
Molecular Landscape and Validation of New Genomic Classification in 2668 Adult AML Patients: Real Life Data from the PETHEMA Registry

 , , , , , , , , ,
, , , , , , , , ,  , ,
, ,  , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Development of the Diagnostic Platform
2.2. Study Design and Reference Laboratories
2.3. Consensus Genes Establishment
2.4. NGS Standardization Procedures and Cross–Validation Rounds
2.5. Patients and Inclusion Criteria
2.6. Clinical Validation
2.7. Statistics
3. Results
3.1. Third Cross Validation Round
3.2. Baseline Demographics and Molecular Profile in NGS–AML Protocol Cohort
3.3. Summary Mutation Profile
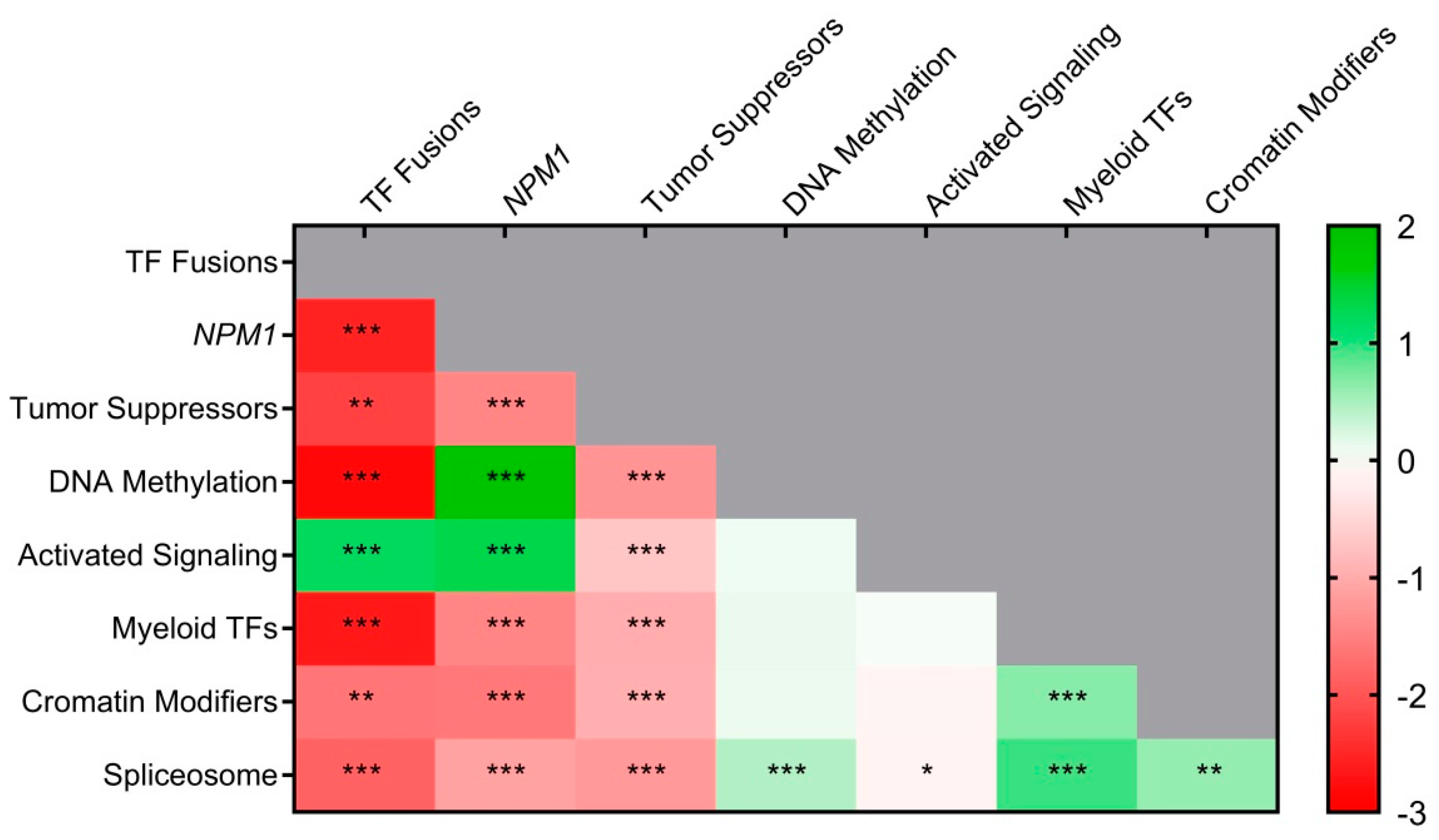
3.4. Co–Mutations and Exclusivity Patterns
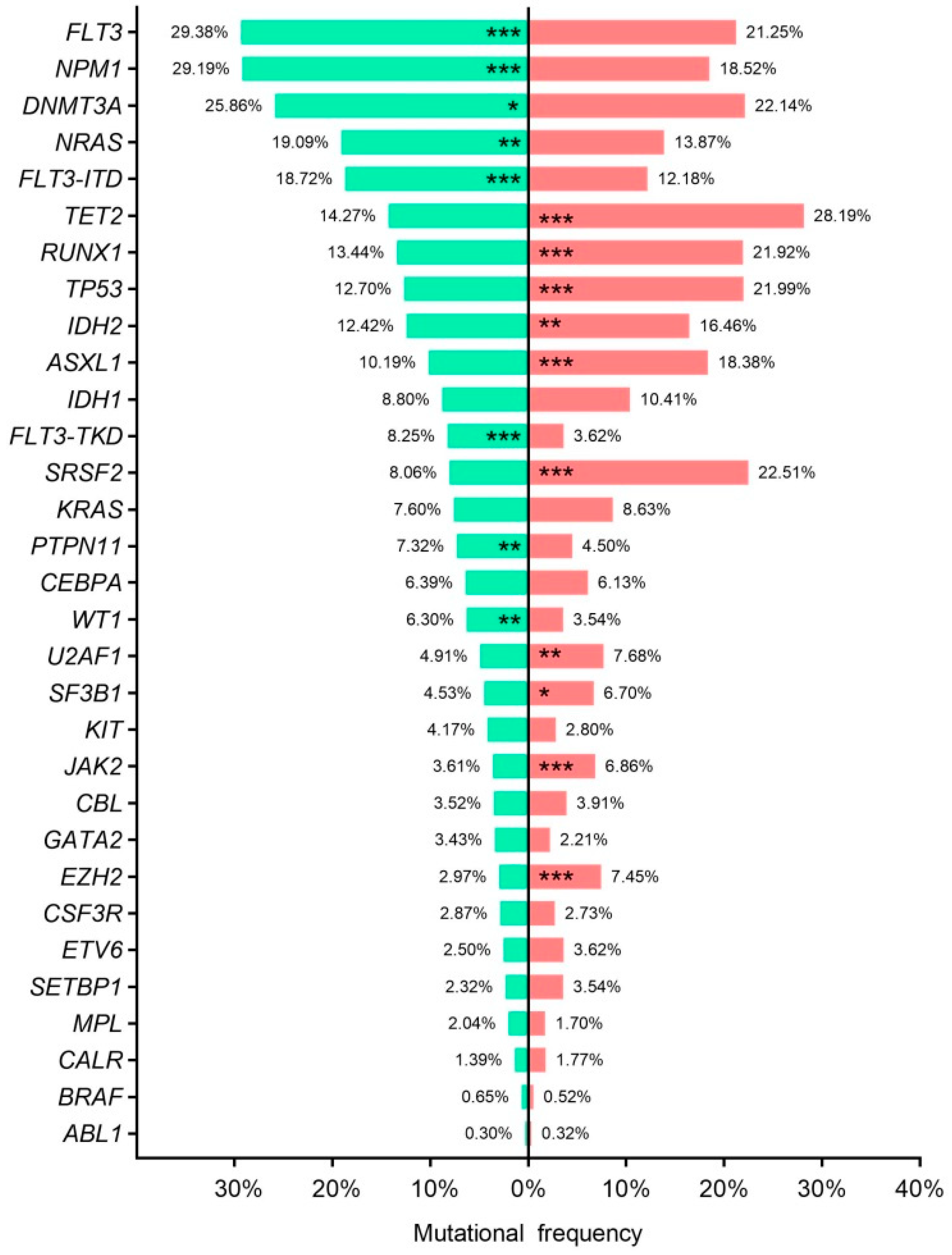
3.5. Disease Stage Mutational Profile
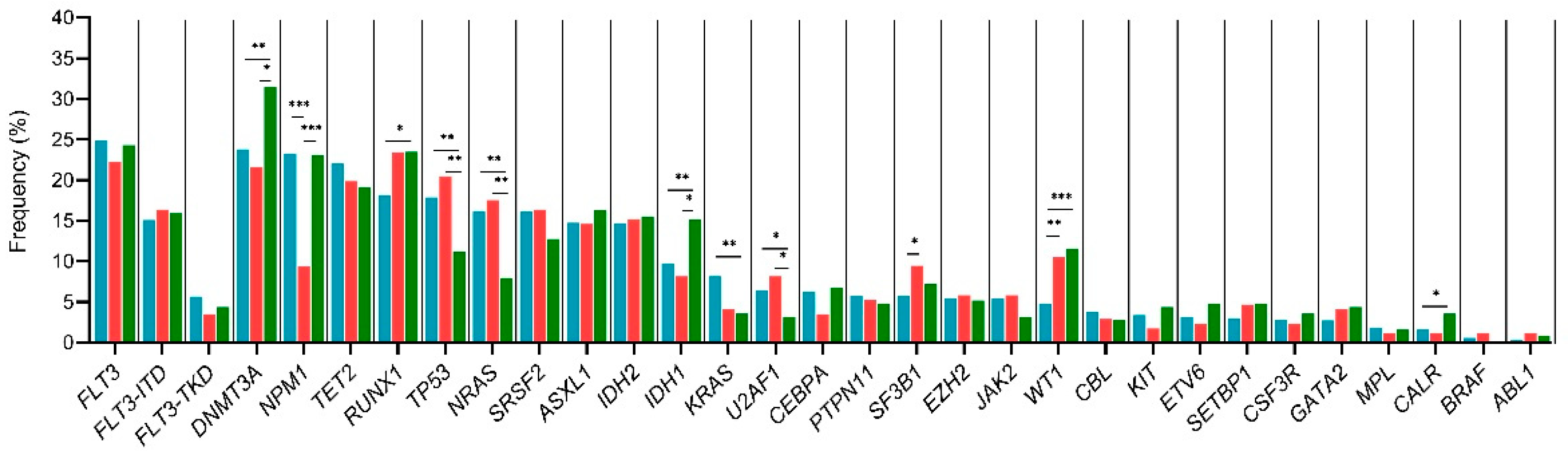
3.6. Age–Related Mutational Profile
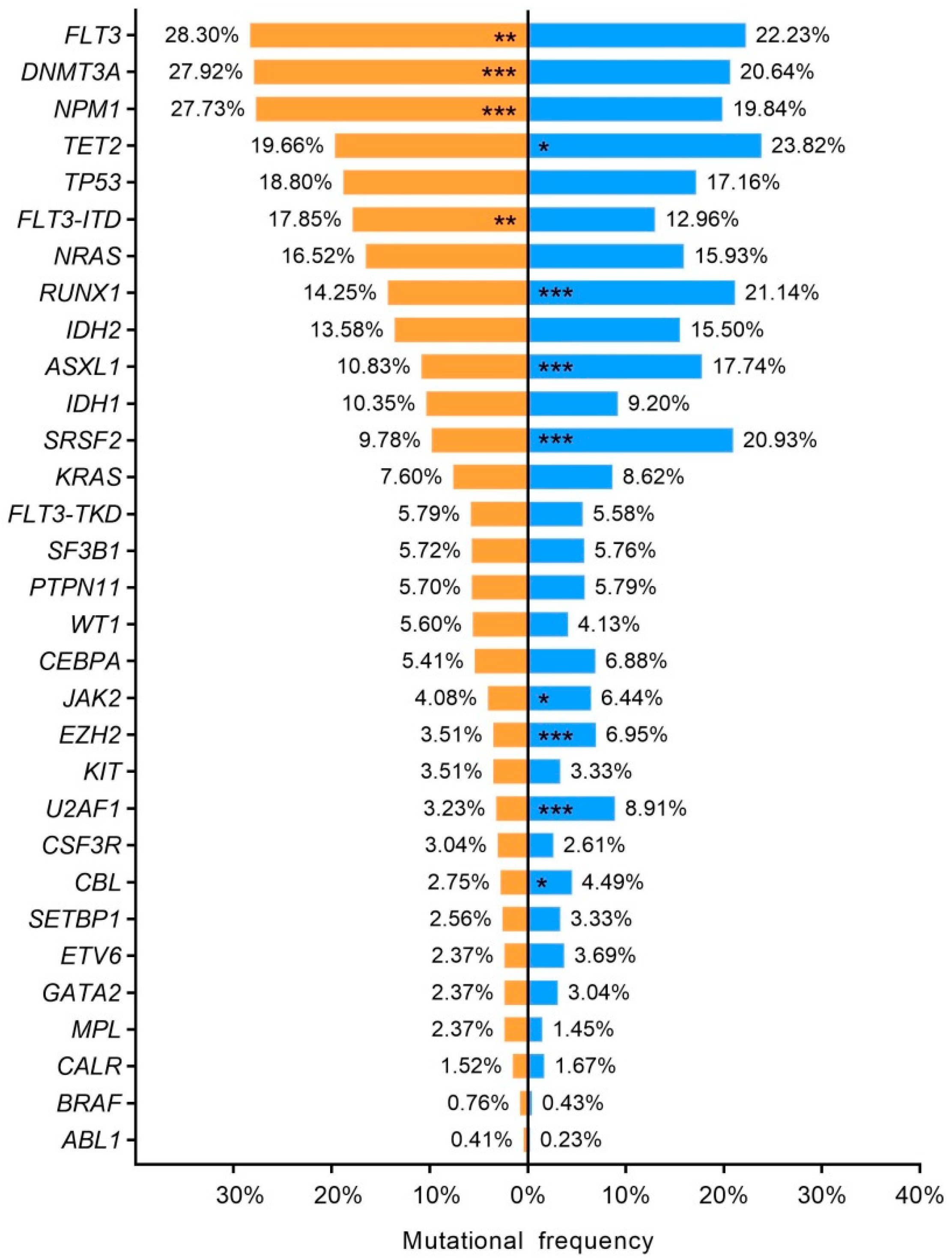
3.7. Sex–Related Mutational Profile
3.8. Paired Samples and Mutation Stability
3.9. New Genomic Classification Applied to PETHEMA–NGS Cohort
3.9.1. Prognosis Value of Molecular Classes
3.9.2. Integrated Risk Score
Comparison between the Integrated Risk Score and 2022 ELN Risk Classification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Tazi, Y.; Arango-Ossa, J.E.; Zhou, Y.; Bernard, E.; Thomas, I.; Gilkes, A.; Freeman, S.; Pradat, Y.; Johnson, S.J.; Hills, R.; et al. Unified Classification and Risk-Stratification in Acute Myeloid Leukemia. medRxiv 2022, 2022.03.09.22271087. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemia: Integrating Morphological, Clinical, and Genomic Data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and Management of AML in Adults: 2022 ELN Recommendations from an International Expert Panel. Blood 2022, 129, 424–447. [Google Scholar] [CrossRef] [Green Version]
