
Merkel Cell Polyomavirus: Infection, Genome, Transcripts and Its Role in Development of Merkel Cell Carcinoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Merkel Cell Polyomavirus (MCPyV) as a Member of the Polyomavirus Family
2. MCPyV Is an Omnipresent Virus
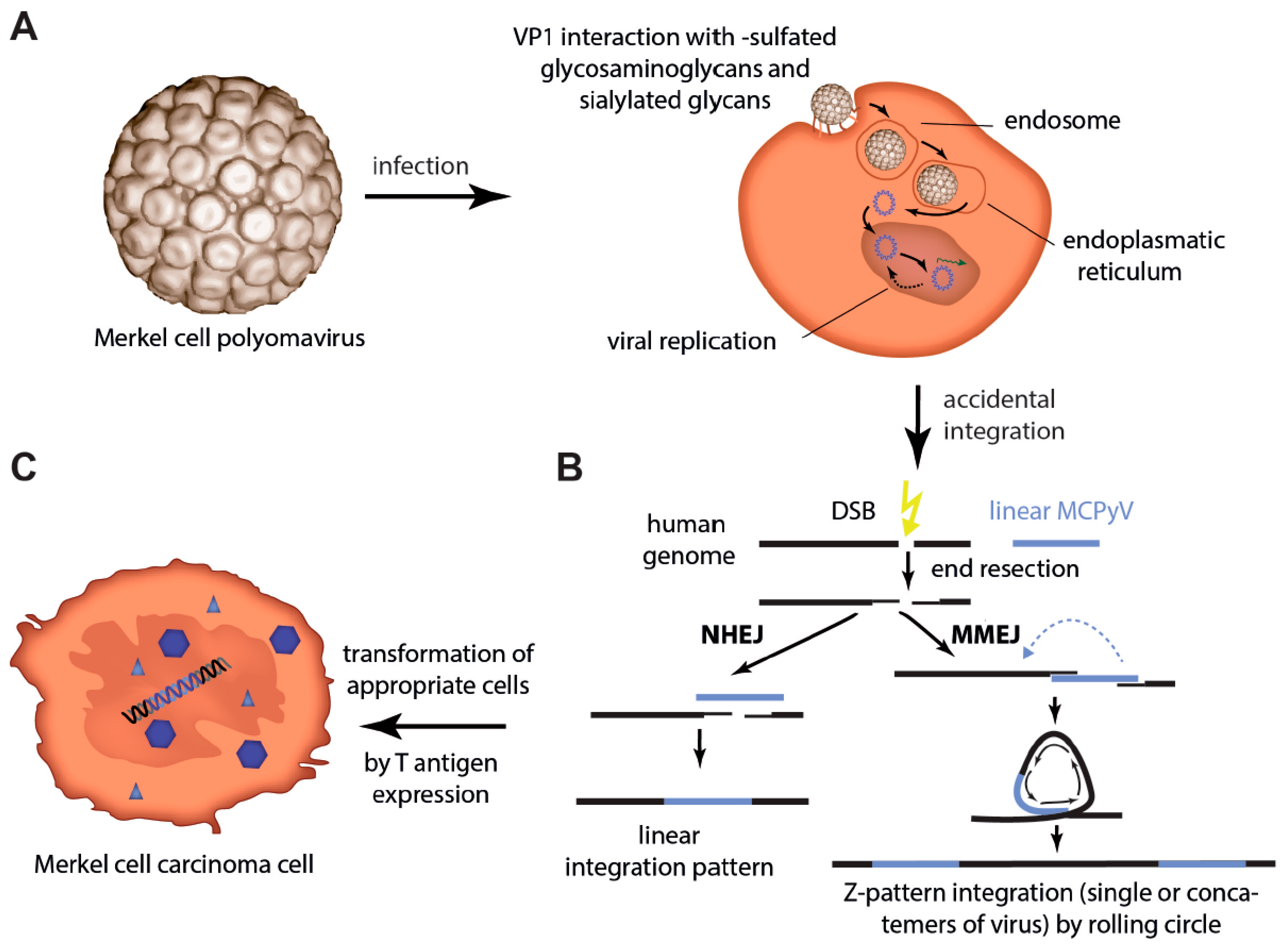
3. Infection of Host Cells and Integration in Merkel Cell Carcinoma
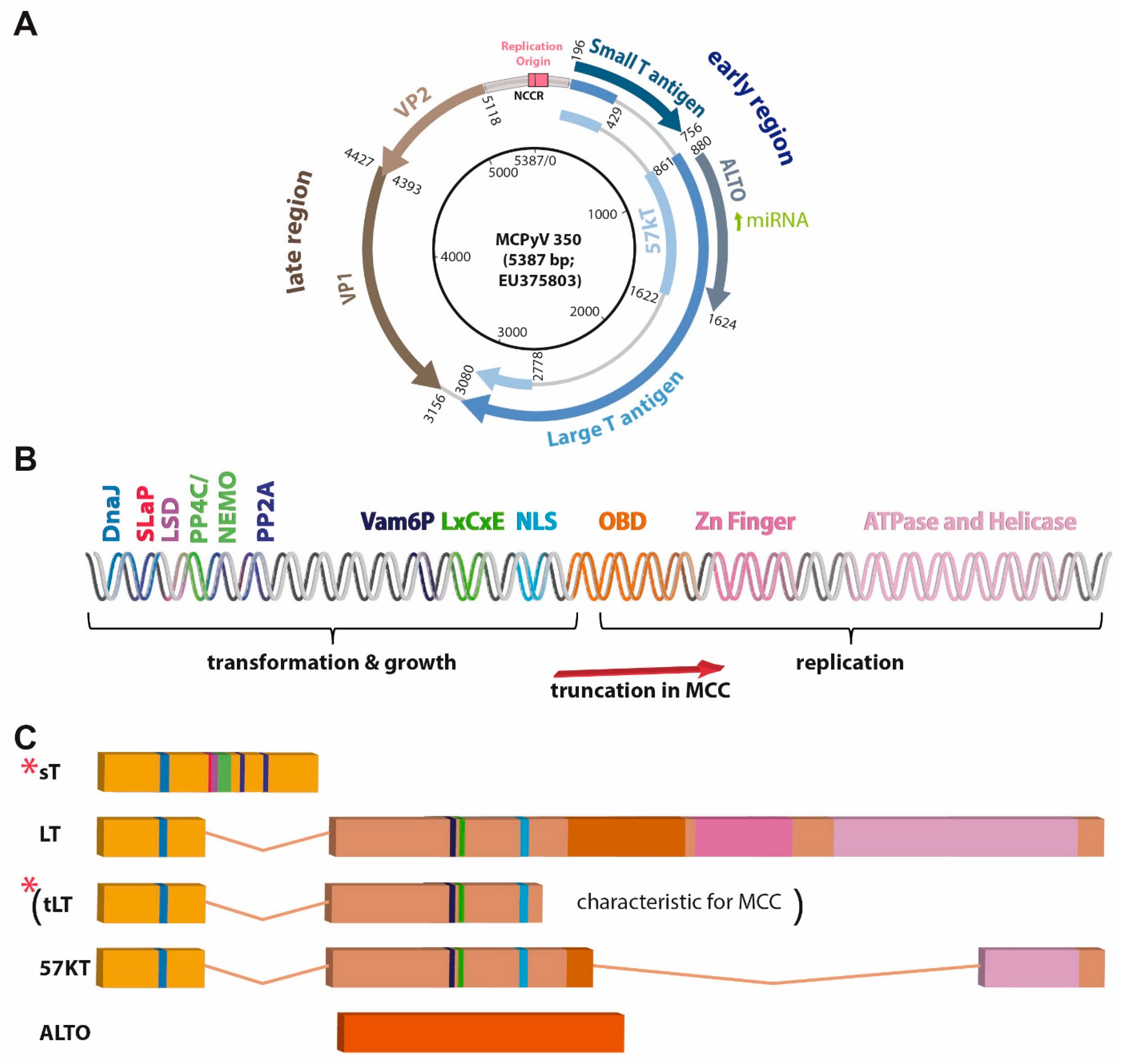
4. Viral Gene Products
4.1. Capsid Proteins
4.2. T Antigens
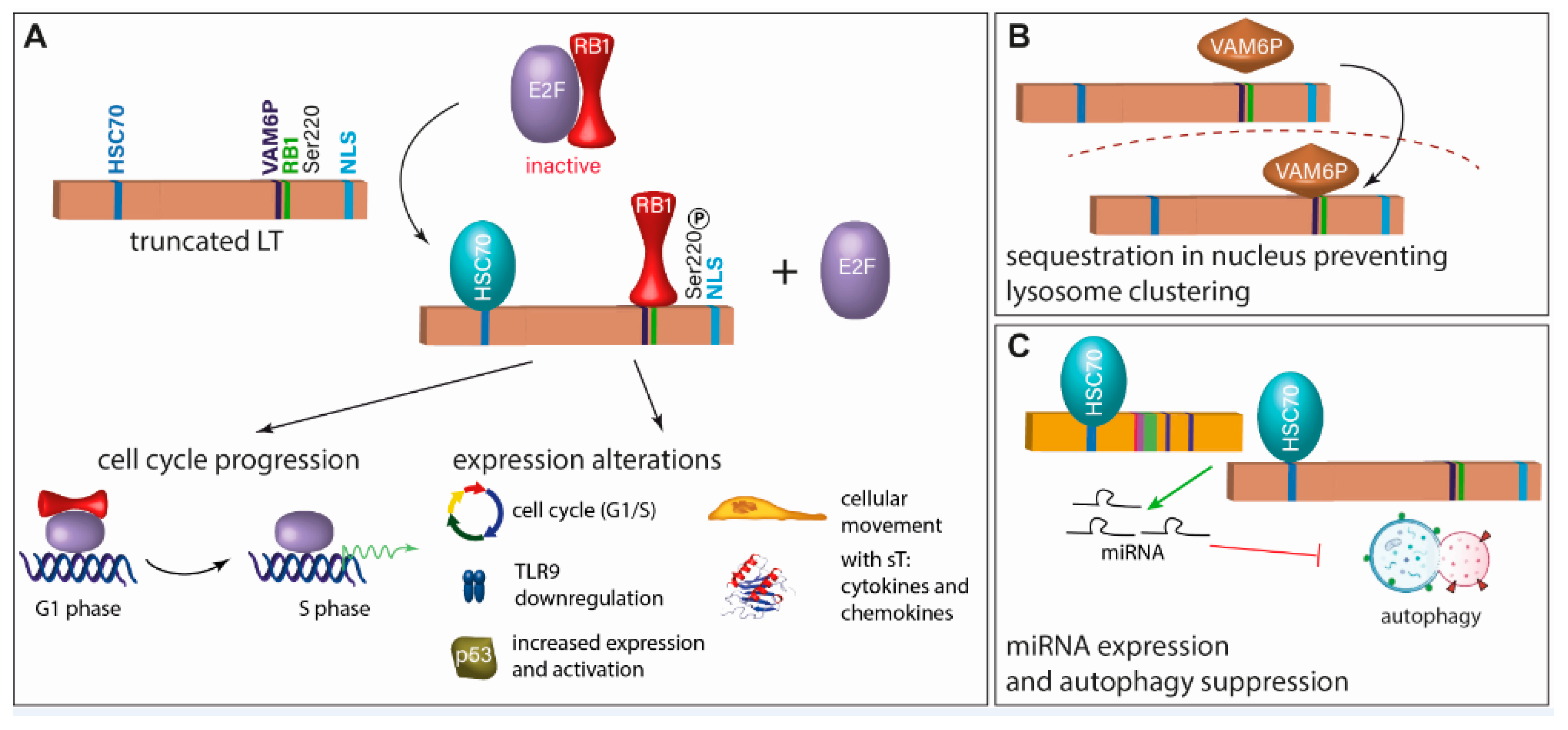
4.2.1. Large T Antigen
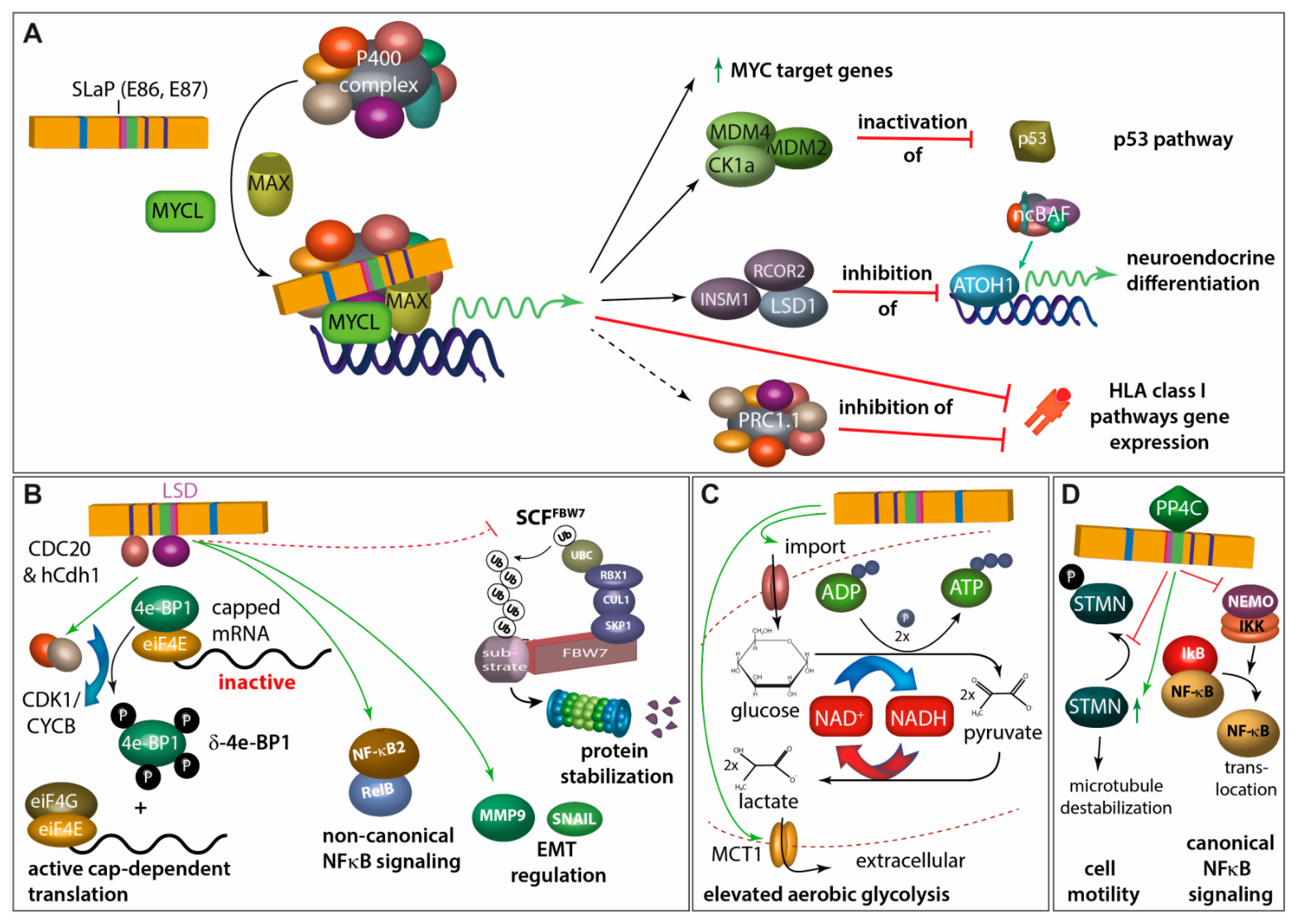
4.2.2. Small T Antigen
4.2.3. ALTO, Circular RNAs and Viral miRNA
5. Merkel Cell Carcinoma
5.1. Merkel Cells
5.2. MCC: Two Different Tumor Entities
5.3. MCPyV-Positive MCC: A Virus-Induced Tumor
5.4. The Cellular Origin of MCC
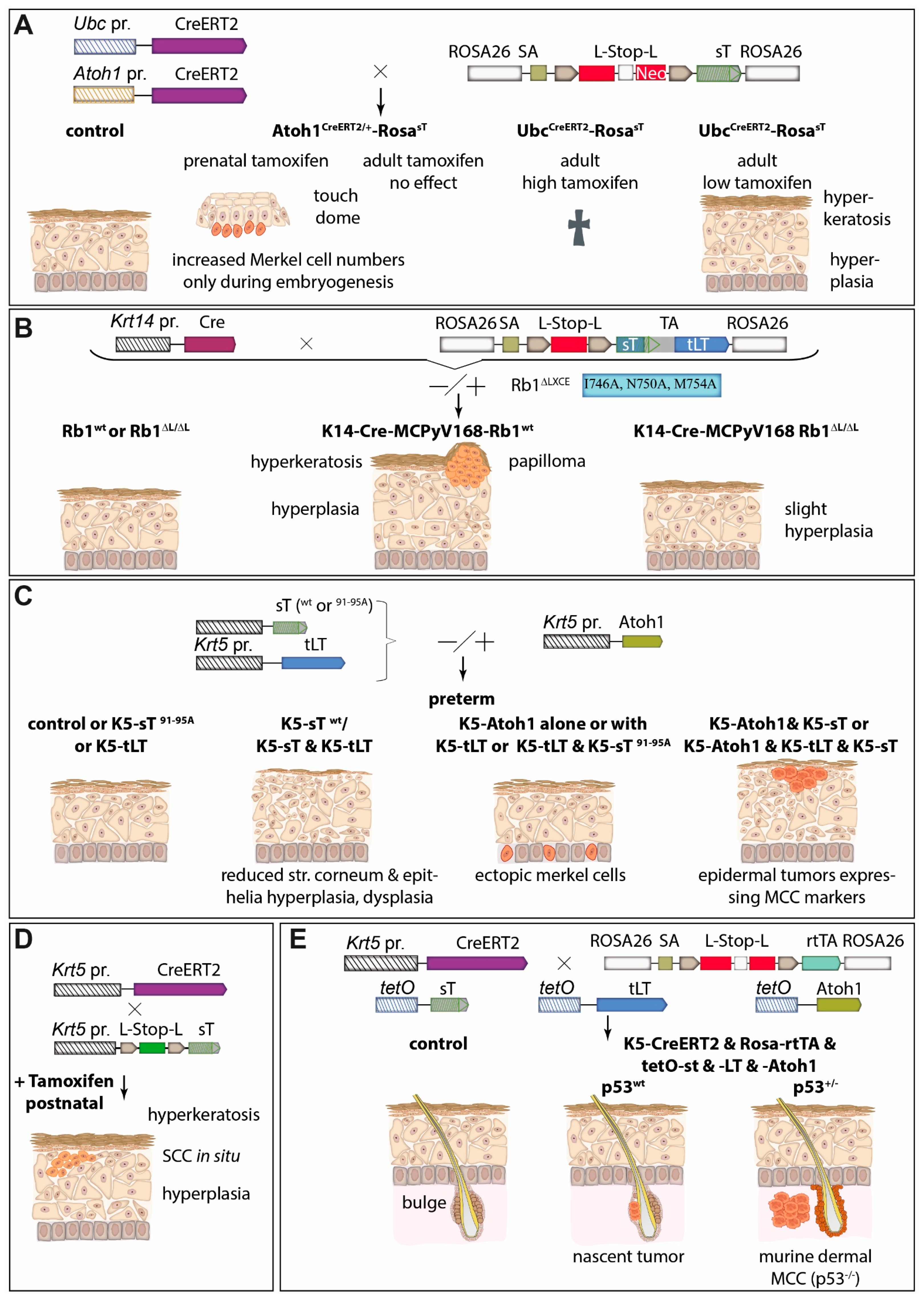
5.5. MCC Mouse Models
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Stewart, S.E.; Eddy, B.E.; Gochenour, A.M.; Borgese, N.G.; Grubbs, G.E. The induction of neoplasms with a substance released from mouse tumors by tissue culture. Virology 1957, 3, 380–400. [Google Scholar] [CrossRef] [PubMed]
- Gross, L. Neck tumors, or leukemia, developing in adult C3H mice following inoculation, in early infancy, with filtered (Berkefeld N), or centrifugated (144,000× g), Ak-leukemic extracts. Cancer 1953, 6, 948–958. [Google Scholar] [CrossRef] [PubMed]
- Eddy, B.E.; Rowe, W.P.; Hartley, J.W.; Stewart, S.E.; Huebner, R.J. Hemagglutination with the SE polyoma virus. Virology 1958, 6, 290–291. [Google Scholar] [CrossRef] [PubMed]
- Stewart, S.E.; Eddy, B.E.; Borgese, N. Neoplasms in mice inoculated with a tumor agent carried in tissue culture. J. Natl. Cancer Inst. 1958, 20, 1223–1243. [Google Scholar] [CrossRef] [PubMed]
- Moens, U.; Prezioso, C.; Pietropaolo, V. Functional Domains of the Early Proteins and Experimental and Epidemiological Studies Suggest a Role for the Novel Human Polyomaviruses in Cancer. Front. Microbiol. 2022, 13, 834368. [Google Scholar] [CrossRef]
- Moens, U.; Prezioso, C.; Pietropaolo, V. Genetic Diversity of the Noncoding Control Region of the Novel Human Polyomaviruses. Viruses 2020, 12, 1406. [Google Scholar] [CrossRef]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [Green Version]
- Gardner, S.D.; Field, A.M.; Coleman, D.V.; Hulme, B. New human papovavirus (B.K.) isolated from urine after renal transplantation. Lancet 1971, 1, 1253–1257. [Google Scholar] [CrossRef]
- Sirohi, D.; Vaske, C.; Sanborn, Z.; Smith, S.C.; Don, M.D.; Lindsey, K.G.; Federman, S.; Vankalakunti, M.; Koo, J.; Bose, S.; et al. Polyoma virus-associated carcinomas of the urologic tract: A clinicopathologic and molecular study. Mod. Pathol. 2018, 31, 1429–1441. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Zhou, Y.; Deng, W.; Wang, Y.; Lee, R.J.; Liu, Y.; Elias, N.; Hu, Y.; Luo, M.H.; Liu, R.; et al. Genome-wide profiling of BK polyomavirus integration in bladder cancer of kidney transplant recipients reveals mechanisms of the integration at the nucleotide level. Oncogene 2021, 40, 46–54. [Google Scholar] [CrossRef]
- Baker, S.C.; Mason, A.S.; Slip, R.G.; Skinner, K.T.; Macdonald, A.; Masood, O.; Harris, R.S.; Fenton, T.R.; Periyasamy, M.; Ali, S.; et al. Induction of APOBEC3-mediated genomic damage in urothelium implicates BK polyomavirus (BKPyV) as a hit-and-run driver for bladder cancer. Oncogene 2022, 41, 2139–2151. [Google Scholar] [CrossRef]
- Foulongne, V.; Courgnaud, V.; Champeau, W.; Segondy, M. Detection of Merkel cell polyomavirus on environmental surfaces. J. Med. Virol. 2011, 83, 1435–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foulongne, V.; Kluger, N.; Dereure, O.; Mercier, G.; Moles, J.P.; Guillot, B.; Segondy, M. Merkel cell polyomavirus in cutaneous swabs. Emerg. Infect. Dis. 2010, 16, 685–687. [Google Scholar] [CrossRef] [PubMed]
- Pastrana, D.V.; Wieland, U.; Silling, S.; Buck, C.B.; Pfister, H. Positive correlation between Merkel cell polyomavirus viral load and capsid-specific antibody titer. Med. Microbiol. Immunol. 2012, 201, 17–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampras, S.S.; Giuliano, A.R.; Lin, H.Y.; Fisher, K.J.; Abrahamsen, M.E.; McKay-Chopin, S.; Gheit, T.; Tommasino, M.; Rollison, D.E. Natural history of polyomaviruses in men: The HPV infection in men (HIM) study. J. Infect. Dis. 2015, 211, 1437–1446. [Google Scholar] [CrossRef] [Green Version]
- Bopp, L.; Wieland, U.; Hellmich, M.; Kreuter, A.; Pfister, H.; Silling, S. Natural History of Cutaneous Human Polyomavirus Infection in Healthy Individuals. Front. Microbiol. 2021, 12, 740947. [Google Scholar] [CrossRef]
