
Synergistic Effects of Metformin and Trastuzumab on HER2 Positive Gastroesophageal Adenocarcinoma Cells In Vitro and In Vivo

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Line and Reagents
2.2. Droplet Digital PCR
2.3. Cell Proliferation Assay
2.4. Growth in Soft Agar
2.5. Western Blot Analysis
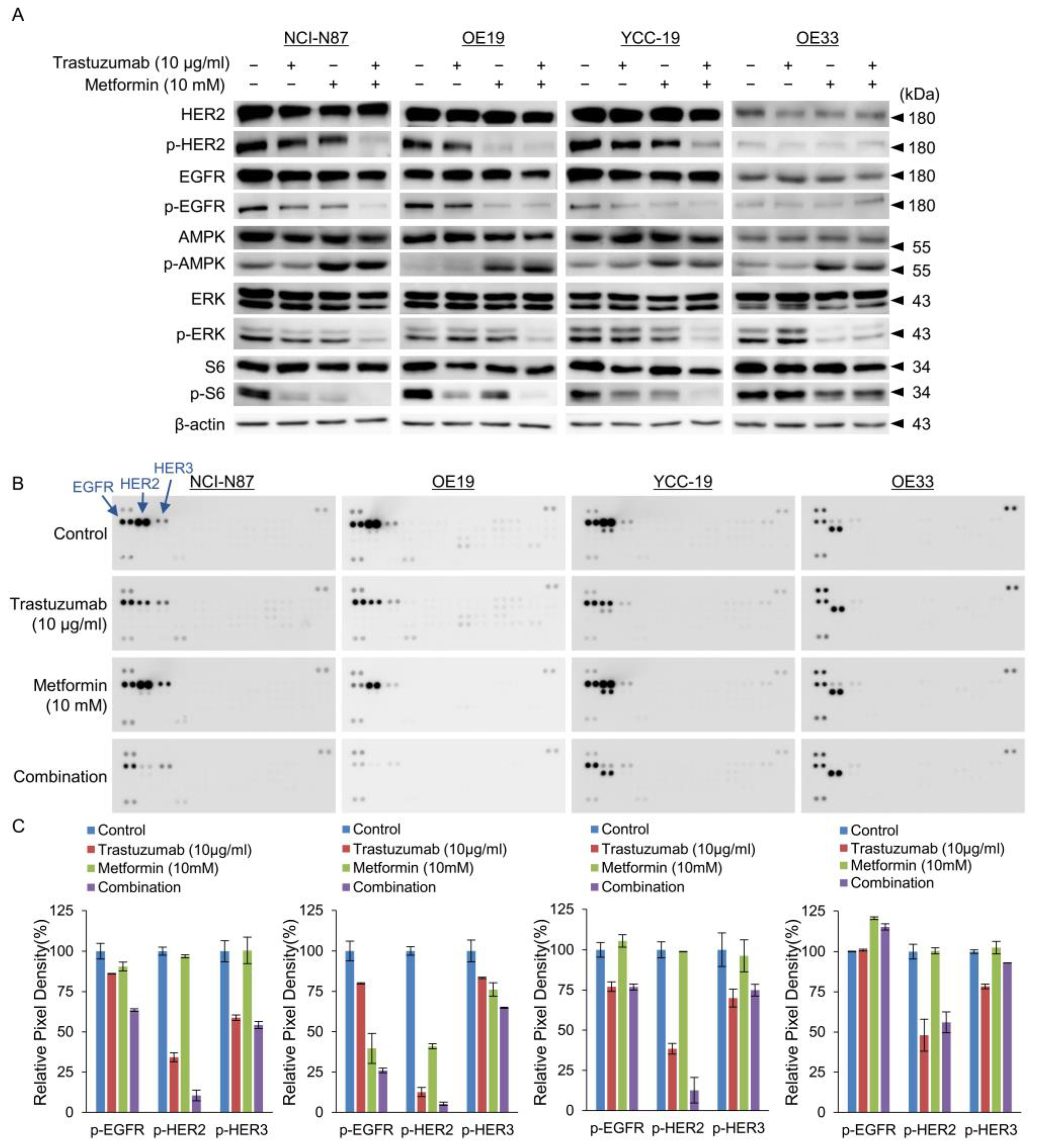
2.6. Proteome Profiling with Phospho-RTK Array
2.7. Xenograft Mouse Model
2.8. Statistical Analysis
3. Results
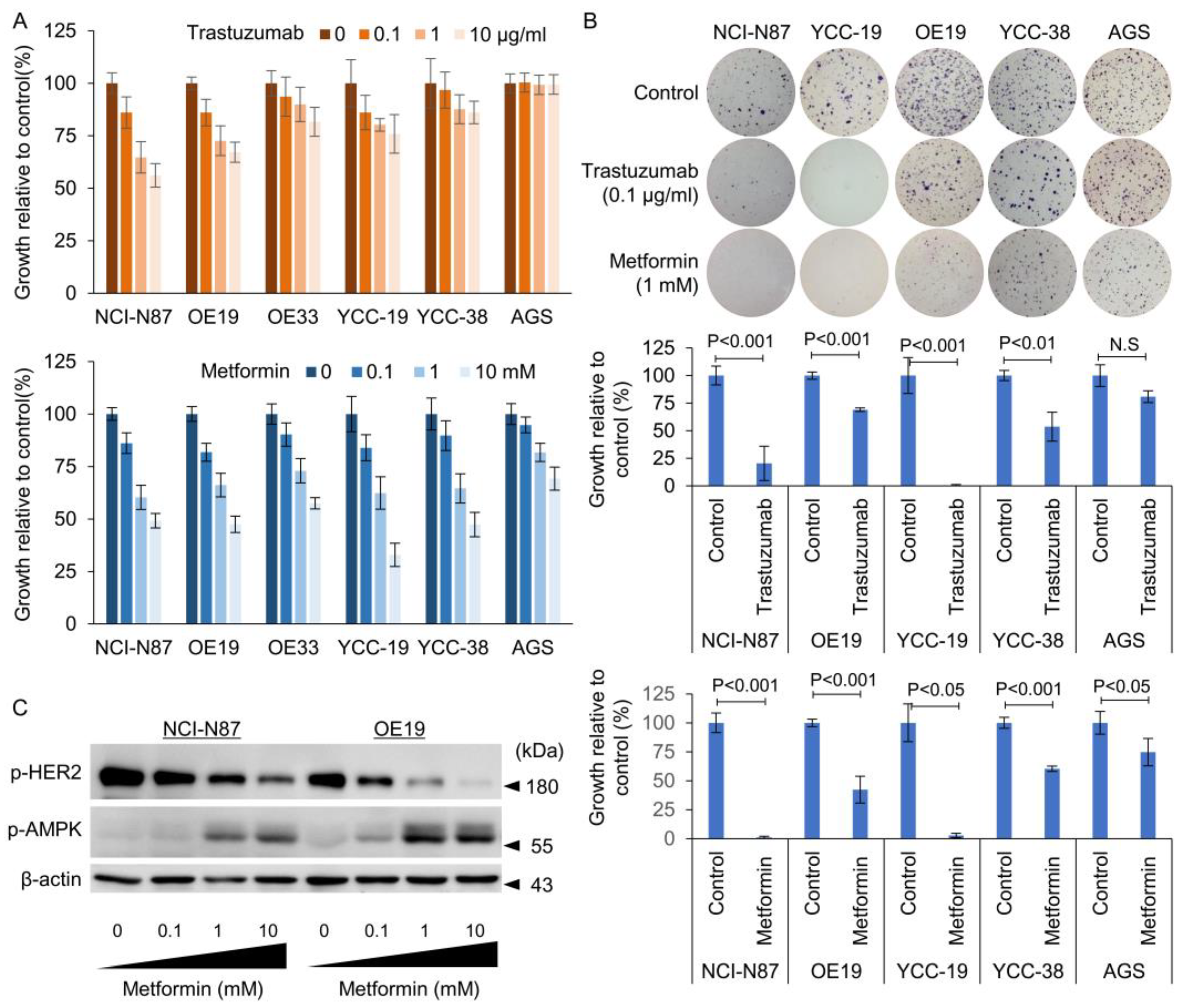
3.1. Identification of HER2 Positivity and Determination of Sensitivity to Tmab
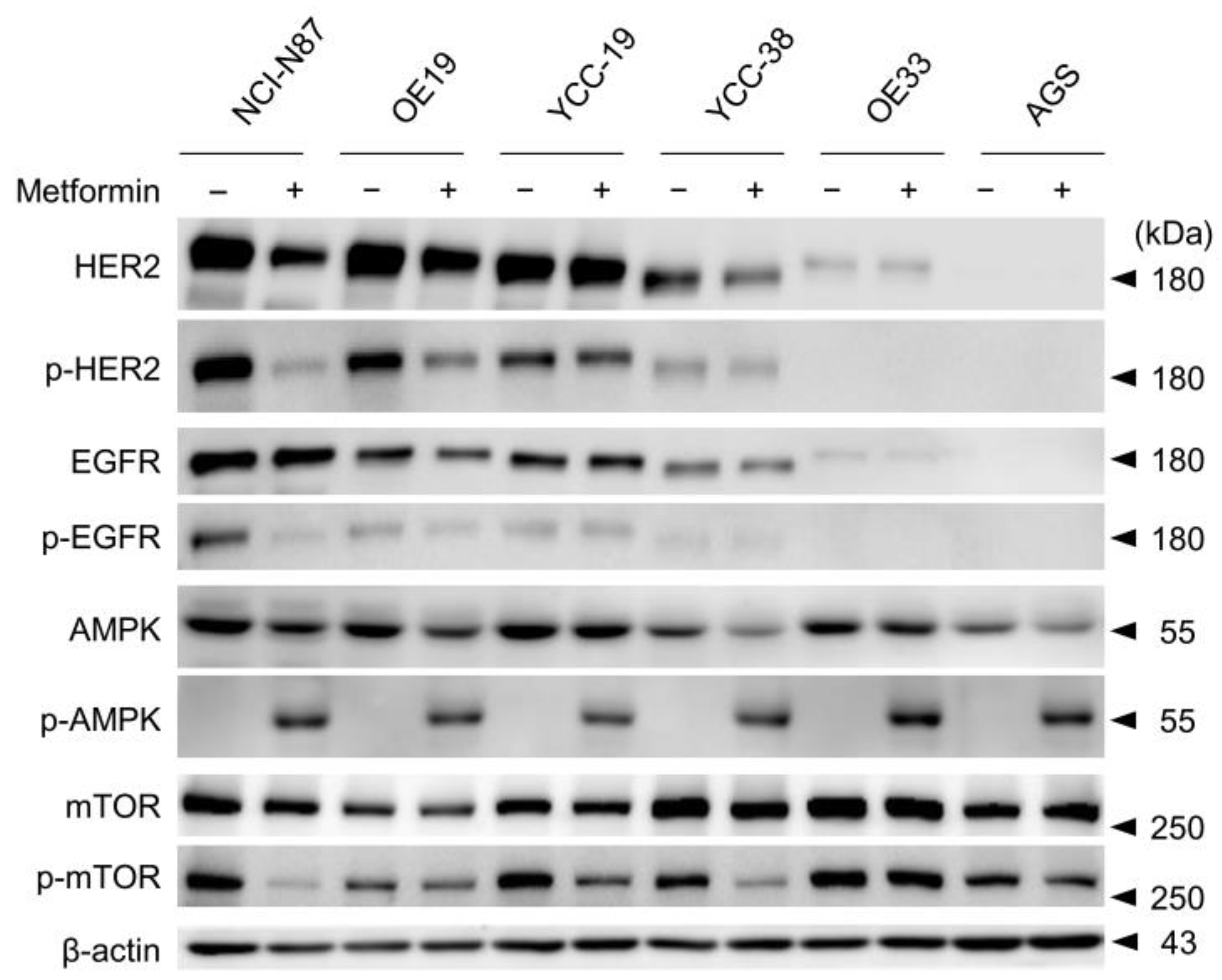
3.2. Metformin Induced Phosphorylation of AMP Kinase and Dephosphorylation of mTOR in Tmab Sensitive GC Cells
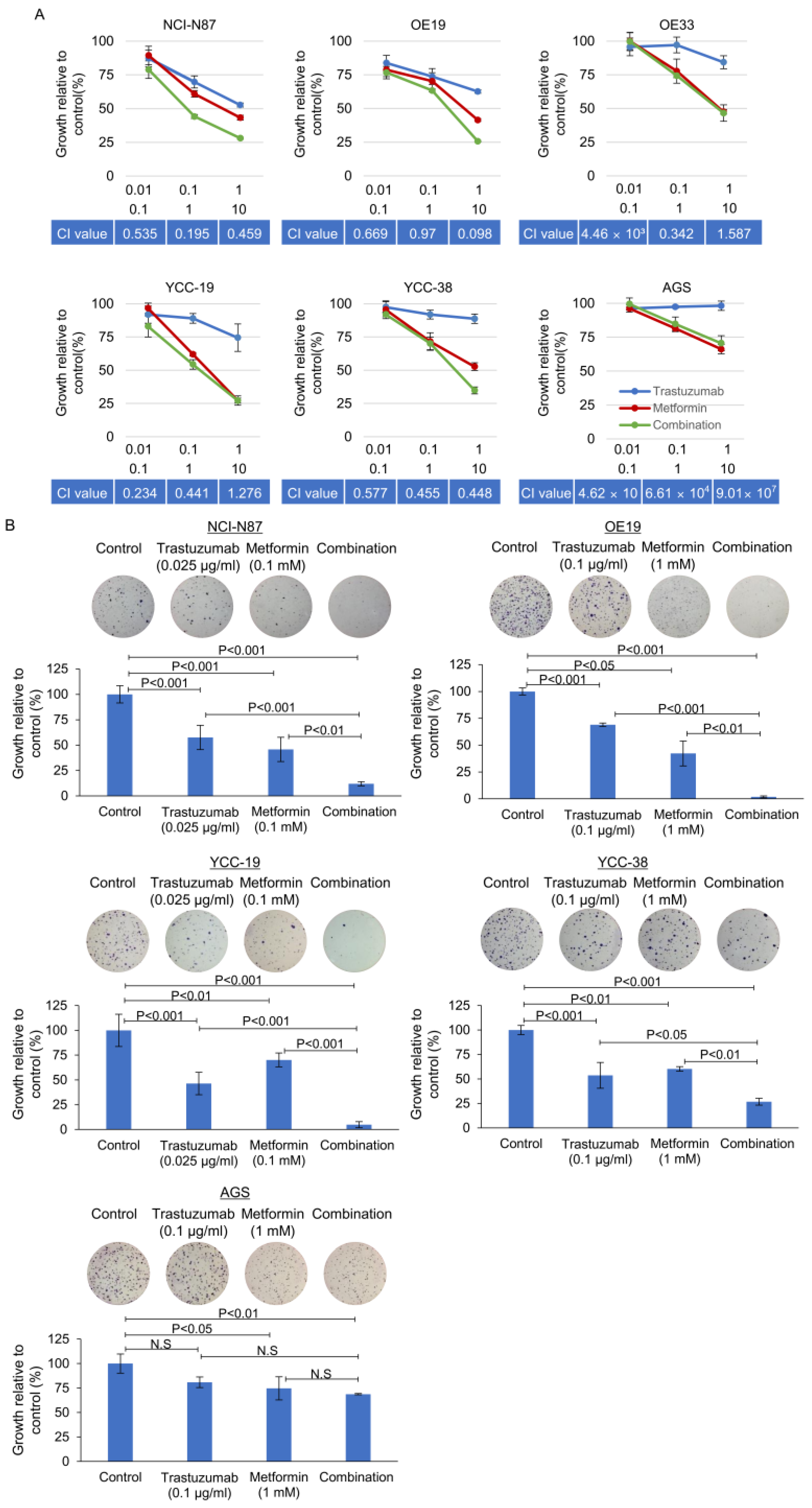
3.3. Effect of Metformin in Combination with Tmab on GC Proliferation and Anchorage Independent Colony Forming Ability
3.4. Tmab and Metformin Are Synergistic in Tmab-Sensitive HER2+ GC Cells
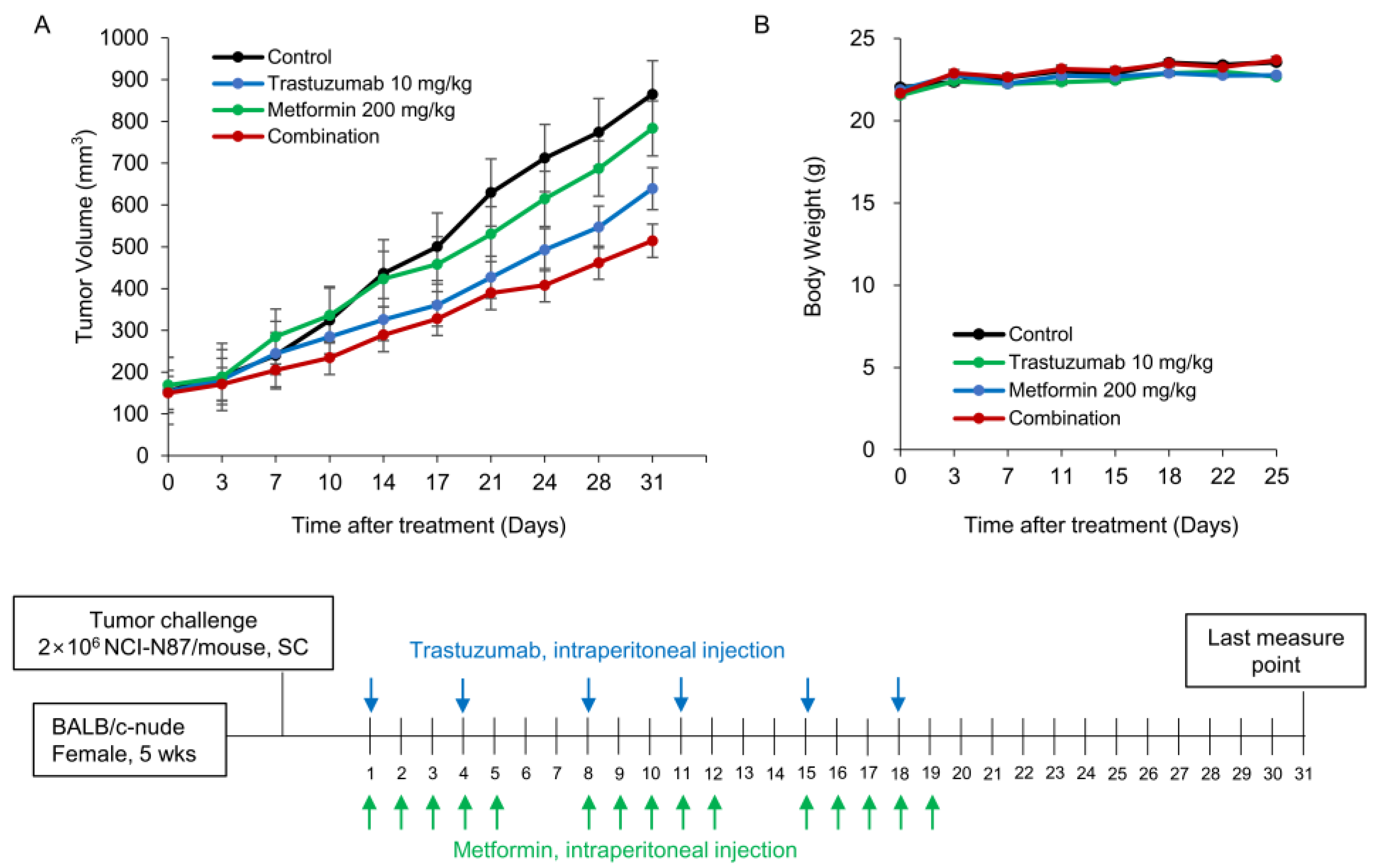