- Levine, R.L.; Valk, P.J.M. Next-Generation Sequencing in the Diagnosis and Minimal Residual Disease Assessment of Acute Myeloid Leukemia. Haematologica 2019, 104, 868. [Google Scholar] [CrossRef] [PubMed]
- Bacher, U.; Shumilov, E.; Flach, J.; Porret, N.; Joncourt, R.; Wiedemann, G.; Fiedler, M.; Novak, U.; Amstutz, U.; Pabst, T. Challenges in the Introduction of Next-Generation Sequencing (NGS) for Diagnostics of Myeloid Malignancies into Clinical Routine Use. Blood Cancer J. 2018, 8, 113. [Google Scholar] [CrossRef] [Green Version]
- Haferlach, T. Advancing Leukemia Diagnostics: Role of Next Generation Sequencing (NGS) in Acute Myeloid Leukemia. Hematol. Rep. 2020, 12, 8957. [Google Scholar] [CrossRef]
- Sargas, C.; Ayala, R.; Chillón, M.C.; Larráyoz, M.J.; Carrillo-Cruz, E.; Bilbao, C.; Yébenes-Ramírez, M.; Llop, M.; Rapado, I.; García-Sanz, R.; et al. Networking for Advanced Molecular Diagnosis in Acute Myeloid Leukemia Patients Is Possible: The PETHEMA NGS-AML Project. Haematologica 2021, 106, 3079–3089. [Google Scholar] [CrossRef]
- Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, G.; Hoadley, K.; Triche, T.J.; Laird, P.W.; Baty, J.D.; et al. Genomic and Epigenomic Landscapes of Adult de Novo Acute Myeloid Leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef]
- Leisch, M.; Jansko, B.; Zaborsky, N.; Greil, R.; Pleyer, L. Next Generation Sequencing in AML-On the Way to Becoming a New Standard for Treatment Initiation and/or Modulation? Cancers 2019, 11, 252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luthra, R.; Patel, K.P.; Routbort, M.J.; Broaddus, R.R.; Yau, J.; Simien, C.; Chen, W.; Hatfield, D.Z.; Medeiros, L.J.; Singh, R.R. A Targeted High-Throughput Next-Generation Sequencing Panel for Clinical Screening of Mutations, Gene Amplifications, and Fusions in Solid Tumors. J. Mol. Diagn. 