- Hashida, Y.; Kamioka, M.; Tanaka, M.; Hosokawa, S.; Murakami, M.; Nakajima, K.; Kikuchi, H.; Fujieda, M.; Sano, S.; Daibata, M. Ecology of Merkel Cell Polyomavirus in Healthy Skin Among Individuals in an Asian Cohort. J. Infect. Dis. 2016, 213, 1708–1716. [Google Scholar] [CrossRef] [Green Version]
- Husseiny, M.I.; Anastasi, B.; Singer, J.; Lacey, S.F. A comparative study of Merkel cell, BK and JC polyomavirus infections in renal transplant recipients and healthy subjects. J. Clin. Virol. 2010, 49, 137–140. [Google Scholar] [CrossRef] [Green Version]
- Bialasiewicz, S.; Lambert, S.B.; Whiley, D.M.; Nissen, M.D.; Sloots, T.P. Merkel cell polyomavirus DNA in respiratory specimens from children and adults. Emerg. Infect. Dis. 2009, 15, 492–494. [Google Scholar] [CrossRef]
- Pancaldi, C.; Corazzari, V.; Maniero, S.; Mazzoni, E.; Comar, M.; Martini, F.; Tognon, M. Merkel cell polyomavirus DNA sequences in the buffy coats of healthy blood donors. Blood 2011, 117, 7099–7101. [Google Scholar] [CrossRef]
- Mertz, K.D.; Junt, T.; Schmid, M.; Pfaltz, M.; Kempf, W. Inflammatory monocytes are a reservoir for Merkel cell polyomavirus. J. Investig. Dermatol. 2010, 130, 1146–1151. [Google Scholar] [CrossRef] [Green Version]
- Viscidi, R.P.; Rollison, D.E.; Sondak, V.K.; Silver, B.; Messina, J.L.; Giuliano, A.R.; Fulp, W.; Ajidahun, A.; Rivanera, D. Age-specific seroprevalence of Merkel cell polyomavirus, BK virus, and JC virus. Clin. Vaccine Immunol. 2011, 18, 1737–1743. [Google Scholar] [CrossRef] [Green Version]
- Nicol, J.T.; Robinot, R.; Carpentier, A.; Carandina, G.; Mazzoni, E.; Tognon, M.; Touze, A.; Coursaget, P. Age-specific seroprevalences of merkel cell polyomavirus, human polyomaviruses 6, 7, and 9, and trichodysplasia spinulosa-associated polyomavirus. Clin. Vaccine Immunol. 2013, 20, 363–368. [Google Scholar] [CrossRef]
- Tolstov, Y.L.; Knauer, A.; Chen, J.G.; Kensler, T.W.; Kingsley, L.A.; Moore, P.S.; Chang, Y. Asymptomatic primary Merkel cell polyomavirus infection among adults. Emerg. Infect. Dis. 2011, 17, 1371–1380. [Google Scholar] [CrossRef]
- Schowalter, R.M.; Pastrana, D.V.; Buck, C.B. Glycosaminoglycans and sialylated glycans sequentially facilitate Merkel cell polyomavirus infectious entry. PLoS Pathog. 2011, 7, e1002161. [Google Scholar] [CrossRef] [Green Version]
- Neu, U.; Hengel, H.; Blaum, B.S.; Schowalter, R.M.; Macejak, D.; Gilbert, M.; Wakarchuk, W.W.; Imamura, A.; Ando, H.; Kiso, M.; et al. Structures of Merkel cell polyomavirus VP1 complexes define a sialic acid binding site required for infection. PLoS Pathog. 2012, 8, e1002738. [Google Scholar] [CrossRef]
- Erickson, K.D.; Garcea, R.L.; Tsai, B. Ganglioside GT1b is a putative host cell receptor for the Merkel cell polyomavirus. J. Virol. 2009, 83, 10275–10279. [Google Scholar] [CrossRef] [Green Version]
- Becker, M.; Dominguez, M.; Greune, L.; Soria-Martinez, L.; Pfleiderer, M.M.; Schowalter, R.; Buck, C.B.; Blaum, B.S.; Schmidt, M.A.; Schelhaas, M. Infectious Entry of Merkel Cell Polyomavirus. J. Virol. 2019, 93, e02004-18. [Google Scholar] [CrossRef] [Green Version]
- Giannecchini, S. Evidence of the Mechanism by Which Polyomaviruses Exploit the Extracellular Vesicle Delivery System during Infection. Viruses 2020, 12, 585. [Google Scholar] [CrossRef]
- Kwun, H.J.; Chang, Y.; Moore, P.S. Protein-mediated viral latency is a novel mechanism for Merkel cell polyomavirus persistence. Proc. Natl. Acad. Sci USA 2017, 114, E4040–E4047. [Google Scholar] [CrossRef]
- Starrett, G.J.; Marcelus, C.; Cantalupo, P.G.; Katz, J.P.; Cheng, J.; Akagi, K.; Thakuria, M.; Rabinowits, G.; Wang, L.C.; Symer, D.E.; et al. Merkel Cell Polyomavirus Exhibits Dominant Control of the Tumor Genome and Transcriptome in Virus-Associated Merkel Cell Carcinoma. mBio 2017, 8, e02079-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czech-Sioli, M.; Gunther, T.; Therre, M.; Spohn, M.; Indenbirken, D.; Theiss, J.; Riethdorf, S.; Qi, M.; Alawi, M.; Wulbeck, C.; et al. High-resolution analysis of Merkel Cell Polyomavirus in Merkel Cell Carcinoma reveals distinct integration patterns and suggests NHEJ and MMBIR as underlying mechanisms. PLoS Pathog. 2020, 16, e1008562. [Google Scholar] [CrossRef] [PubMed]
- Schowalter, R.M.; Reinhold, W.C.; Buck, C.B. Entry tropism of BK and Merkel cell polyomaviruses in cell culture. PLoS ONE 2012, 7, e42181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Yang, R.; Payne, A.S.; Schowalter, R.M.; Spurgeon, M.E.; Lambert, P.F.; Xu, X.; Buck, C.B.; You, J. Identifying the Target Cells and Mechanisms of Merkel Cell Polyomavirus Infection. Cell Host Microbe 2016, 19, 775–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeCaprio, J.A. Molecular Pathogenesis of Merkel Cell Carcinoma. Annu. Rev. Pathol. 2021, 16, 69–91. [Google Scholar] [CrossRef]
