
EZH2: An Accomplice of Gastric Cancer
Abstract
:Simple Summary
Abstract
1. Introduction
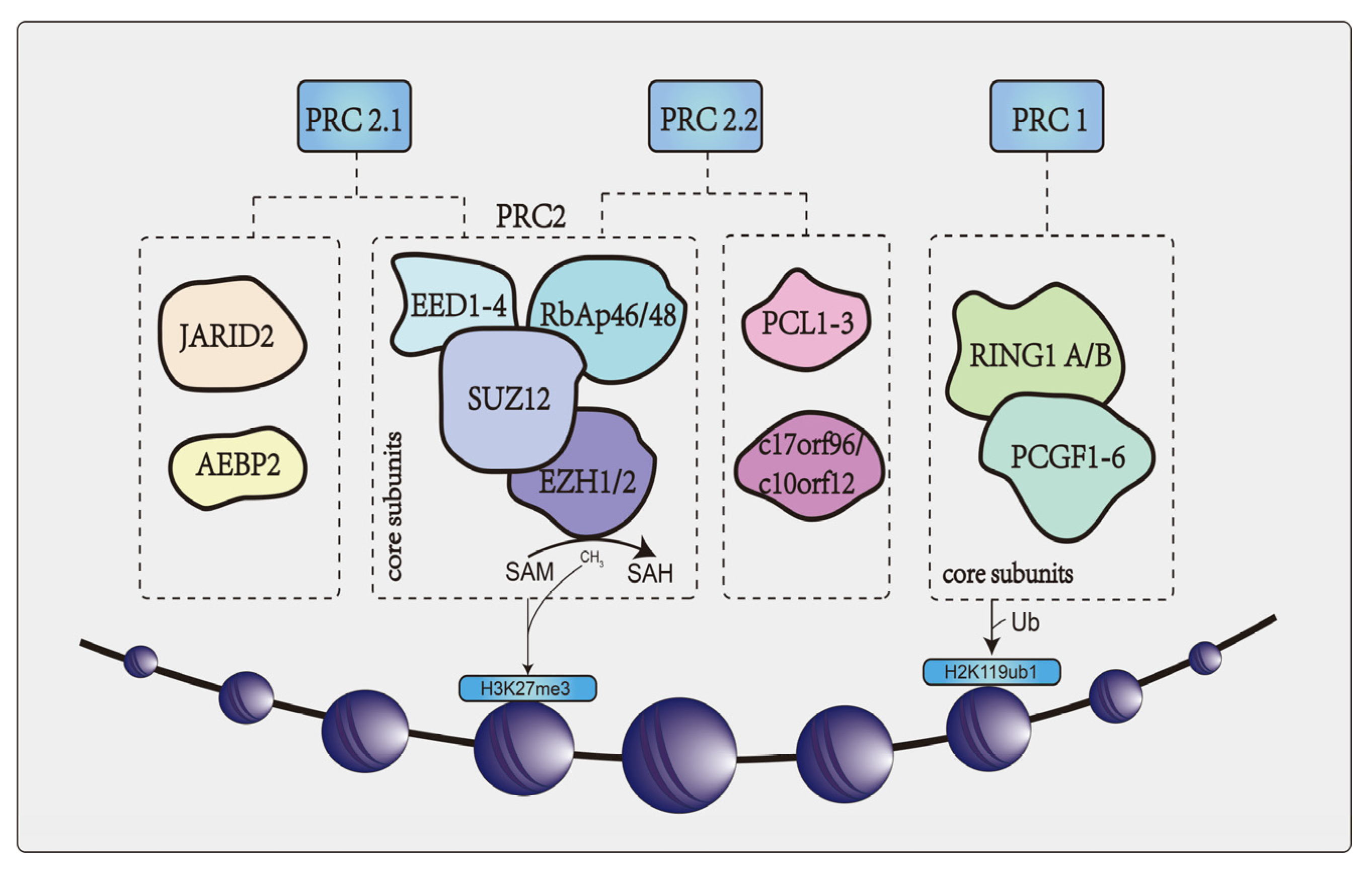
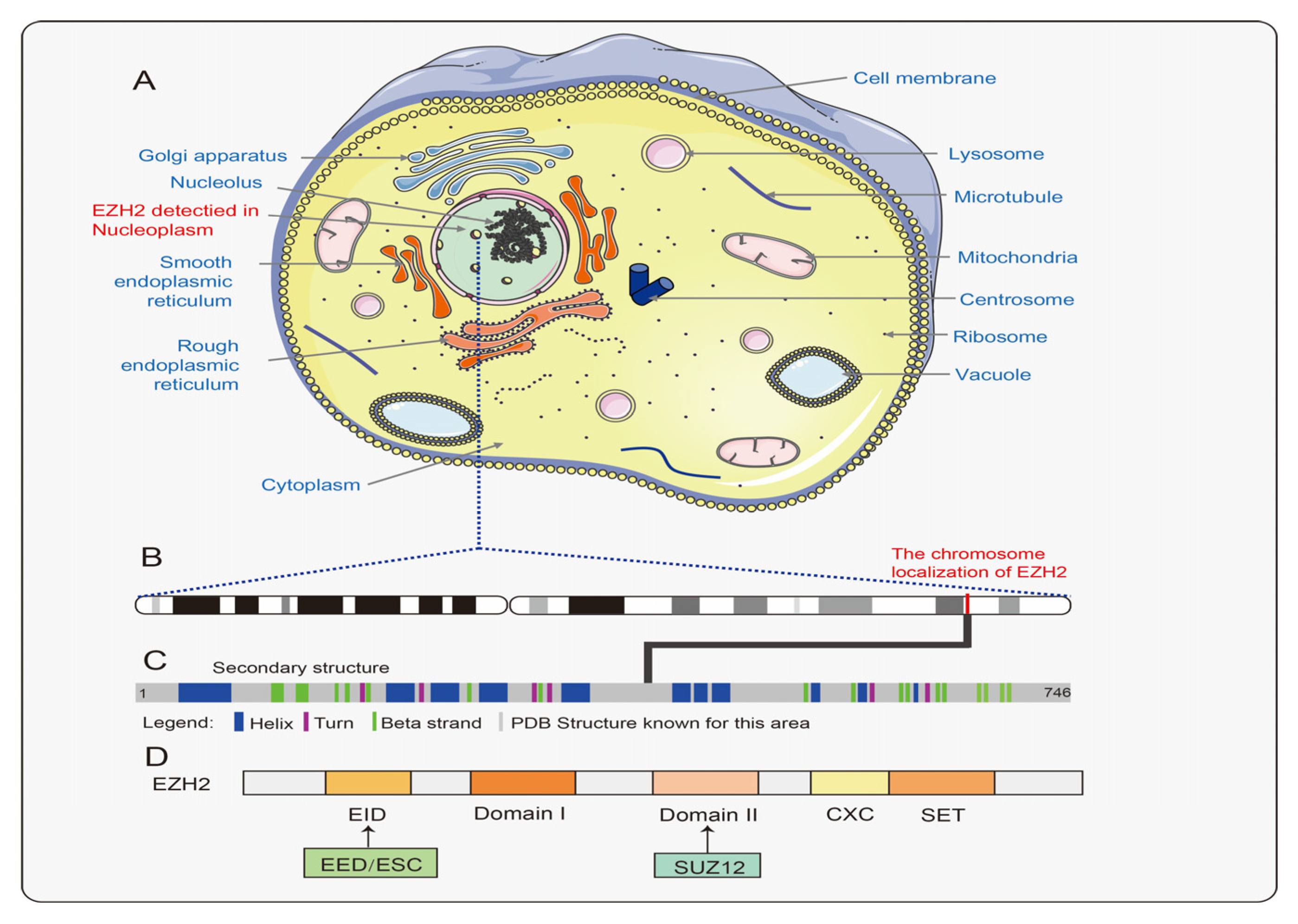
2. Overview of EZH2
3. Histone Modification of EZH2
4. DNA Methylation of EZH2
5. Mechanism of Abnormal Expression of EZH2 in Cancer
6. Regulation of EZH2 in Transcription Levels
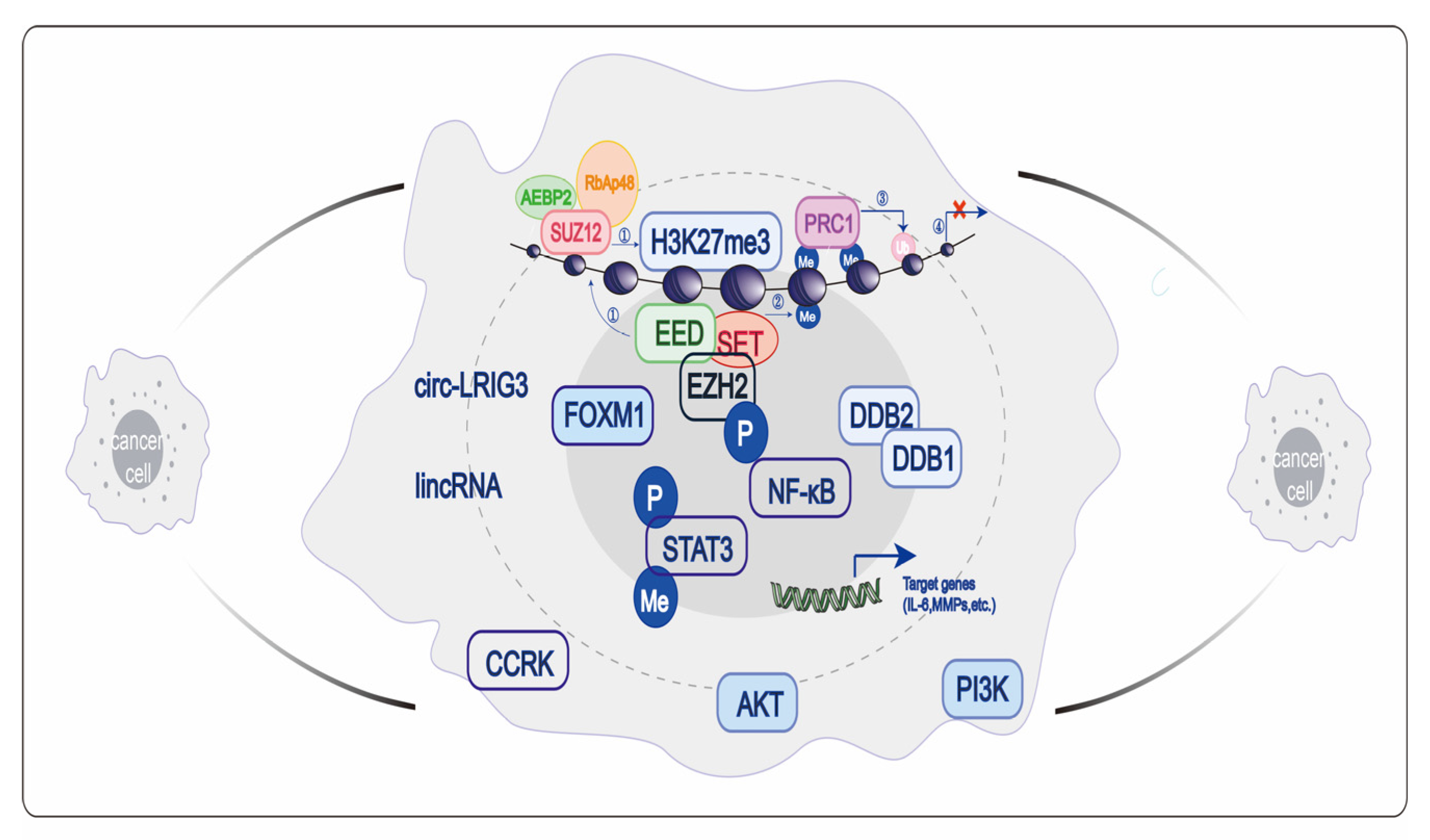
6.1. Transcription Inhibition Dependent on PcG Family
6.2. Transcription Inhibition Independent of the PcG Family
7. Regulation of EZH2 Translation and Post-Translational Modifications
8. Role of EZH2 in GC
9. Association of the EZH2 Gene with Genetic Susceptibility to GC
10. Correlation of EZH2 Gene with Gastric Carcinogenesis and Invasive Metastasis
11. EZH2 Mediates Resistance to Chemotherapeutic Drugs of GC
12. High Expression of EZH2 Leads to Poor Prognosis for GC Patients
13. Current Status of EZH2 Inhibitor Research
14. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Smyth, E.C.; Nilsson, M.; Grabsch, H.I.; van Grieken, N.C.; Lordick, F. Gastric cancer. Lancet 2020, 396, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Liu, Q. Understanding the Global Cancer Statistics 2018: Implications for cancer control. Sci. China Life Sci. 2021, 64, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Canale, M.; Casadei-Gardini, A.; Ulivi, P.; Arechederra, M.; Berasain, C.; Lollini, P.L.; Fernández-Barrena, M.G.; Avila, M.A. Epigenetic Mechanisms in Gastric Cancer: Potential New Therapeutic Opportunities. Int. J. Mol. Sci. 2020, 21, 5500. [Google Scholar] [CrossRef]
- Ramezankhani, R.; Solhi, R.; Es, H.A.; Vosough, M.; Hassan, M. Novel molecular targets in gastric adenocarcinoma. Pharmacol. Ther. 2021, 220, 107714. [Google Scholar] [CrossRef] [PubMed]
- Papale, M.; Ferretti, E.; Battaglia, G.; Bellavia, D.; Mai, A.; Tafani, M. EZH2, HIF-1, and Their Inhibitors: An Overview on Pediatric Cancers. Front. Pediatr. 2018, 6, 328. [Google Scholar] [CrossRef] [PubMed]
- He, L.J.; Cai, M.Y.; Xu, G.L.; Li, J.J.; Weng, Z.J.; Xu, D.Z.; Luo, G.Y.; Zhu, S.L.; Xie, D. Prognostic significance of overexpression of EZH2 and H3k27me3 proteins in gastric cancer. Asian Pac. J. Cancer Prev. APJCP 2012, 13, 3173–3178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, G.G. No Easy Way Out for EZH2: Its Pleiotropic, Noncanonical Effects on Gene Regulation and Cellular Function. Int. J. Mol. Sci. 2020, 21, 9501. [Google Scholar] [CrossRef]
- Stairiker, C.J.; Thomas, G.D.; Salek-Ardakani, S. EZH2 as a Regulator of CD8+ T Cell Fate and Function. Front. Immunol. 2020, 11, 593203. [Google Scholar] [CrossRef]
- Chittock, E.C.; Latwiel, S.; Miller, T.C.; Müller, C.W. Molecular architecture of polycomb repressive complexes. Biochem. Soc. Trans. 2017, 45, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Kuzmichev, A.; Nishioka, K.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002, 16, 2893–2905. [Google Scholar] [CrossRef]
- Margueron, R.; Li, G.; Sarma, K.; Blais, A.; Zavadil, J.; Woodcock, C.L.; Dynlacht, B.D.; Reinberg, D. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell 2008, 32, 503–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, J.A.; Lange, C.A. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat. Res. 2008, 647, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Duan, R.; Du, W.; Guo, W. EZH2: A novel target for cancer treatment. J. Hematol. Oncol. 2020, 13, 104. [Google Scholar] [CrossRef] [PubMed]
- Millán-Zambrano, G.; Burton, A.; Bannister, A.J.; Schneider, R. Histone post-translational modifications—Cause and consequence of genome function. Nat. Rev. Genet. 2022, 23, 563–580. [Google Scholar] [CrossRef]
- Chen, Y.; Ren, B.; Yang, J.; Wang, H.; Yang, G.; Xu, R.; You, L.; Zhao, Y. The role of histone methylation in the development of digestive cancers: A potential direction for cancer management. Signal Transduct. Target. Ther. 2020, 5, 143. [Google Scholar] [CrossRef]