3.5. Combination of Tmab and Metformin Shows Superior Efficacy in Suppression of Tumor Growth In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bang, Y.-J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Xu, R.H.; Chung, H.C.; Sun, G.P.; Doi, T.; Xu, J.M.; Bang, Y.J. Lapatinib plus paclitaxel versus paclitaxel alone in the second-line treatment of HER2-amplified advanced gastric cancer in Asian populations: TyTAN—A randomized, phase III study. J. Clin. Oncol. 2014, 32, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Hecht, J.R.; Bang, Y.J.; Qin, S.K.; Chung, H.C.; Xu, J.M.; Park, J.O.; Shparyk, Y.; Hoff, P.M.; Sobrero, A.; Salman, P.; et al. Lapatinib in Combination With Capecitabine Plus Oxaliplatin in Human Epidermal Growth Factor Receptor 2-Positive Advanced or Metastatic Gastric, Esophageal, or Gastroesophageal Adenocarcinoma: TRIO-013/LOGiC—A Randomized Phase III Trial. J. Clin. Oncol. 2016, 34, 443–451. [Google Scholar] [CrossRef]
- Tabernero, J.; Hoff, P.M.; Shen, L.; Ohtsu, A.; A Shah, M.A.; Cheng, K.; Song, C.; Wu, H.; Eng-Wong, J.; Kim, K.; et al. Pertuzumab plus trastuzumab and chemotherapy for HER2-positive metastatic gastric or gastro-oesophageal junction cancer (JACOB): Final analysis of a double-blind, randomised, placebo-controlled phase 3 study. Lancet Oncol. 2018, 19, 1372–1384. [Google Scholar] [CrossRef] [PubMed]
