertollano, R.; Correas, I.; Alonso, M.A. An essential role for the MAL protein in targeting Lck to the plasma membrane of human T lymphocytes. J. Exp. Med. 2008, 205, 3201–3213. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Ajina, A.; Maher, J. Strategies to Address Chimeric Antigen Receptor Tonic Signaling. Mol. Cancer Ther. 2018, 17, 1795–1815. [Google Scholar] [CrossRef]
- Gomes-Silva, D.; Mukherjee, M.; Srinivasan, M.; Krenciute, G.; Dakhova, O.; Zheng, Y.; Cabral, J.M.; Rooney, C.M.; Orange, J.S.; Brenner, M.K.; et al. Tonic 4-1BB Costimulation in Chimeric Antigen Receptors Impedes T Cell Survival and Is Vector-Dependent. Cell Rep. 2017, 21, 17–26. [Google Scholar] [CrossRef]
- Kouro, T.; Himuro, H.; Sasada, T. Exhaustion of CAR T cells: Potential causes and solutions. J. Transl. Med. 2022, 20, 239. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-N.; Yang, J.-F.; Huang, D.-J.; Ni, H.-H.; Zhang, C.-X.; Zhang, L.; He, J.; Gu, J.-M.; Chen, H.-X.; Mai, H.-Q.; et al. Hypoxia Induces Mitochondrial Defect that Promotes T Cell Exhaustion in Tumor Microenvironment Through MYC-Regulated Pathways. Front. Immunol. 2020, 11, 1906. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shoshan, J.; Maysel-Auslender, S.; Mor, A.; Keren, G.; George, J. Hypoxia controls CD4+CD25+ regulatory T-cell homeostasis via hypoxia-inducible factor-1alpha. Eur. J. Immunol. 2008, 38, 2412–2418. [Google Scholar] [CrossRef] [PubMed]
- Valiullina, A.K.; Zmievskaya, E.A.; Ganeeva, I.A.; Zhuravleva, M.N.; Garanina, E.E.; Rizvanov, A.A.; Petukhov, A.V.; Bulatov, E.R. Evaluation of CAR-T Cells’ Cytotoxicity against Modified Solid Tumor Cell Lines. Biomedicines 2023, 11, 626. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Conditions or Cohorts | Number of Cells | Surface Markers | Type of Analyses |
|---|---|---|---|---|
| Pre-xenograft MKN-45 | Pre-xenograft | 3300 | ICAM-1, EpCAM, HT1 | GEX, Surface |
| Xenograft MKN-45 | No T cohort | 3300 | ICAM-1, EpCAM, HT2 | GEX, Surface |
| NT (donor 9) | 3300 | ICAM-1, EpCAM, HT3 | GEX, Surface | |
| UBS54 (donor 9) | 3300 | ICAM-1, EpCAM, HT4 | GEX, Surface | |
| Bi-dual (donor 9) | 3300 | ICAM-1, EpCAM, HT5 | GEX, Surface | |
| Tan-dual (donor 8) | 3300 | ICAM-1, EpCAM, HT6 | GEX, Surface | |
| T cell products | NT cell (donor 9) | 2500 | CD3, CD4, CD8, CD25, CD127, HT8 | GEX, Surface, V(D)J |
| NT cell (donor 8) | 2500 | CD3, CD4, CD8, CD25, CD127, HT1, HT2 | GEX, Surface, V(D)J | |
| Bi-dual CAR T cell (donor 9) | 2500 | CD3, CD4, CD8, CD25, CD127, HT1, HT3 | GEX, Surface, V(D)J | |
| Tan-dual CAR T cell (donor 8) | 2500 | CD3, CD4, CD8, CD25, CD127, HT1, HT4 | GEX, Surface, V(D)J | |
| TILs | UBS54 TIL (donor 9) | 3300 | CD3, CD4, CD8, CD25, CD127, HT1, HT5 | GEX, Surface, V(D)J |
| Bi-dual TIL (donor 9) | 3300 | CD3, CD4, CD8, CD25, CD127, HT1, HT6 | GEX, Surface, V(D)J | |
| Tan-dual TIL (donor 8) | 3300 | CD3, CD4, CD8, CD25, CD127, HT1, HT7 | GEX, Surface, V(D)J |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Louie, R.; Puc, J.; Vedvyas, Y.; Alcaina, Y.; Min, I.M.; Britz, M.; Luciani, F.; Jin, M.M. Chimeric Antigen Receptor T Cell Therapy Targeting Epithelial Cell Adhesion Molecule in Gastric Cancer: Mechanisms of Tumor Resistance. Cancers 2023, 15, 5552. https://doi.org/10.3390/cancers15235552
Yang Y, Louie R, Puc J, Vedvyas Y, Alcaina Y, Min IM, Britz M, Luciani F, Jin MM. Chimeric Antigen Receptor T Cell Therapy Targeting Epithelial Cell Adhesion Molecule in Gastric Cancer: Mechanisms of Tumor Resistance. Cancers. 2023; 15(23):5552. https://doi.org/10.3390/cancers15235552
Chicago/Turabian StyleYang, Yanping, Raymond Louie, Janusz Puc, Yogindra Vedvyas, Yago Alcaina, Irene M. Min, Matt Britz, Fabio Luciani, and Moonsoo M. Jin. 2023. "Chimeric Antigen Receptor T Cell Therapy Targeting Epithelial Cell Adhesion Molecule in Gastric Cancer: Mechanisms of Tumor Resistance" Cancers 15, no. 23: 5552. https://doi.org/10.3390/cancers15235552