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications—Writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef]
- Viré, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef]
- Cyrus, S.; Burkardt, D.; Weaver, D.D.; Gibson, W.T. PRC2-complex related dysfunction in overgrowth syndromes: A review of EZH2, EED, and SUZ12 and their syndromic phenotypes. Am. J. Med. Genetics. Part C Semin. Med. Genet. 2019, 181, 519–531. [Google Scholar] [CrossRef]
- Hsu, J.H.; Rasmusson, T.; Robinson, J.; Pachl, F.; Read, J.; Kawatkar, S.; O’Donovan, D.H.; Bagal, S.; Code, E.; Rawlins, P.; et al. EED-Targeted PROTACs Degrade EED, EZH2, and SUZ12 in the PRC2 Complex. Cell Chem. Biol. 2020, 27, 41–46.e17. [Google Scholar] [CrossRef]
- Zhu, K.; Du, D.; Yang, R.; Tao, H.; Zhang, H. Identification and Assessments of Novel and Potent Small-Molecule Inhibitors of EED-EZH2 Interaction of Polycomb Repressive Complex 2 by Computational Methods and Biological Evaluations. Chem. Pharm. Bull. 2020, 68, 58–63. [Google Scholar] [CrossRef]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.H.; Roberts, C.W. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanaki, S.; Shimada, M. Targeting EZH2 as cancer therapy. J. Biochem. 2021, 170, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Kleine-Kohlbrecher, D.; Dietrich, N.; Pasini, D.; Gargiulo, G.; Beekman, C.; Theilgaard-Mönch, K.; Minucci, S.; Porse, B.T.; Marine, J.C.; et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007, 21, 525–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Chen, S.; Yu, J.; Gao, Z.; Sun, Z.; Yi, Y.; Long, T.; Zhang, C.; Li, Y.; Pan, Y.; et al. Interplay of m(6) A and histone modifications contributes to temozolomide resistance in glioblastoma. Clin. Transl. Med. 2021, 11, e553. [Google Scholar] [CrossRef] [PubMed]
- Eich, M.L.; Athar, M.; Ferguson, J.E., 3rd; Varambally, S. EZH2-Targeted Therapies in Cancer: Hype or a Reality. Cancer Res. 2020, 80, 5449–5458. [Google Scholar] [CrossRef]
- Han Li, C.; Chen, Y. Targeting EZH2 for cancer therapy: Progress and perspective. Curr. Protein Pept. Sci. 2015, 16, 559–570. [Google Scholar] [CrossRef]
- Timp, W.; Feinberg, A.P. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nature Rev. Cancer 2013, 13, 497–510. [Google Scholar] [CrossRef] [Green Version]
- Cedar, H.; Bergman, Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nat. Rev. Genet. 2009, 10, 295–304. [Google Scholar] [CrossRef]
- Lee, J.; Son, M.J.; Woolard, K.; Donin, N.M.; Li, A.; Cheng, C.H.; Kotliarova, S.; Kotliarov, Y.; Walling, J.; Ahn, S.; et al. Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell 2008, 13, 69–80. [Google Scholar] [CrossRef]
- Lu, C.; Han, H.D.; Mangala, L.S.; Ali-Fehmi, R.; Newton, C.S.; Ozbun, L.; Armaiz-Pena, G.N.; Hu, W.; Stone, R.L.; Munkarah, A.; et al. Regulation of tumor angiogenesis by EZH2. Cancer Cell 2010, 18, 185–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezaei, S.; Hosseinpourfeizi, M.A.; Moaddab, Y.; Safaralizadeh, R. Contribution of DNA methylation and EZH2 in SRBC down-regulation in gastric cancer. Mol. Biol. Rep. 2020, 47, 5721–5727. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Teng, J.; Li, H.; Chen, F.; Zheng, J. The Emerging Roles of RASSF5 in Human Malignancy. Anti-Cancer Agents Med. Chem. 2018, 18, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Tiffen, J.; Gallagher, S.J.; Filipp, F.; Gunatilake, D.; Emran, A.A.; Cullinane, C.; Dutton-Register, K.; Aoude, L.; Hayward, N.; Chatterjee, A.; et al. EZH2 Cooperates with DNA Methylation to Downregulate Key Tumor Suppressors and IFN Gene Signatures in Melanoma. J. Investig. Dermatol. 2020, 140, 2442–2454.e2445. [Google Scholar] [CrossRef]
- Asano, T. Drug Resistance in Cancer Therapy and the Role of Epigenetics. J. Nippon. Med. Sch. Nippon. Ika Daigaku Zasshi 2020, 87, 244–251. [Google Scholar] [CrossRef]