- Thuss-Patience, P.C.; A Shah, M.A.; Ohtsu, A.; Van Cutsem, E.; A Ajani, J.A.; Castro, H.; Mansoor, W.; Chung, H.C.; Bodoky, G.; Shitara, K.; et al. Trastuzumab emtansine versus taxane use for previously treated HER2-positive locally advanced or metastatic gastric or gastro-oesophageal junction adenocarcinoma (GATSBY): An international randomised, open-label, adaptive, phase 2/3 study. Lancet Oncol. 2017, 18, 640–653. [Google Scholar] [CrossRef]
- Shitara, K.; Bang, Y.-J.; Iwasa, S.; Sugimoto, N.; Ryu, M.-H.; Sakai, D.; Chung, H.-C.; Kawakami, H.; Yabusaki, H.; Lee, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Gastric Cancer. N. Engl. J. Med. 2020, 382, 2419–2430. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef]
- Liu, W.; Chang, J.; Liu, M.; Yuan, J.; Zhang, J.; Qin, J.; etXia, X.; Walng, Y. Quantitative proteomics profiling reveals activation of mTOR pathway in trastuzumab resistance. Oncotarget 2017, 8, 45793–45806. [Google Scholar] [CrossRef]
- Wang, K.; Yuen, S.T.; Xu, J.; Lee, S.P.; Yan, H.H.; Shi, S.T.; Siu, H.C.; Deng, S.; Chu, K.M.; Law, S.; et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet. 2014, 46, 573–582. [Google Scholar] [CrossRef]