- Yang, J.F.; You, J. Merkel cell polyomavirus and associated Merkel cell carcinoma. Tumour Virus Res. 2022, 13, 200232. [Google Scholar] [CrossRef]
- Ahmed, M.M.; Cushman, C.H.; De Caprio, J.A. Merkel Cell Polyomavirus: Oncogenesis in a Stable Genome. Viruses 2021, 14, 58. [Google Scholar] [CrossRef]
- Sastre-Garau, X.; Peter, M.; Avril, M.F.; Laude, H.; Couturier, J.; Rozenberg, F.; Almeida, A.; Boitier, F.; Carlotti, A.; Couturaud, B.; et al. Merkel cell carcinoma of the skin: Pathological and molecular evidence for a causative role of MCV in oncogenesis. J. Pathol. 2009, 218, 48–56. [Google Scholar] [CrossRef]
- Schrama, D.; Sarosi, E.M.; Adam, C.; Ritter, C.; Kaemmerer, U.; Klopocki, E.; Konig, E.M.; Utikal, J.; Becker, J.C.; Houben, R. Characterization of six Merkel cell polyomavirus-positive Merkel cell carcinoma cell lines: Integration pattern suggest that large T antigen truncating events occur before or during integration. Int. J. Cancer 2019, 145, 1020–1032. [Google Scholar] [CrossRef]
- Starrett, G.J.; Thakuria, M.; Chen, T.; Marcelus, C.; Cheng, J.; Nomburg, J.; Thorner, A.R.; Slevin, M.K.; Powers, W.; Burns, R.T.; et al. Clinical and molecular characterization of virus-positive and virus-negative Merkel cell carcinoma. Genome Med. 2020, 12, 30. [Google Scholar] [CrossRef]
- Shuda, M.; Feng, H.; Kwun, H.J.; Rosen, S.T.; Gjoerup, O.; Moore, P.S.; Chang, Y. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc. Natl. Acad. Sci. USA 2008, 105, 16272–16277. [Google Scholar] [CrossRef] [Green Version]
- Shuda, M.; Kwun, H.J.; Feng, H.; Chang, Y.; Moore, P.S. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J. Clin. Investig. 2011, 121, 3623–3634. [Google Scholar] [CrossRef]
- Houben, R.; Shuda, M.; Weinkam, R.; Schrama, D.; Feng, H.; Chang, Y.; Moore, P.S.; Becker, J.C. Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expression of viral T antigens. J. Virol. 2010, 84, 7064–7072. [Google Scholar] [CrossRef] [Green Version]
- Verhaegen, M.E.; Mangelberger, D.; Harms, P.W.; Eberl, M.; Wilbert, D.M.; Meireles, J.; Bichakjian, C.K.; Saunders, T.L.; Wong, S.Y.; Dlugosz, A.A. Merkel Cell Polyomavirus Small T Antigen Initiates Merkel Cell Carcinoma-like Tumor Development in Mice. Cancer Res. 2017, 77, 3151–3157. [Google Scholar] [CrossRef] [Green Version]
- Spurgeon, M.E.; Cheng, J.; Bronson, R.T.; Lambert, P.F.; De Caprio, J.A. Tumorigenic activity of merkel cell polyomavirus T antigens expressed in the stratified epithelium of mice. Cancer Res. 2015, 75, 1068–1079. [Google Scholar] [CrossRef] [Green Version]
- Harms, P.W.; Vats, P.; Verhaegen, M.E.; Robinson, D.R.; Wu, Y.M.; Dhanasekaran, S.M.; Palanisamy, N.; Siddiqui, J.; Cao, X.; Su, F.; et al. The Distinctive Mutational Spectra of Polyomavirus-Negative Merkel Cell Carcinoma. Cancer Res. 2015, 75, 3720–3727. [Google Scholar] [CrossRef] [Green Version]
- Goh, G.; Walradt, T.; Markarov, V.; Blom, A.; Riaz, N.; Doumani, R.; Stafstrom, K.; Moshiri, A.; Yelistratova, L.; Levinsohn, J.; et al. Mutational landscape of MCPyV-positive and MCPyV-negative Merkel cell carcinomas with implications for immunotherapy. Oncotarget 2016, 7, 3403–3415. [Google Scholar] [CrossRef] [Green Version]
- De Caprio, J.A.; Garcea, R.L. A cornucopia of human polyomaviruses. Nat. Rev. Microbiol. 2013, 11, 264–276. [Google Scholar] [CrossRef]
- Moens, U.; Krumbholz, A.; Ehlers, B.; Zell, R.; Johne, R.; Calvignac-Spencer, S.; Lauber, C. Biology, evolution, and medical importance of polyomaviruses: An update. Infect. Genet. Evol. 2017, 54, 18–38. [Google Scholar] [CrossRef]
- Johne, R.; Buck, C.B.; Allander, T.; Atwood, W.J.; Garcea, R.L.; Imperiale, M.J.; Major, E.O.; Ramqvist, T.; Norkin, L.C. Taxonomical developments in the family Polyomaviridae. Arch. Virol. 2011, 156, 1627–1634. [Google Scholar] [CrossRef]
- Schowalter, R.M.; Buck, C.B. The Merkel cell polyomavirus minor capsid protein. PLoS Pathog. 2013, 9, e1003558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johne, R.; Paul, G.; Enderlein, D.; Stahl, T.; Grund, C.; Muller, H. Avian polyomavirus mutants with deletions in the VP4-encoding region show deficiencies in capsid assembly and virus release, and have reduced infectivity in chicken. J. Gen. Virol. 2007, 88, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, C.S.; Pipas, J.M. T antigens of simian virus 40: Molecular chaperones for viral replication and tumorigenesis. Microbiol. Mol. Biol. Rev. 2002, 66, 179–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moens, U.; Van Ghelue, M.; Ludvigsen, M.; Korup-Schulz, S.; Ehlers, B. Early and late promoters of BK polyomavirus, Merkel cell polyomavirus, Trichodysplasia spinulosa-associated polyomavirus and human polyomavirus 12 are among the strongest of all known human polyomaviruses in 10 different cell lines. J. Gen. Virol. 2015, 96, 2293–2303. [Google Scholar] [CrossRef] [PubMed]