- Batool, A.; Jin, C.; Liu, Y.X. Role of EZH2 in cell lineage determination and relative signaling pathways. Front. Biosci. (Landmark Ed.) 2019, 24, 947–960. [Google Scholar] [CrossRef]
- Lu, H.; Li, G.; Zhou, C.; Jin, W.; Qian, X.; Wang, Z.; Pan, H.; Jin, H.; Wang, X. Regulation and role of post-translational modifications of enhancer of zeste homologue 2 in cancer development. Am. J. Cancer Res. 2016, 6, 2737–2754. [Google Scholar]
- Ku, M.; Koche, R.P.; Rheinbay, E.; Mendenhall, E.M.; Endoh, M.; Mikkelsen, T.S.; Presser, A.; Nusbaum, C.; Xie, X.; Chi, A.S.; et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 2008, 4, e1000242. [Google Scholar] [CrossRef]
- Leeb, M.; Pasini, D.; Novatchkova, M.; Jaritz, M.; Helin, K.; Wutz, A. Polycomb complexes act redundantly to repress genomic repeats and genes. Genes Dev. 2010, 24, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Pan, X.; Zhang, W.; Guo, H.; Cheng, S.; He, Q.; Yang, B.; Ding, L. Epigenetic strategies synergize with PD-L1/PD-1 targeted cancer immunotherapies to enhance antitumor responses. Acta Pharm. Sin. B 2020, 10, 723–733. [Google Scholar] [CrossRef]
- Sun, S.; Yu, F.; Xu, D.; Zheng, H.; Li, M. EZH2, a prominent orchestrator of genetic and epigenetic regulation of solid tumor microenvironment and immunotherapy. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188700. [Google Scholar] [CrossRef] [PubMed]
- Sanches, J.G.P.; Song, B.; Zhang, Q.; Cui, X.; Yabasin, I.B.; Ntim, M.; Li, X.; He, J.; Zhang, Y.; Mao, J.; et al. The Role of KDM2B and EZH2 in Regulating the Stemness in Colorectal Cancer Through the PI3K/AKT Pathway. Front. Oncol. 2021, 11, 637298. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Kim, M.; Woo, D.H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y.T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef] [Green Version]
- Yan, K.S.; Lin, C.Y.; Liao, T.W.; Peng, C.M.; Lee, S.C.; Liu, Y.J.; Chan, W.P.; Chou, R.H. EZH2 in Cancer Progression and Potential Application in Cancer Therapy: A Friend or Foe? Int. J. Mol. Sci. 2017, 18, 1172. [Google Scholar] [CrossRef] [PubMed]
- Tumes, D.J.; Onodera, A.; Suzuki, A.; Shinoda, K.; Endo, Y.; Iwamura, C.; Hosokawa, H.; Koseki, H.; Tokoyoda, K.; Suzuki, Y.; et al. The polycomb protein Ezh2 regulates differentiation and plasticity of CD4(+) T helper type 1 and type 2 cells. Immunity 2013, 39, 819–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.; Wu, Y.; Guo, W.; Yu, F.; Kong, L.; Ren, Y.; Wang, Y.; Yao, X.; Jing, C.; Zhang, C.; et al. STAT3/HOTAIR Signaling Axis Regulates HNSCC Growth in an EZH2-dependent Manner. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 2665–2677. [Google Scholar] [CrossRef] [Green Version]
- Hao, A.; Wang, Y.; Stovall, D.B.; Wang, Y.; Sui, G. Emerging Roles of LncRNAs in the EZH2-regulated Oncogenic Network. Int. J. Biol. Sci. 2021, 17, 3268–3280. [Google Scholar] [CrossRef]
- Vu, L.D.; Gevaert, K.; De Smet, I. Protein Language: Post-Translational Modifications Talking to Each Other. Trends Plant Sci. 2018, 23, 1068–1080. [Google Scholar] [CrossRef]
- Nie, L.; Wei, Y.; Zhang, F.; Hsu, Y.H.; Chan, L.C.; Xia, W.; Ke, B.; Zhu, C.; Deng, R.; Tang, J.; et al. CDK2-mediated site-specific phosphorylation of EZH2 drives and maintains triple-negative breast cancer. Nat. Commun. 2019, 10, 5114. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Y.; Qiu, R.; Yang, Y.; Gao, T.; Zheng, Y.; Huang, W.; Gao, J.; Zhang, K.; Liu, R.; Wang, S.; et al. Regulation of EZH2 by SMYD2-Mediated Lysine Methylation Is Implicated in Tumorigenesis. Cell Rep. 2019, 29, 1482–1498.e1484. [Google Scholar] [CrossRef]