- Ohtsu, A.; Ajani, J.A.; Bai, Y.X.; Bang, Y.J.; Chung, H.C.; Pan, H.M.; Sahmoud, T.; Shen, L.; Yeh, K.-H.; Chin, K.; et al. Everolimus for previously treated advanced gastric cancer: Results of the randomized, double-blind, phase III GRANITE-1 study. J. Clin. Oncol. 2013, 31, 3935–3943. [Google Scholar] [CrossRef]
- Kourelis, T.V.; Siegel, R.D. Metformin and cancer: New applications for an old drug. Med. Oncol. 2012, 29, 1314–1327. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Gong, J.; Iwama, H.; Kitanaka, A.; Tani, J.; Miyoshi, H.; Nomura, K.; Mimura, S.; Kobayashi, M.; Aritomo, Y.; et al. The antidiabetic drug metformin inhibits gastric cancer cell proliferation in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Gong, H.; Wang, Y.; Guo, S.; Liu, K. AMPK/mTOR-mediated inhibition of survivin partly contributes to metformin-induced apoptosis in human gastric cancer cell. Cancer Biol. Ther. 2015, 16, 77–87. [Google Scholar] [CrossRef]
- Shackelford, D.B.; Abt, E.; Gerken, L.; Vasquez, D.S.; Seki, A.; Leblanc, M.; Wei, L.; Fishbein, M.C.; Czernin, J.; Mischel, P.S.; et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell. 