- Ajuh, E.T.; Wu, Z.; Kraus, E.; Weissbach, F.H.; Bethge, T.; Gosert, R.; Fischer, N.; Hirsch, H.H. Novel Human Polyomavirus Noncoding Control Regions Differ in Bidirectional Gene Expression according to Host Cell, Large T-Antigen Expression, and Clinically Occurring Rearrangements. J. Virol. 2018, 92, e02231-17. [Google Scholar] [CrossRef] [Green Version]
- Houben, R.; Hesbacher, S.; Sarma, B.; Schulte, C.; Sarosi, E.M.; Popp, S.; Adam, C.; Kervarrec, T.; Schrama, D. Inhibition of T-antigen expression promoting glycogen synthase kinase 3 impairs merkel cell carcinoma cell growth. Cancer Lett. 2022, 524, 259–267. [Google Scholar] [CrossRef]
- Baez, C.F.; Brandao Varella, R.; Villani, S.; Delbue, S. Human Polyomaviruses: The Battle of Large and Small Tumor Antigens. Virology 2017, 8, 1178122X17744785. [Google Scholar] [CrossRef]
- Pietropaolo, V.; Prezioso, C.; Moens, U. Merkel Cell Polyomavirus and Merkel Cell Carcinoma. Cancers 2020, 12, 1774. [Google Scholar] [CrossRef]
- Carter, J.J.; Daugherty, M.D.; Qi, X.; Bheda-Malge, A.; Wipf, G.C.; Robinson, K.; Roman, A.; Malik, H.S.; Galloway, D.A. Identification of an overprinting gene in Merkel cell polyomavirus provides evolutionary insight into the birth of viral genes. Proc. Natl. Acad. Sci. USA 2013, 110, 12744–12749. [Google Scholar] [CrossRef] [Green Version]
- van der Meijden, E.; Feltkamp, M. The Human Polyomavirus Middle and Alternative T-Antigens; Thoughts on Roles and Relevance to Cancer. Front. Microbiol. 2018, 9, 398. [Google Scholar] [CrossRef]
- Cheng, J.; DeCaprio, J.A.; Fluck, M.M.; Schaffhausen, B.S. Cellular transformation by Simian Virus 40 and Murine Polyoma Virus T antigens. Semin. Cancer Biol. 2009, 19, 218–228. [Google Scholar] [CrossRef] [Green Version]
- De Caprio, J.A. How the Rb tumor suppressor structure and function was revealed by the study of Adenovirus and SV40. Virology 2009, 384, 274–284. [Google Scholar] [CrossRef] [Green Version]
- Lane, D.P.; Crawford, L.V. T antigen is bound to a host protein in SV40-transformed cells. Nature 1979, 278, 261–263. [Google Scholar] [CrossRef]
- Cheng, J.; Rozenblatt-Rosen, O.; Paulson, K.G.; Nghiem, P.; DeCaprio, J.A. Merkel cell polyomavirus large T antigen has growth-promoting and inhibitory activities. J. Virol. 2013, 87, 6118–6126. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, X.; Diaz, J.; Tsang, S.H.; Buck, C.B.; You, J. Merkel cell polyomavirus large T antigen disrupts host genomic integrity and inhibits cellular proliferation. J. Virol. 2013, 87, 9173–9188. [Google Scholar] [CrossRef] [Green Version]
- Borchert, S.; Czech-Sioli, M.; Neumann, F.; Schmidt, C.; Wimmer, P.; Dobner, T.; Grundhoff, A.; Fischer, N. High-affinity Rb binding, p53 inhibition, subcellular localization, and transformation by wild-type or tumor-derived shortened Merkel cell polyomavirus large T antigens. J. Virol. 2014, 88, 3144–3160. [Google Scholar] [CrossRef] [Green Version]
- Houben, R.; Dreher, C.; Angermeyer, S.; Borst, A.; Utikal, J.; Haferkamp, S.; Peitsch, W.K.; Schrama, D.; Hesbacher, S. Mechanisms of p53 restriction in Merkel cell carcinoma cells are independent of the Merkel cell polyoma virus T antigens. J. Investig. Dermatol. 2013, 133, 2453–2460. [Google Scholar] [CrossRef] [Green Version]
- Park, D.E.; Cheng, J.; Berrios, C.; Montero, J.; Cortes-Cros, M.; Ferretti, S.; Arora, R.; Tillgren, M.L.; Gokhale, P.C.; De Caprio, J.A. Dual inhibition of MDM2 and MDM4 in virus-positive Merkel cell carcinoma enhances the p53 response. Proc. Natl. Acad. Sci. USA 2019, 116, 1027–1032. [Google Scholar] [CrossRef] [Green Version]
- Czech-Sioli, M.; Siebels, S.; Radau, S.; Zahedi, R.P.; Schmidt, C.; Dobner, T.; Grundhoff, A.; Fischer, N. The Ubiquitin-Specific Protease Usp7, a Novel Merkel Cell Polyomavirus Large T-Antigen Interaction Partner, Modulates Viral DNA Replication. J. Virol. 2020, 94, e01638-19. [Google Scholar] [CrossRef]
- Hesbacher, S.; Pfitzer, L.; Wiedorfer, K.; Angermeyer, S.; Borst, A.; Haferkamp, S.; Scholz, C.J.; Wobser, M.; Schrama, D.; Houben, R. RB1 is the crucial target of the Merkel cell polyomavirus Large T antigen in Merkel cell carcinoma cells. Oncotarget 2016, 7, 32956–32968. [Google Scholar] [CrossRef]