- Lo, P.W.; Shie, J.J.; Chen, C.H.; Wu, C.Y.; Hsu, T.L.; Wong, C.H. O-GlcNAcylation regulates the stability and enzymatic activity of the histone methyltransferase EZH2. Proc. Natl. Acad. Sci. USA 2018, 115, 7302–7307. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Liu, S.; Li, B.; Xie, Y.; Izban, M.G.; Ballard, B.R.; Sathyanarayana, S.A.; Adunyah, S.E.; Matusik, R.J.; Chen, Z. SKP2 loss destabilizes EZH2 by promoting TRAF6-mediated ubiquitination to suppress prostate cancer. Oncogene 2017, 36, 1364–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, Y.; Jiang, X.; Lee, S.T.; Karuturi, R.K.; Hooi, S.C.; Yu, Q. FOXQ1 regulates epithelial-mesenchymal transition in human cancers. Cancer Res. 2011, 71, 3076–3086. [Google Scholar] [CrossRef] [Green Version]
- Gan, L.; Xu, M.; Hua, R.; Tan, C.; Zhang, J.; Gong, Y.; Wu, Z.; Weng, W.; Sheng, W.; Guo, W. The polycomb group protein EZH2 induces epithelial-mesenchymal transition and pluripotent phenotype of gastric cancer cells by binding to PTEN promoter. J. Hematol. Oncol. 2018, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Chen, J.; Ma, M.; Cai, M.; Xu, F.; Wang, G.; Tao, K.; Shuai, X. Inhibiting enhancer of zeste homolog 2 promotes cellular senescence in gastric cancer cells SGC-7901 by activation of p21 and p16. DNA Cell Biol. 2014, 33, 337–344. [Google Scholar] [CrossRef]
- Ito, T.; Teo, Y.V.; Evans, S.A.; Neretti, N.; Sedivy, J.M. Regulation of Cellular Senescence by Polycomb Chromatin Modifiers through Distinct DNA Damage- and Histone Methylation-Dependent Pathways. Cell Rep. 2018, 22, 3480–3492. [Google Scholar] [CrossRef] [Green Version]
- Jie, B.; Weilong, C.; Ming, C.; Fei, X.; Xinghua, L.; Junhua, C.; Guobin, W.; Kaixiong, T.; Xiaoming, S. Enhancer of zeste homolog 2 depletion induces cellular senescence via histone demethylation along the INK4/ARF locus. Int. J. Biochem. Cell Biol. 2015, 65, 104–112. [Google Scholar] [CrossRef]
- Ling, Z.; You, Z.; Hu, L.; Zhang, L.; Wang, Y.; Zhang, M.; Zhang, G.; Chen, S.; Xu, B.; Chen, M. Effects of four single nucleotide polymorphisms of EZH2 on cancer risk: A systematic review and meta-analysis. OncoTargets Ther. 2018, 11, 851–865. [Google Scholar] [CrossRef]
- Breyer, J.P.; McReynolds, K.M.; Yaspan, B.L.; Bradley, K.M.; Dupont, W.D.; Smith, J.R. Genetic variants and prostate cancer risk: Candidate replication and exploration of viral restriction genes. Cancer Epidemiol. Biomark. Prev. 2009, 18, 2137–2144. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Du, W.D.; Wu, Q.; Liu, Y.; Chen, G.; Ruan, J.; Xu, S.; Yang, F.; Zhou, F.S.; Tang, X.F.; et al. EZH2 genetic variants affect risk of gastric cancer in the Chinese Han population. Mol. Carcinog. 2014, 53, 589–597. [Google Scholar] [CrossRef]
- Lee, S.W.; Park, D.Y.; Kim, M.Y.; Kang, C. Synergistic triad epistasis of epigenetic H3K27me modifier genes, EZH2, KDM6A, and KDM6B, in gastric cancer susceptibility. Gastric Cancer 2019, 22, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Lin, Y.; Wang, X.; Lan, F.; Yu, Y.; Huang, Q. Single Nucleotide Polymorphism of the Enhancer of Zeste Homolog 2 Gene rs2072408 is Associated with Lymph Node Metastasis and Depth of Primary Tumor Invasion in Gastric Cancer. Clin. Lab. 2016, 62, 2099–2105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hu, Z.; Horta, C.A.; Yang, J. Regulation of epithelial-mesenchymal transition by tumor microenvironmental signals and its implication in cancer therapeutics. Semin. Cancer Biol. 2022, 88, 46–66. [Google Scholar] [CrossRef]
- Marrelli, D.; Marano, L.; Ambrosio, M.R.; Carbone, L.; Spagnoli, L.; Petrioli, R.; Ongaro, A.; Piccioni, S.; Fusario, D.; Roviello, F. Immunohistochemical Markers of the Epithelial-to-Mesenchymal Transition (EMT) Are Related to Extensive Lymph Nodal Spread, Peritoneal Dissemination, and Poor Prognosis in the Microsatellite-Stable Diffuse Histotype of Gastric Cancer. Cancers 2022, 14, 6023. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Ochiai, A. Enhancer of zeste homolog 2 downregulates E-cadherin by mediating histone H3 methylation in gastric cancer cells. Cancer Sci. 2008, 99, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.W.; Sun, M.; Xia, R.; Zhang, E.B.; Liu, X.H.; Zhang, Z.H.; Xu, T.P.; De, W.; Liu, B.R.; Wang, Z.X. LincHOTAIR epigenetically silences miR34a by binding to PRC2 to promote the epithelial-to-mesenchymal transition in human gastric cancer. Cell Death Dis. 2015, 6, e1802. [Google Scholar] [CrossRef] [Green Version]