2017, 19, 255–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrackova, A.; Vasinek, M.; Sedlarikova, L.; Dyskova, T.; Schneiderova, P.; Novosad, T.; Papajik, T.; Kriegova, E. Standardization of Sequencing Coverage Depth in NGS: Recommendation for Detection of Clonal and Subclonal Mutations in Cancer Diagnostics. Front. Oncol. 2019, 9, 851. [Google Scholar] [CrossRef] [PubMed]
- Endrullat, C.; Glökler, J.; Franke, P.; Frohme, M. Standardization and Quality Management in Next-Generation Sequencing. Appl. Transl. Genom. 2016, 10, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Pandzic, T.; Ladenvall, C.; Engvall, M.; Mattsson, M.; Hermanson, M.; Cavelier, L.; Ljungström, V.; Baliakas, P. Five Percent Variant Allele Frequency Is a Reliable Reporting Threshold for TP53 Variants Detected by Next Generation Sequencing in Chronic Lymphocytic Leukemia in the Clinical Setting. Hemasphere 2022, 6, e761. [Google Scholar] [CrossRef]
- Jennings, L.J.; Arcila, M.E.; Corless, C.; Kamel-Reid, S.; Lubin, I.M.; Pfeifer, J.; Temple-Smolkin, R.L.; Voelkerding, K.V.; Nikiforova, M.N. Guidelines for Validation of Next-Generation Sequencing-Based Oncology Panels: A Joint Consensus Recommendation of the Association for Molecular Pathology and College of American Pathologists. J. Mol. Diagn. 2017, 19, 341–365. [Google Scholar] [CrossRef] [Green Version]
- Guillermin, Y.; Lopez, J.; Chabane, K.; Hayette, S.; Bardel, C.; Salles, G.; Sujobert, P.; Huet, S. What Does This Mutation Mean? The Tools and Pitfalls of Variant Interpretation in Lymphoid Malignancies. Int. J. Mol. Sci. 2018, 19, 1251. [Google Scholar] [CrossRef] [Green Version]