2013, 23, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Gutkind, J.S.; Molinolo, A.A.; Wu, X.; Wang, Z.; Nachmanson, D.; Harismendy, O.; Alexandrov, L.B.; Wuertz, B.R.; Ondrey, F.G.; Laronde, D.M.; et al. Inhibition of mTOR signaling and clinical activity of metformin in oral premalignant lesions. JCI Insight. 2021, 6, e147096. [Google Scholar] [CrossRef]
- Chung, W.S.; Le, P.H.; Kuo, C.J.; Chen, T.H.; Kuo, C.F.; Chiou, M.J.; Chou, W.-C.; Yeh, T.-S.; Hsu, J.-T. Impact of Metformin Use on Survival in Patients with Gastric Cancer and Diabetes Mellitus Following Gastrectomy. Cancers 2020, 12, 2013. [Google Scholar] [CrossRef]
- Zheng, J.; Santoni, G.; Xie, S.H.; Lagergren, J. Improved prognosis in gastric adenocarcinoma among metformin users in a population-based study. Br. J. Cancer. 2021, 125, 277–283. [Google Scholar] [CrossRef]
- Kim, H.J.; Kwon, H.; Lee, J.W.; Kim, H.J.; Lee, S.B.; Park, H.S.; Sohn, G.; Lee, Y.; Koh, B.S.; Yu, J.H.; et al. Metformin increases survival in hormone receptor-positive, HER2-positive breast cancer patients with diabetes. Breast Cancer Res. 2015, 17, 64. [Google Scholar] [CrossRef]