- He, Z.C.; Yang, F.; Guo, L.L.; Wei, Z.; Dong, X. LncRNA TP73-AS1 promotes the development of Epstein-Barr virus associated gastric cancer by recruiting PRC2 complex to regulate WIF1 methylation. Cell. Signal. 2021, 110094. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.J.; Ruan, H.J.; He, X.J.; Ma, Y.Y.; Jiang, X.T.; Xia, Y.J.; Ye, Z.Y.; Tao, H.Q. MicroRNA-101 is down-regulated in gastric cancer and involved in cell migration and invasion. Eur. J. Cancer 2010, 46, 2295–2303. [Google Scholar] [CrossRef]
- Tiwari, N.; Tiwari, V.K.; Waldmeier, L.; Balwierz, P.J.; Arnold, P.; Pachkov, M.; Meyer-Schaller, N.; Schübeler, D.; van Nimwegen, E.; Christofori, G. Sox4 is a master regulator of epithelial-mesenchymal transition by controlling Ezh2 expression and epigenetic reprogramming. Cancer Cell 2013, 23, 768–783. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wang, C.; Chen, Z.; Jin, Y.; Wang, Y.; Kolokythas, A.; Dai, Y.; Zhou, X. MicroRNA-138 suppresses epithelial-mesenchymal transition in squamous cell carcinoma cell lines. Biochem. J. 2011, 440, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Yu, F.; Zhang, L.; Zhou, X. EZH2, an on-off valve in signal network of tumor cells. Cell. Signal. 2016, 28, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Kudo-Saito, C.; Shirako, H.; Takeuchi, T.; Kawakami, Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell 2009, 15, 195–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashrafizadeh, M.; Zarrabi, A.; Hushmandi, K.; Kalantari, M.; Mohammadinejad, R.; Javaheri, T.; Sethi, G. Association of the Epithelial-Mesenchymal Transition (EMT) with Cisplatin Resistance. Int. J. Mol. Sci. 2020, 21, 4002. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tong, T. FOXA1 antagonizes EZH2-mediated CDKN2A repression in carcinogenesis. Biochem. Biophys. Res. Commun. 2014, 453, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Wang, Y.; Sreekumar, A.; Buckley, K.M.; Shi, H.; Jillella, A.; Ustun, C.; Rao, R.; Fernandez, P.; Chen, J.; et al. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood 2009, 114, 2733–2743. [Google Scholar] [CrossRef] [Green Version]
- Engeland, K. Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef]
- Lu, H.; Sun, J.; Wang, F.; Feng, L.; Ma, Y.; Shen, Q.; Jiang, Z.; Sun, X.; Wang, X.; Jin, H. Enhancer of zeste homolog 2 activates wnt signaling through downregulating CXXC finger protein 4. Cell Death Dis. 2013, 4, e776. [Google Scholar] [CrossRef] [Green Version]
- Fujii, S.; Ito, K.; Ito, Y.; Ochiai, A. Enhancer of zeste homologue 2 (EZH2) down-regulates RUNX3 by increasing histone H3 methylation. J. Biol. Chem. 2008, 283, 17324–17332. [Google Scholar] [CrossRef] [Green Version]
- O’Driscoll, M. INK4a/ARF-dependent senescence upon persistent replication stress. Cell Cycle 2013, 12, 1997–1998. [Google Scholar] [CrossRef] [Green Version]
- Hirosue, A.; Ishihara, K.; Tokunaga, K.; Watanabe, T.; Saitoh, N.; Nakamoto, M.; Chandra, T.; Narita, M.; Shinohara, M.; Nakao, M. Quantitative assessment of higher-order chromatin structure of the INK4/ARF locus in human senescent cells. Aging Cell 2012, 11, 553–556. [Google Scholar] [CrossRef]
- Qi, L.N.; Xiang, B.D.; Wu, F.X.; Ye, J.Z.; Zhong, J.H.; Wang, Y.Y.; Chen, Y.Y.; Chen, Z.S.; Ma, L.; Chen, J.; et al. Circulating Tumor Cells Undergoing EMT Provide a Metric for Diagnosis and Prognosis of Patients with Hepatocellular Carcinoma. Cancer Res. 2018, 78, 4731–4744. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Wang, L.; Zuo, Z.; Bei, Y.; Liu, K. The role of surgery and radiation in advanced gastric cancer: A population-based study of Surveillance, Epidemiology, and End Results database. PLoS ONE 2019, 14, e0213596. [Google Scholar] [CrossRef] [PubMed]