- Strom, S.P. Current Practices and Guidelines for Clinical Next-Generation Sequencing Oncology Testing. Cancer Biol. Med. 2016, 13, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Qin, G.; Ilya, S.; Kim, T.-K.; Tercan, B.; Martins, T.J.; Dai, J.; Chien, S.; Carson, A.; Patay, B.; Estey, E.H.; et al. Co-Occurring Mutation Clusters Predict Drug Sensitivity in Acute Myeloid Leukemia. Blood 2020, 136, 12–13. [Google Scholar] [CrossRef]
- Lachowiez, C.A.; Reville, P.K.; Kantarjian, H.; Jabbour, E.; Borthakur, G.; Daver, N.; Issa, G.; Furudate, K.; Tanaka, T.; Pierce, S.; et al. Contemporary Outcomes in IDH-Mutated Acute Myeloid Leukemia: The Impact of Co-Occurring NPM1 Mutations and Venetoclax-Based Treatment. Am. J. Hematol. 2022, 97, 1443–1452. [Google Scholar] [CrossRef]
- Bullinger, L.; Döhner, K.; Dohner, H. Genomics of Acute Myeloid Leukemia Diagnosis and Pathways. J. Clin. Oncol. 2017, 35, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal Evolution in Relapsed Acute Myeloid Leukaemia Revealed by Whole-Genome Sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, J.; Peng, D.; Liu, L. Drug Resistance Mechanisms of Acute Myeloid Leukemia Stem Cells. Front. Oncol. 2022, 12, 896426. [Google Scholar] [CrossRef] [PubMed]
- van Gils, N.; Denkers, F.; Smit, L. Escape From Treatment; the Different Faces of Leukemic Stem Cells and Therapy Resistance in Acute Myeloid Leukemia. Front. Oncol. 2021, 11, 659253. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gu, Y.; Chen, B. Mechanisms of Drug Resistance in Acute Myeloid Leukemia. Onco. Targets Ther. 2019, 12, 1937–1945. [Google Scholar] [CrossRef] [Green Version]