- Liu, B.; Fan, Z.; Edgerton, S.M.; Yang, X.; Lind, S.E.; Thor, A.D. Potent anti-proliferative effects of metformin on trastuzumab-resistant breast cancer cells via inhibition of erbB2/IGF-1 receptor interactions. Cell Cycle 2011, 10, 2959–2966. [Google Scholar] [CrossRef]
- Cufi, S.; Corominas-Faja, B.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Dorca, J.; Bosch-Barrera, J.; Menendez, J.A. Metformin-induced preferential killing of breast cancer initiating CD44+CD24-/low cells is sufficient to overcome primary resistance to trastuzumab in HER2+ human breast cancer xenografts. Oncotarget 2012, 3, 395–398. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; Menendez, J.A. The antidiabetic drug metformin suppresses HER2 (erbB-2) oncoprotein overexpression via inhibition of the mTOR effector p70S6K1 in human breast carcinoma cells. Cell Cycle 2009, 8, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Yam, C.; Esteva, F.J.; Patel, M.M.; Raghavendra, A.S.; Ueno, N.T.; Moulder, S.L.; Hess, K.R.; Shroff, G.S.; Hodge, S.; Koenig, K.H.; et al. Efficacy and safety of the combination of metformin, everolimus and exemestane in overweight and obese postmenopausal patients with metastatic, hormone