- Marin, J.J.; Al-Abdulla, R.; Lozano, E.; Briz, O.; Bujanda, L.; Banales, J.M.; Macias, R.I. Mechanisms of Resistance to Chemotherapy in Gastric Cancer. Anti-Cancer Agents Med. Chem. 2016, 16, 318–334. [Google Scholar] [CrossRef]
- Zhou, W.; Wang, J.; Man, W.Y.; Zhang, Q.W.; Xu, W.G. siRNA silencing EZH2 reverses cisplatin-resistance of human non-small cell lung and gastric cancer cells. Asian Pac. J. Cancer Prev. APJCP 2015, 16, 2425–2430. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Li, Z.; Liu, H.; Zhou, D.; Fu, A.; Zhang, E. MicroRNA-126 increases chemosensitivity in drug-resistant gastric cancer cells by targeting EZH2. Biochem. Biophys. Res. Commun. 2016, 479, 91–96. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Cai, Q.; Hu, L.; He, C.Y.; Li, J.F.; Quan, Z.W.; Liu, B.Y.; Li, C.; Zhu, Z.G. Long noncoding RNA UCA1 induced by SP1 promotes cell proliferation via recruiting EZH2 and activating AKT pathway in gastric cancer. Cell Death Dis. 2017, 8, e2839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Q.; Zhang, T.; Pan, J.; Li, C. LncRNA UCA1 promotes cisplatin resistance in gastric cancer via recruiting EZH2 and activating PI3K/AKT pathway. J. Cancer 2020, 11, 3882–3892. [Google Scholar] [CrossRef]
- Li, H.; Ma, X.; Yang, D.; Suo, Z.; Dai, R.; Liu, C. PCAT-1 contributes to cisplatin resistance in gastric cancer through epigenetically silencing PTEN via recruiting EZH2. J. Cell. Biochem. 2020, 121, 1353–1361. [Google Scholar] [CrossRef]
- Guo, Y.; Yue, P.; Wang, Y.; Chen, G.; Li, Y. PCAT-1 contributes to cisplatin resistance in gastric cancer through miR-128/ZEB1 axis. Biomed. Pharmacother. Biomed. Pharmacother. 2019, 118, 109255. [Google Scholar] [CrossRef]
- Lee, H.; Yoon, S.O.; Jeong, W.Y.; Kim, H.K.; Kim, A.; Kim, B.H. Immunohistochemical analysis of polycomb group protein expression in advanced gastric cancer. Hum. Pathol. 2012, 43, 1704–1710. [Google Scholar] [CrossRef]
- Pan, Y.M.; Wang, C.G.; Zhu, M.; Xing, R.; Cui, J.T.; Li, W.M.; Yu, D.D.; Wang, S.B.; Zhu, W.; Ye, Y.J.; et al. STAT3 signaling drives EZH2 transcriptional activation and mediates poor prognosis in gastric cancer. Mol. Cancer 2016, 15, 79. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Yang, T.F.; Liang, S.C.; Guo, J.X.; Wang, Q. Role of EZH2 protein expression in gastric carcinogenesis among Asians: A meta-analysis. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2014, 35, 6649–6656. [Google Scholar] [CrossRef] [PubMed]
- Chi, P.; Allis, C.D.; Wang, G.G. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 2010, 10, 457–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knutson, S.K.; Wigle, T.J.; Warholic, N.M.; Sneeringer, C.J.; Allain, C.J.; Klaus, C.R.; Sacks, J.D.; Raimondi, A.; Majer, C.R.; Song, J.; et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat. Chem. Biol. 2012, 8, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Hayden, A.; Johnson, P.W.; Packham, G.; Crabb, S.J. S-adenosylhomocysteine hydrolase inhibition by 3-deazaneplanocin A analogues induces anti-cancer effects in breast cancer cell lines and synergy with both histone deacetylase and HER2 inhibition. Breast Cancer Res. Treat. 2011, 127, 109–119. [Google Scholar] [CrossRef] [Green Version]
- Kemp, C.D.; Rao, M.; Xi, S.; Inchauste, S.; Mani, H.; Fetsch, P.; Filie, A.; Zhang, M.; Hong, J.A.; Walker, R.L.; et al. Polycomb repressor complex-2 is a novel target for mesothelioma therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Crea, F.; Hurt, E.M.; Mathews, L.A.; Cabarcas, S.M.; Sun, L.; Marquez, V.E.; Danesi, R.; Farrar, W.L. Pharmacologic disruption of Polycomb Repressive Complex 2 inhibits tumorigenicity and tumor progression in prostate cancer. Mol. Cancer 2011, 10, 40. [Google Scholar] [CrossRef] [Green Version]