- Schrama, D.; Hesbacher, S.; Angermeyer, S.; Schlosser, A.; Haferkamp, S.; Aue, A.; Adam, C.; Weber, A.; Schmidt, M.; Houben, R. Serine 220 phosphorylation of the Merkel cell polyomavirus large T antigen crucially supports growth of Merkel cell carcinoma cells. Int. J. Cancer 2016, 138, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Houben, R.; Angermeyer, S.; Haferkamp, S.; Aue, A.; Goebeler, M.; Schrama, D.; Hesbacher, S. Characterization of functional domains in the Merkel cell polyoma virus Large T antigen. Int. J. Cancer 2015, 136, E290–E300. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hein, J.; Richardson, S.C.; Basse, P.H.; Toptan, T.; Moore, P.S.; Gjoerup, O.V.; Chang, Y. Merkel cell polyomavirus large T antigen disrupts lysosome clustering by translocating human Vam6p from the cytoplasm to the nucleus. J. Biol. Chem. 2011, 286, 17079–17090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Xie, H.; Shi, H.; Gao, J.; Juhlin, C.C.; Bjornhagen, V.; Hoog, A.; Lee, L.; Larsson, C.; Lui, W.O. Merkel cell polyomavirus oncoproteins induce microRNAs that suppress multiple autophagy genes. Int. J. Cancer 2020, 146, 1652–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caplan, S.; Hartnell, L.M.; Aguilar, R.C.; Naslavsky, N.; Bonifacino, J.S. Human Vam6p promotes lysosome clustering and fusion in vivo. J. Cell. Biol. 2001, 154, 109–122. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Sato, Y.; Watanabe, D.; Ito, H.; Shimonohara, N.; Tsuji, T.; Nakajima, N.; Suzuki, Y.; Matsuo, K.; Nakagawa, H.; et al. Nuclear localization of Merkel cell polyomavirus large T antigen in Merkel cell carcinoma. Virology 2010, 398, 273–279. [Google Scholar] [CrossRef]
- Nwogu, N.; Ortiz, L.E.; Kwun, H.J. Merkel Cell Polyomavirus Large T Antigen Unique Domain Regulates Its Own Protein Stability and Cell Growth. Viruses 2020, 12, 1043. [Google Scholar] [CrossRef]
- Kumar, S.H.; Rangarajan, A. Simian virus 40 small T antigen activates AMPK and triggers autophagy to protect cancer cells from nutrient deprivation. J. Virol. 2009, 83, 8565–8574. [Google Scholar] [CrossRef] [Green Version]
- Sablina, A.A.; Hahn, W.C. SV40 small T antigen and PP2A phosphatase in cell transformation. Cancer Metastasis Rev. 2008, 27, 137–146. [Google Scholar] [CrossRef]
- Dye, K.N.; Welcker, M.; Clurman, B.E.; Roman, A.; Galloway, D.A. Merkel cell polyomavirus Tumor antigens expressed in Merkel cell carcinoma function independently of the ubiquitin ligases Fbw7 and beta-TrCP. PLoS Pathog. 2019, 15, e1007543. [Google Scholar] [CrossRef]
- Zhao, J.; Jia, Y.; Shen, S.; Kim, J.; Wang, X.; Lee, E.; Brownell, I.; Cho-Vega, J.H.; Lewis, C.; Homsi, J.; et al. Merkel Cell Polyomavirus Small T Antigen Activates Noncanonical NF-kappaB Signaling to Promote Tumorigenesis. Mol. Cancer Res. 2020, 18, 1623–1637. [Google Scholar] [CrossRef] [PubMed]
- Nwogu, N.; Ortiz, L.E.; Whitehouse, A.; Kwun, H.J. Merkel Cell Polyomavirus Small Tumor Antigen Activates Matrix Metallopeptidase-9 Gene Expression for Cell Migration and Invasion. J. Virol. 2020, 94, e00786-20. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Park, D.E.; Berrios, C.; White, E.A.; Arora, R.; Yoon, R.; Branigan, T.; Xiao, T.; Westerling, T.; Federation, A.; et al. Merkel cell polyomavirus recruits MYCL to the EP400 complex to promote oncogenesis. PLoS Pathog. 2017, 13, e1006668. [Google Scholar] [CrossRef] [PubMed]
- Park, D.E.; Cheng, J.; McGrath, J.P.; Lim, M.Y.; Cushman, C.; Swanson, S.K.; Tillgren, M.L.; Paulo, J.A.; Gokhale, P.C.; Florens, L.; et al. Merkel cell polyomavirus activates LSD1-mediated blockade of non-canonical BAF to regulate transformation and tumorigenesis. Nat. Cell Biol. 2020, 22, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.C.; Klaeger, S.; Le, P.M.; Korthauer, K.; Cheng, J.; Ananthapadmanabhan, V.; Frost, T.C.; Stevens, J.D.; Wong, A.Y.; Iorgulescu, J.B.; et al. Reversal of viral and epigenetic HLA class I repression in Merkel cell carcinoma. J. Clin. Investig. 2022, 132, e151666. [Google Scholar] [CrossRef]
- Shuda, M.; Velasquez, C.; Cheng, E.; Cordek, D.G.; Kwun, H.J.; Chang, Y.; Moore, P.S. CDK1 substitutes for mTOR kinase to activate mitotic cap-dependent protein translation. Proc. Natl. Acad. Sci. USA 2015, 112, 5875–5882. [Google Scholar] [CrossRef] [Green Version]
- Kwun, H.J.; Shuda, M.; Feng, H.; Camacho, C.J.; Moore, P.S.; Chang, Y. Merkel cell polyomavirus small T antigen controls viral replication and oncoprotein expression by targeting the cellular ubiquitin ligase SCFFbw7. Cell Host Microbe 2013, 14, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Nwogu, N.; Ortiz, L.E.; Kwun, H.J. Surface charge of Merkel cell polyomavirus small T antigen determines cell transformation through allosteric FBW7 WD40 domain targeting. Oncogenesis 2020, 9, 53. [Google Scholar] [CrossRef]