receptor-positive, HER2-negative breast cancer: A phase II study. Invest. New Drugs 2019, 37, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Nanni, O.; Amadori, D.; De Censi, A.; Rocca, A.; Freschi, A.; Bologna, A.; Gianni, L.; Rosetti, F.; Amaducci, L.; Cavanna, L.; et al. Metformin plus chemotherapy versus chemotherapy alone in the first-line treatment of HER2-negative metastatic breast cancer. The MYME randomized, phase 2 clinical trial. Breast Cancer Res. Treat. 2019, 174, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Fujihara, S.; Kato, K.; Morishita, A.; Iwama, H.; Nishioka, T.; Chiyo, T.; Nishiyama, N.; Miyoshi, H.; Kobayashi, M.; Kobara, H.; et al. Antidiabetic drug metformin inhibits esophageal adenocarcinoma cell proliferation in vitro and in vivo. Int. J. Oncol. 2015, 46, 2172–2180. [Google Scholar] [CrossRef]
- Martin-Castillo, B.; Pernas, S.; Dorca, J.; AÁlvarez, I.; Martiínez, S.; Peérez-Garcia, J.M.; Batista-López, N.; Rodríguez-Sánchez, C.A.; Amillano, K.; Domínguez, S.; et al. A phase 2 trial of neoadjuvant metformin in combination with trastuzumab and chemotherapy in women with early HER2-positive breast cancer: The METTEN study. Oncotarget 2018, 9, 35687–35704. [Google Scholar] [CrossRef]