- Sha, M.; Mao, G.; Wang, G.; Chen, Y.; Wu, X.; Wang, Z. DZNep inhibits the proliferation of colon cancer HCT116 cells by inducing senescence and apoptosis. Acta Pharm. Sin. B 2015, 5, 188–193. [Google Scholar] [CrossRef] [Green Version]
- Miranda, T.B.; Cortez, C.C.; Yoo, C.B.; Liang, G.; Abe, M.; Kelly, T.K.; Marquez, V.E.; Jones, P.A. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol. Cancer Ther. 2009, 8, 1579–1588. [Google Scholar] [CrossRef] [Green Version]
- Dockerill, M.; Gregson, C.; DH, O.D. Targeting PRC2 for the treatment of cancer: An updated patent review (2016–2020). Expert Opin. Ther. Pat. 2021, 31, 119–135. [Google Scholar] [CrossRef]
- Fioravanti, R.; Stazi, G.; Zwergel, C.; Valente, S.; Mai, A. Six Years (2012–2018) of Researches on Catalytic EZH2 Inhibitors: The Boom of the 2-Pyridone Compounds. Chem. Rec. 2018, 18, 1818–1832. [Google Scholar] [CrossRef] [PubMed]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Graves, A.P.; Della Pietra, A., 3rd; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef]
- Yap, T.A.; Winter, J.N.; Giulino-Roth, L.; Longley, J.; Lopez, J.; Michot, J.M.; Leonard, J.P.; Ribrag, V.; McCabe, M.T.; Creasy, C.L.; et al. Phase I Study of the Novel Enhancer of Zeste Homolog 2 (EZH2) Inhibitor GSK2816126 in Patients with Advanced Hematologic and Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 7331–7339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Wang, Z.; Zhou, J.; Huang, J.; Zhou, L.; Luo, J.; Wan, Y.Y.; Long, H.; Zhu, B. EZH2 Inhibitor GSK126 Suppresses Antitumor Immunity by Driving Production of Myeloid-Derived Suppressor Cells. Cancer Res. 2019, 79, 2009–2020. [Google Scholar] [CrossRef]
- Konze, K.D.; Ma, A.; Li, F.; Barsyte-Lovejoy, D.; Parton, T.; Macnevin, C.J.; Liu, F.; Gao, C.; Huang, X.P.; Kuznetsova, E.; et al. An orally bioavailable chemical probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chem. Biol. 2013, 8, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; On, D.M.; Ma, A.; Parton, T.; Konze, K.D.; Pattenden, S.G.; Allison, D.F.; Cai, L.; Rockowitz, S.; Liu, S.; et al. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood 2015, 125, 346–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knutson, S.K.; Warholic, N.M.; Wigle, T.J.; Klaus, C.R.; Allain, C.J.; Raimondi, A.; Porter Scott, M.; Chesworth, R.; Moyer, M.P.; Copeland, R.A.; et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl. Acad. Sci. USA 2013, 110, 7922–7927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izutsu, K.; Ando, K.; Nishikori, M.; Shibayama, H.; Teshima, T.; Kuroda, J.; Kato, K.; Imaizumi, Y.; Nosaka, K.; Sakai, R.; et al. Phase II study of tazemetostat for relapsed or refractory B-cell non-Hodgkin lymphoma with EZH2 mutation in Japan. Cancer Sci. 2021, 112, 3627–3635. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Soria, J.C.; Toulmonde, M.; Michot, J.M.; Lucchesi, C.; Varga, A.; Coindre, J.M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: A first-in-human, open-label, phase 1 study. Lancet Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef]
- Ribrag, V.; Soria, J.C.; Michot, J.M.; Schmitt, A.; Postel-Vinay, S.; Bijou, F.; Thomson, B.; Keilhack, H.; Blakemore, S.J.; Reyderman, L.; et al. Phase 1 Study of Tazemetostat (EPZ-6438), an Inhibitor of Enhancer of Zeste-Homolog 2 (EZH2): Preliminary Safety and Activity in Relapsed or Refractory Non Hodgkin Lymphoma (NHL) Patients. Blood 2015, 126, 3. [Google Scholar] [CrossRef]