- Benard, B.A.; Leak, L.B.; Azizi, A.; Thomas, D.; Gentles, A.J.; Majeti, R. Clonal Architecture Predicts Clinical Outcomes and Drug Sensitivity in Acute Myeloid Leukemia. Nat. Commun. 2021, 12, 7244. [Google Scholar] [CrossRef]
- Thol, F.; Heuser, M. Treatment for Relapsed/Refractory Acute Myeloid Leukemia. Hemasphere 2021, 5, E572. [Google Scholar] [CrossRef]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 Mutations in AML: Review of Current Knowledge and Evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Tarlock, K.; Zhong, S.; He, Y.; Ries, R.; Severson, E.; Bailey, M.; Morley, S.; Balasubramanian, S.; Erlich, R.; Lipson, D.; et al. Distinct Age-Associated Molecular Profiles in Acute Myeloid Leukemia Defined by Comprehensive Clinical Genomic Profiling. Oncotarget 2018, 9, 26417–26430. [Google Scholar] [CrossRef] [Green Version]
- Grimwade, D.; Ivey, A.; Huntly, B.J.P. Molecular Landscape of Acute Myeloid Leukemia in Younger Adults and Its Clinical Relevance. Blood 2016, 127, 29. [Google Scholar] [CrossRef]
- di Nardo, C.D.; Cortes, J.E. Mutations in AML: Prognostic and Therapeutic Implications. Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 348. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Jiang, P.Y.Z.; Sun, H.; Zhang, X.; Jiang, Z.; Li, Y.; Song, Y. Advances in Targeted Therapy for Acute Myeloid Leukemia. Biomark. Res. 2020, 8, 17. [Google Scholar] [CrossRef]
- Ranieri, R.; Pianigiani, G.; Sciabolacci, S.; Perriello, V.M.; Marra, A.; Cardinali, V.; Pierangeli, S.; Milano, F.; Gionfriddo, I.; Brunetti, L.; et al. Current Status and Future Perspectives in Targeted Therapy of NPM1-Mutated AML. Leukemia 2022, 36, 2351–2367. [Google Scholar] [CrossRef] [PubMed]