- Berrios, C.; Padi, M.; Keibler, M.A.; Park, D.E.; Molla, V.; Cheng, J.; Lee, S.M.; Stephanopoulos, G.; Quackenbush, J.; DeCaprio, J.A. Merkel Cell Polyomavirus Small T Antigen Promotes Pro-Glycolytic Metabolic Perturbations Required for Transformation. PLoS Pathog. 2016, 12, e1006020. [Google Scholar] [CrossRef] [Green Version]
- Knight, L.M.; Stakaityte, G.; Wood, J.J.; Abdul-Sada, H.; Griffiths, D.A.; Howell, G.J.; Wheat, R.; Blair, G.E.; Steven, N.M.; Macdonald, A.; et al. Merkel cell polyomavirus small T antigen mediates microtubule destabilization to promote cell motility and migration. J. Virol. 2015, 89, 35–47. [Google Scholar] [CrossRef]
- Stakaityte, G.; Nwogu, N.; Lippiat, J.D.; Blair, G.E.; Poterlowicz, K.; Boyne, J.R.; Macdonald, A.; Mankouri, J.; Whitehouse, A. The cellular chloride channels CLIC1 and CLIC4 contribute to virus-mediated cell motility. J. Biol. Chem. 2018, 293, 4582–4590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stakaityte, G.; Nwogu, N.; Dobson, S.J.; Knight, L.M.; Wasson, C.W.; Salguero, F.J.; Blackbourn, D.J.; Blair, G.E.; Mankouri, J.; Macdonald, A.; et al. Merkel Cell Polyomavirus Small T Antigen Drives Cell Motility via Rho-GTPase-Induced Filopodium Formation. J. Virol. 2018, 92, e00940-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, D.A.; Abdul-Sada, H.; Knight, L.M.; Jackson, B.R.; Richards, K.; Prescott, E.L.; Peach, A.H.; Blair, G.E.; Macdonald, A.; Whitehouse, A. Merkel cell polyomavirus small T antigen targets the NEMO adaptor protein to disrupt inflammatory signaling. J. Virol. 2013, 87, 13853–13867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, R.; Lee, E.E.; Kim, J.; Choi, J.H.; Kolitz, E.; Chen, Y.; Crewe, C.; Salisbury, N.J.H.; Scherer, P.E.; Cockerell, C.; et al. Characterization of ALTO-encoding circular RNAs expressed by Merkel cell polyomavirus and trichodysplasia spinulosa polyomavirus. PLoS Pathog. 2021, 17, e1009582. [Google Scholar] [CrossRef] [PubMed]
- Abere, B.; Zhou, H.; Li, J.; Cao, S.; Toptan, T.; Grundhoff, A.; Fischer, N.; Moore, P.S.; Chang, Y. Merkel Cell Polyomavirus Encodes Circular RNAs (circRNAs) Enabling a Dynamic circRNA/microRNA/mRNA Regulatory Network. mBio 2020, 11, e03059-20. [Google Scholar] [CrossRef]
- Zou, W.; Imperiale, M.J. Biology of Polyomavirus miRNA. Front. Microbiol. 2021, 12, 662892. [Google Scholar] [CrossRef]
- Seo, G.J.; Chen, C.J.; Sullivan, C.S. Merkel cell polyomavirus encodes a microRNA with the ability to autoregulate viral gene expression. Virology 2009, 383, 183–187. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Paulson, K.G.; Murchison, E.P.; Afanasiev, O.K.; Alkan, C.; Leonard, J.H.; Byrd, D.R.; Hannon, G.J.; Nghiem, P. Identification and validation of a novel mature microRNA encoded by the Merkel cell polyomavirus in human Merkel cell carcinomas. J. Clin. Virol. 2011, 52, 272–275. [Google Scholar] [CrossRef] [Green Version]
- Theiss, J.M.; Gunther, T.; Alawi, M.; Neumann, F.; Tessmer, U.; Fischer, N.; Grundhoff, A. A Comprehensive Analysis of Replicating Merkel Cell Polyomavirus Genomes Delineates the Viral Transcription Program and Suggests a Role for mcv-miR-M1 in Episomal Persistence. PLoS Pathog. 2015, 11, e1004974. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhbari, P.; Tobin, D.; Poterlowicz, K.; Roberts, W.; Boyne, J.R. MCV-miR-M1 Targets the Host-Cell Immune Response Resulting in the Attenuation of Neutrophil Chemotaxis. J. Investig. Dermatol. 2018, 138, 2343–2354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harms, K.L.; Healy, M.A.; Nghiem, P.; Sober, A.J.; Johnson, T.M.; Bichakjian, C.K.; Wong, S.L. Analysis of Prognostic Factors from 9387 Merkel Cell Carcinoma Cases Forms the Basis for the New 8th Edition AJCC Staging System. Ann. Surg. Oncol. 2016, 23, 3564–3571. [Google Scholar] [CrossRef] [PubMed]
- Eisemann, N.; Jansen, L.; Castro, F.A.; Chen, T.; Eberle, A.; Nennecke, A.; Zeissig, S.R.; Brenner, H.; Katalinic, A. Survival with nonmelanoma skin cancer in Germany. Br. J. Dermatol. 2016, 174, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Toker, C. Trabecular carcinoma of the skin. Arch. Dermatol. 1972, 105, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Rywlin, A.M. Malignant Merkel-cell tumor is a more accurate description than trabecular carcinoma. Am. J. Dermatopathol. 1982, 4, 513–515. [Google Scholar] [CrossRef]