- Hoy, S.M. Tazemetostat: First Approval. Drugs 2020, 80, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Tomassi, S.; Romanelli, A.; Zwergel, C.; Valente, S.; Mai, A. Polycomb Repressive Complex 2 Modulation through the Development of EZH2-EED Interaction Inhibitors and EED Binders. J. Med. Chem. 2021, 64, 11774–11797. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Zhao, K.; Gu, J.; Huang, Y.; Wang, Y.; Zhang, H.; Zhang, M.; Zhang, J.; Yu, Z.; Li, L.; et al. An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat. Chem. Biol. 2017, 13, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Sendzik, M.; Zhang, J.; Gao, Z.; Sun, Y.; Wang, L.; Gu, J.; Zhao, K.; Yu, Z.; Zhang, L.; et al. Discovery of the Clinical Candidate MAK683: An EED-Directed, Allosteric, and Selective PRC2 Inhibitor for the Treatment of Advanced Malignancies. J. Med. Chem. 2022, 65, 5317–5333. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, Y.; Gong, Y.; Zhang, T.; Huang, J.; Tan, Z.; Xue, L. Finding an easy way to harmonize: A review of advances in clinical research and combination strategies of EZH2 inhibitors. Clin. Epigenet. 2021, 13, 62. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Genes | Mechanism of Action of EZH2 | The Role of Genes | Reference |
|---|---|---|---|
| METTL3 | EZH2 overexpression leads to increases in H3K27me3, up-regulating the expression of METTL3 | Drug resistance | [25] |
| P16 | EZH2 overexpression leads to increases in H3K27me3, inhibiting the expression of P16 | Inhibition of tumor growth; Drive cellular differentiation | [26] |
| E-cadherin | EZH2 overexpression leads to increases in H3K27me3, inhibiting E-cadherin | Inhibition of tumor growth | [26] |
| HIF-1α | EZH2 stabilizes the expression of HIF-1α | Promotion of tumor growth and metabolism favoring glycolysis | [5] |
| INK4B-ARF-INK4A | EZH2 suppresses the expression of INK4B–ARF–INK4A | Induce cell cycle progression and inhibit cell senescence | [27] |
| Genes | Mechanism of Action of EZH2 | The Role of Genes | Reference |
|---|---|---|---|
| BMPR1B | EZH2 represses the expression of BMPR1B | Inhibition of growth of tumors | [30] |
| VASH1 | EZH2 represses VASH1 | Promotion of the angiogenesis of tumors | [31] |
| SRBC | EZH2 plays a substantial role in silencing SRBC | Inhibition of tumor growth; involved in tumor resistance against chemotherapeutic agents | [32] |
| RASSF5 | EZH2 inhibits RASSF5 | Suppression of cell growth | [33] |
| ITGB2 | EZH2 inhibits ITGB2 | Contribute to natural killer cell development and function | [34] |
| Genes | Mechanism of Action of EZH2 | The Role of Genes | Reference |
|---|---|---|---|
| E-cadherin | EZH2 causes the silencing of the E-cadherin gene | E-cadherin is involved in epithelial–mesenchymal transition, causing GC metastasis | [53] |
| PTEN | EZH2 downregulates PTEN expression | PTEN causes GC metastasis | [54] |
| P21 | p21 increases when EZH2 is knocked down | p21 inhibits proliferation and invasion of GC cells | [55] |
| P16 | p16 increases when EZH2 is knocked down | p16 promotes GC cellular senescence | [56] |
| INK4/ARF | EZH2 silencing results in the activation of INK4/ARF | INK4/ARF causes cell cycle arrest and induces senescence in GC cells | [57] |
| Drug | Role | Phase | Reference(s) |
|---|---|---|---|
| DZNep | SAH hydrolase inhibitor | pre-clinical | [94,99] |
| EPZ005687 | Inhibitor of EZH2 T641 and A677 mutants | pre-clinical | [101] |
| GSK126 (GSK2816126) | SAM-competitive inhibitors of EZH2 | Phase I | [102] |
| Tazemetostat (EPZ-6438, E7438) | SAM-competitive inhibitors of EZH2 | Phase I/II | [107] |
| UNC1999 | SAM-competitive inhibitors of EZH2 and EZH1 | pre-clinical | [105] |
| MAK683 (EED226) | Selective EED inhibitor | Phase I/II | [113] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, W.; Liu, N.; Song, X.; Chen, L.; Wang, M.; Xiao, G.; Li, T.; Wang, Z.; Zhang, Y. EZH2: An Accomplice of Gastric Cancer. Cancers 2023, 15, 425. https://doi.org/10.3390/cancers15020425
Yu W, Liu N, Song X, Chen L, Wang M, Xiao G, Li T, Wang Z, Zhang Y. EZH2: An Accomplice of Gastric Cancer. Cancers. 2023; 15(2):425. https://doi.org/10.3390/cancers15020425
Chicago/Turabian StyleYu, Wuhan, Ning Liu, Xiaogang Song, Lang Chen, Mancai Wang, Guohui Xiao, Tengfei Li, Zheyuan Wang, and Youcheng Zhang. 2023. "EZH2: An Accomplice of Gastric Cancer" Cancers 15, no. 2: 425. https://doi.org/10.3390/cancers15020425