- Ayala, R.; Rapado, I.; Onecha, E.; Martínez-Cuadrón, D.; Carreño-Tarragona, G.; Bergua, J.M.; Vives, S.; Algarra, J.L.; Tormo, M.; Martinez, P.; et al. The Mutational Landscape of Acute Myeloid Leukaemia Predicts Responses and Outcomes in Elderly Patients from the PETHEMA-FLUGAZA Phase 3 Clinical Trial. Cancers 2021, 13, 2458. [Google Scholar] [CrossRef] [PubMed]
- Prassek, V.V.; Rothenberg-Thurley, M.; Sauerland, M.C.; Herold, T.; Janke, H.; Ksienzyk, B.; Konstandin, N.P.; Goerlich, D.; Krug, U.; Faldum, A.; et al. Genetics of Acute Myeloid Leukemia in the Elderly: Mutation Spectrum and Clinical Impact in Intensively Treated Patients Aged 75 Years or Older. Haematologica 2018, 103, 1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, J.; Hong, J.; Long, Z.; Li, Q.; Xia, R.; Zeng, Q. Mutation Profile and Prognostic Relevance in Elderly Patients with de Novo Acute Myeloid Leukemia Treated with Decitabine-Based Chemotherapy. Int. J. Lab. Hematol. 2020, 42, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Hellesøy, M.; Engen, C.; Grob, T.; Löwenberg, B.; Valk, P.J.M.; Gjertsen, B.T. Sex Disparity in Acute Myeloid Leukaemia with FLT3 Internal Tandem Duplication Mutations: Implications for Prognosis. Mol. Oncol. 2021, 15, 2285–2299. [Google Scholar] [CrossRef]
- De-Morgan, A.; Meggendorfer, M.; Haferlach, C.; Shlush, L. Male Predominance in AML Is Associated with Specific Preleukemic Mutations. Leukemia 2021, 35, 867–870. [Google Scholar] [CrossRef]
- Bullinger, L. CEBPA Mutations in AML: Site Matters. Blood 2022, 139, 6–7. [Google Scholar] [CrossRef]
- Wakita, S.; Sakaguchi, M.; Oh, I.; Kako, S.; Toya, T.; Najima, Y.; Doki, N.; Kanda, J.; Kuroda, J.; Mori, S.; et al. Prognostic Impact of CEBPA BZIP Domain Mutation in Acute Myeloid Leukemia. Blood Adv. 2022, 6, 238–247. [Google Scholar] [CrossRef]