- Maricich, S.M.; Wellnitz, S.A.; Nelson, A.M.; Lesniak, D.R.; Gerling, G.J.; Lumpkin, E.A.; Zoghbi, H.Y. Merkel cells are essential for light-touch responses. Science 2009, 324, 1580–1582. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, R.; Cha, M.; Ling, J.; Jia, Z.; Coyle, D.; Gu, J.G. Merkel cells transduce and encode tactile stimuli to drive Abeta-afferent impulses. Cell 2014, 157, 664–675. [Google Scholar] [CrossRef] [Green Version]
- Halata, Z.; Grim, M.; Bauman, K.I. Friedrich Sigmund Merkel and his “Merkel cell”, morphology, development, and physiology: Review and new results. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 2003, 271, 225–239. [Google Scholar] [CrossRef]
- Xiao, Y.; Williams, J.S.; Brownell, I. Merkel cells and touch domes: More than mechanosensory functions? Exp. Dermatol. 2014, 23, 692–695. [Google Scholar] [CrossRef]
- Ranade, S.S.; Woo, S.H.; Dubin, A.E.; Moshourab, R.A.; Wetzel, C.; Petrus, M.; Mathur, J.; Begay, V.; Coste, B.; Mainquist, J.; et al. Piezo2 is the major transducer of mechanical forces for touch sensation in mice. Nature 2014, 516, 121–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moll, I.; Kuhn, C.; Moll, R. Cytokeratin 20 is a general marker of cutaneous Merkel cells while certain neuronal proteins are absent. J. Investig. Dermatol. 1995, 104, 910–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fradette, J.; Godbout, M.J.; Michel, M.; Germain, L. Localization of Merkel cells at hairless and hairy human skin sites using keratin 18. Biochem. Cell Biol. 1995, 73, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Ben-Arie, N.; Hassan, B.A.; Bermingham, N.A.; Malicki, D.M.; Armstrong, D.; Matzuk, M.; Bellen, H.J.; Zoghbi, H.Y. Functional conservation of atonal and Math1 in the CNS and PNS. Development 2000, 127, 1039–1048. [Google Scholar] [CrossRef]
- Morrison, K.M.; Miesegaes, G.R.; Lumpkin, E.A.; Maricich, S.M. Mammalian Merkel cells are descended from the epidermal lineage. Dev. Biol. 2009, 336, 76–83. [Google Scholar] [CrossRef] [Green Version]
- Nirenberg, A.; Steinman, H.; Dixon, J.; Dixon, A. Merkel cell carcinoma update: The case for two tumours. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 1425–1431. [Google Scholar] [CrossRef]
- Wijaya, W.A.; Liu, Y.; Qing, Y.; Li, Z. Prevalence of Merkel Cell Polyomavirus in Normal and Lesional Skin: A Systematic Review and Meta-Analysis. Front. Oncol. 2022, 12, 868781. [Google Scholar] [CrossRef]
- Sunshine, J.C.; Jahchan, N.S.; Sage, J.; Choi, J. Are there multiple cells of origin of Merkel cell carcinoma? Oncogene 2018, 37, 1409–1416. [Google Scholar] [CrossRef]
- Gambichler, T.; Abu Rached, N.; Tannapfel, A.; Becker, J.C.; Vogt, M.; Skrygan, M.; Wieland, U.; Silling, S.; Susok, L.; Stücker, M.; et al. Expression of Mismatch Repair Proteins in Merkel Cell Carcinoma. Cancers 2021, 13, 2524. [Google Scholar] [CrossRef]
- Merot, Y. Is the neuroendocrine carcinoma of the skin a Merkel cell tumor? What we can learn from immunohistochemical and ultrastructural studies. Int. J. Dermatol. 1990, 29, 102–104. [Google Scholar] [CrossRef]
- Shuda, M.; Guastafierro, A.; Geng, X.; Shuda, Y.; Ostrowski, S.M.; Lukianov, S.; Jenkins, F.J.; Honda, K.; Maricich, S.M.; Moore, P.S.; et al. Merkel Cell Polyomavirus Small T Antigen Induces Cancer and Embryonic Merkel Cell Proliferation in a Transgenic Mouse Model. PLoS ONE 2015, 10, e0142329. [Google Scholar] [CrossRef] [Green Version]
- Tilling, T.; Moll, I. Which are the cells of origin in merkel cell carcinoma? J. Skin. Cancer 2012, 2012, 680410. [Google Scholar] [CrossRef] [Green Version]
- Siebels, S.; Czech-Sioli, M.; Spohn, M.; Schmidt, C.; Theiss, J.; Indenbirken, D.; Gunther, T.; Grundhoff, A.; Fischer, N. Merkel Cell Polyomavirus DNA Replication Induces Senescence in Human Dermal Fibroblasts in a Kap1/Trim28-Dependent Manner. mBio 2020, 11, e00142-20. [Google Scholar] [CrossRef] [Green Version]
- Kervarrec, T.; Samimi, M.; Guyetant, S.; Sarma, B.; Cheret, J.; Blanchard, E.; Berthon, P.; Schrama, D.; Houben, R.; Touze, A. Histogenesis of Merkel Cell Carcinoma: A Comprehensive Review. Front. Oncol. 2019, 9, 451. [Google Scholar] [CrossRef] [Green Version]
- Zur Hausen, A.; Rennspiess, D.; Winnepenninckx, V.; Speel, E.J.; Kurz, A.K. Early B-cell differentiation in Merkel cell carcinomas: Clues to cellular ancestry. Cancer Res. 2013, 73, 4982–4987. [Google Scholar] [CrossRef] [Green Version]
- Sauer, C.M.; Haugg, A.M.; Chteinberg, E.; Rennspiess, D.; Winnepenninckx, V.; Speel, E.J.; Becker, J.C.; Kurz, A.K.; Zur Hausen, A. Reviewing the current evidence supporting early B-cells as the cellular origin of Merkel cell carcinoma. Crit. Rev. Oncol. Hematol. 2017, 116, 99–105. [Google Scholar] [CrossRef]