- Mannelli, F.; Ponziani, V.; Bencini, S.; Bonetti, M.I.; Benelli, M.; Cutini, I.; Gianfaldoni, G.; Scappini, B.; Pancani, F.; Piccini, M.; et al. CEBPA–Double-Mutated Acute Myeloid Leukemia Displays a Unique Phenotypic Profile: A Reliable Screening Method and Insight into Biological Features. Haematologica 2017, 102, 529–540. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Mean | Median | Range | N | (%) |
|---|---|---|---|---|---|
| Age, years | 64.9 | 67.7 | 18–98 | 1268 | 100 |
| <65 | 540 | 42.6 | |||
| ≥65 | 728 | 57.4 | |||
| Sex | 1268 | 100 | |||
| Male | 712 | 56.2 | |||
| Female | 556 | 43.8 | |||
| ECOG | 1075 | 100 | |||
| 0 | 420 | 39.1 | |||
| 1 | 452 | 42.0 | |||
| 2 | 135 | 12.6 | |||
| 3 | 53 | 4.9 | |||
| 4 | 15 | 1.4 | |||
| Not available | 193 | ||||
| WBC (×109/L) | 32.8 | 8.8 | 0.24–407 | 1118 | |
| BM blast cells, % | 53.4 | 52.0 | 0–100 | 1026 | |
| Creatinine, mg/dL | 1.1 | 0.90 | 0.28–10.3 | 1071 | |
| MRC cytogenetic profile | 1011 | 100 | |||
| Favorable | 57 | 5.6 | |||
| Intermediate | 178 | 17.6 | |||
| Unfavorable | 269 | 26.6 | |||
| Normal karyotype | 507 | 50.1 | |||
| Not available | 257 | ||||
| AML FAB subtype | 715 | 100 | |||
| M0 | 88 | 12.3 | |||
| M1 | 144 | 20.1 | |||
| M2 | 126 | 17.6 | |||
| M4 | 173 | 24.2 | |||
| M5 | 144 | 20.1 | |||
| M6 | 31 | 4.3 | |||
| M7 | 9 | 1.3 | |||
| Not available | 553 | ||||
| Therapeutic approach | 1268 | 100 | |||
| Intensive | 695 | 54.8 | |||
| Non–intensive | 513 | 40.5 | |||
| Supportive care only | 60 | 4.7 | |||
| Type of AML | 1268 | 100 | |||
| De novo | 920 | 72.6 | |||
| Secondary | 348 | 27.4 |
| Molecular Classes | OS | (95% CI) | p | |