- Verhaegen, M.E.; Mangelberger, D.; Harms, P.W.; Vozheiko, T.D.; Weick, J.W.; Wilbert, D.M.; Saunders, T.L.; Ermilov, A.N.; Bichakjian, C.K.; Johnson, T.M.; et al. Merkel cell polyomavirus small T antigen is oncogenic in transgenic mice. J. Investig. Dermatol. 2015, 135, 1415–1424. [Google Scholar] [CrossRef] [Green Version]
- Kervarrec, T.; Appenzeller, S.; Samimi, M.; Sarma, B.; Sarosi, E.M.; Berthon, P.; Le Corre, Y.; Hainaut-Wierzbicka, E.; Blom, A.; Benethon, N.; et al. Merkel Cell PolyomavirusNegative Merkel Cell Carcinoma Originating from In Situ Squamous Cell Carcinoma: A Keratinocytic Tumor with Neuroendocrine Differentiation. J. Investig. Dermatol. 2022, 142, 516–527. [Google Scholar] [CrossRef]
- De Coste, R.C.; Walsh, N.M.; Gaston, D.; Ly, T.Y.; Pasternak, S.; Cutler, S.; Nightingale, M.; Carter, M.D. RB1-deficient squamous cell carcinoma: The proposed source of combined Merkel cell carcinoma. Mod. Pathol. 2022, 35, 1829–1836. [Google Scholar] [CrossRef]
- Harms, P.W.; Verhaegen, M.E.; Hu, K.; Hrycaj, S.M.; Chan, M.P.; Liu, C.J.; Grachtchouk, M.; Patel, R.M.; Udager, A.M.; Dlugosz, A.A. Genomic evidence suggests that cutaneous neuroendocrine carcinomas can arise from squamous dysplastic precursors. Mod. Pathol. 2022, 35, 506–514. [Google Scholar] [CrossRef]
- Walsh, N.M. Primary neuroendocrine (Merkel cell) carcinoma of the skin: Morphologic diversity and implications thereof. Hum. Pathol. 2001, 32, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Donizy, P.; Wu, C.L.; Cornejo, K.M.; Rys, J.; Hoang, M.P. Morphologic Diversity of Merkel Cell Carcinoma. Am. J. Dermatopathol. 2020, 42, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Gravemeyer, J.; Spassova, I.; Verhaegen, M.E.; Dlugosz, A.A.; Hoffmann, D.; Lange, A.; Becker, J.C. DNA-methylation patterns imply a common cellular origin of virus- and UV-associated Merkel cell carcinoma. Oncogene 2022, 41, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.C.; Eberl, M.; Vagnozzi, A.N.; Belkadi, A.; Veniaminova, N.A.; Verhaegen, M.E.; Bichakjian, C.K.; Ward, N.L.; Dlugosz, A.A.; Wong, S.Y. Basal cell carcinoma preferentially arises from stem cells within hair follicle and mechanosensory niches. Cell Stem. Cell 2015, 16, 400–412. [Google Scholar] [CrossRef] [Green Version]
- Kervarrec, T.; Aljundi, M.; Appenzeller, S.; Samimi, M.; Maubec, E.; Cribier, B.; Deschamps, L.; Sarma, B.; Sarosi, E.M.; Berthon, P.; et al. Polyomavirus-Positive Merkel Cell Carcinoma Derived from a Trichoblastoma Suggests an Epithelial Origin of this Merkel Cell Carcinoma. J. Investig. Dermatol. 2020, 140, 976–985. [Google Scholar] [CrossRef]
- Syder, A.J.; Karam, S.M.; Mills, J.C.; Ippolito, J.E.; Ansari, H.R.; Farook, V.; Gordon, J.I. A transgenic mouse model of metastatic carcinoma involving transdifferentiation of a gastric epithelial lineage progenitor to a neuroendocrine phenotype. Proc. Natl. Acad. Sci. USA 2004, 101, 4471–4476. [Google Scholar] [CrossRef] [Green Version]
- Purkis, P.E.; Steel, J.B.; Mackenzie, I.C.; Nathrath, W.B.; Leigh, I.M.; Lane, E.B. Antibody markers of basal cells in complex epithelia. J. Cell. Sci. 1990, 97 Pt 1, 39–50. [Google Scholar] [CrossRef]
- Spurgeon, M.E.; Cheng, J.; Ward-Shaw, E.; Dick, F.A.; De Caprio, J.A.; Lambert, P.F. Merkel cell polyomavirus large T antigen binding to pRb promotes skin hyperplasia and tumor development. PLoS Pathog. 2022, 18, e1010551. [Google Scholar] [CrossRef]
- Spurgeon, M.E.; Liem, A.; Buehler, D.; Cheng, J.; DeCaprio, J.A.; Lambert, P.F. The Merkel Cell Polyomavirus T Antigens Function as Tumor Promoters in Murine Skin. Cancers 2021, 13, 222. [Google Scholar] [CrossRef]
- Verhaegen, M.E.; Harms, P.W.; Van Goor, J.J.; Arche, J.; Patrick, M.T.; Wilbert, D.; Zabawa, H.; Grachtchouk, M.; Liu, C.J.; Hu, K.; et al. Direct cellular reprogramming enables development of viral T antigen-driven Merkel cell carcinoma in mice. J. Clin. Investig. 2022, 132, e152069. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Houben, R.; Celikdemir, B.; Kervarrec, T.; Schrama, D. Merkel Cell Polyomavirus: Infection, Genome, Transcripts and Its Role in Development of Merkel Cell Carcinoma. Cancers 2023, 15, 444. https://doi.org/10.3390/cancers15020444
Houben R, Celikdemir B, Kervarrec T, Schrama D. Merkel Cell Polyomavirus: Infection, Genome, Transcripts and Its Role in Development of Merkel Cell Carcinoma. Cancers. 2023; 15(2):444. https://doi.org/10.3390/cancers15020444
Chicago/Turabian StyleHouben, Roland, Büke Celikdemir, Thibault Kervarrec, and David Schrama. 2023. "Merkel Cell Polyomavirus: Infection, Genome, Transcripts and Its Role in Development of Merkel Cell Carcinoma" Cancers 15, no. 2: 444. https://doi.org/10.3390/cancers15020444