|---|---|---|---|---|
| Lower IC | Upper IC | |||
| inv(16) | NR | <0.001 | ||
| CEBPA bZIP | NR | |||
| No events | NR | |||
| NPM1 | 29.0 | 19.9 | 38.0 | |
| Not class defining mutations | 23.3 | 11.0 | 35.6 | |
| DNMT3A/IDH1–2 | 18.5 | 1.7 | 35.3 | |
| sAML1 | 18.1 | 12.5 | 23.7 | |
| t(8;21) | 17.5 | 3.7 | 31.3 | |
| Trisomies | 14.4 | |||
| t(X;11) | 13.2 | 0.0 | 31.3 | |
| sAML2 | 12.1 | 9.9 | 14.2 | |
| TP53–CK | 5.3 | 2.9 | 7.6 | |
| inv(3) | 4.9 | 0.8 | 9.1 | |
| WT1 | 4.0 | 0.0 | 18.4 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sargas, C.; Ayala, R.; Larráyoz, M.J.; Chillón, M.C.; Carrillo-Cruz, E.; Bilbao-Sieyro, C.; Prados de la Torre, E.; Martínez-Cuadrón, D.; Rodríguez-Veiga, R.; Boluda, B.; et al. Molecular Landscape and Validation of New Genomic Classification in 2668 Adult AML Patients: Real Life Data from the PETHEMA Registry. Cancers 2023, 15, 438. https://doi.org/10.3390/cancers15020438
Sargas C, Ayala R, Larráyoz MJ, Chillón MC, Carrillo-Cruz E, Bilbao-Sieyro C, Prados de la Torre E, Martínez-Cuadrón D, Rodríguez-Veiga R, Boluda B, et al. Molecular Landscape and Validation of New Genomic Classification in 2668 Adult AML Patients: Real Life Data from the PETHEMA Registry. Cancers. 2023; 15(2):438. https://doi.org/10.3390/cancers15020438
Chicago/Turabian StyleSargas, Claudia, Rosa Ayala, María José Larráyoz, María Carmen Chillón, Estrella Carrillo-Cruz, Cristina Bilbao-Sieyro, Esther Prados de la Torre, David Martínez-Cuadrón, Rebeca Rodríguez-Veiga, Blanca Boluda, and et al. 2023. "Molecular Landscape and Validation of New Genomic Classification in 2668 Adult AML Patients: Real Life Data from the PETHEMA Registry" Cancers 15, no. 2: 438. https://doi.org/10.3390/cancers15020438