- Kim, J.S.; Lee, S.C.; Min, H.Y.; Park, K.H.; Hyun, S.Y.; Kwon, S.J.; Choi, S.P.; Kim, W.Y.; Lee, H.J.; Lee, H.Y. Activation of insulin-like growth factor receptor signaling mediates resistance to histone deacetylase inhibitors. Cancer Lett. 2015, 361, 197–206. [Google Scholar] [CrossRef]
- Wang, C.; Wang, L.; Yu, X.; Zhang, Y.; Meng, Y.; Wang, H.; Yang, Y.; Gao, J.; Wei, H.; Zhao, J.; et al. Combating acquired resistance to trastuzumab by an anti-ErbB2 fully human antibody. Oncotarget 2017, 8, 42742–42751. [Google Scholar] [CrossRef]
- Howell, J.J.; Hellberg, K.; Turner, M.; Talbott, G.; Kolar, M.J.; Ross, D.S.; Hoxhaj, G.; Saghatetlian, A.L.; Shaw, R.J.; Manning, B.D. Metformin Inhibits Hepatic mTORC1 Signaling via Dose-Dependent Mechanisms Involving AMPK and the TSC Complex. Cell Metab. 2017, 25, 463–471. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Hassan, M.S.; Williams, F.; Awasthi, N.; Schwarz, M.A.; Schwarz, R.E.; Li, J.; von Holzen, U. Combination effect of lapatinib with foretinib in HER2 and MET co-activated experimental esophageal adenocarcinoma. Sci. Rep. 2019, 9, 17608. [Google Scholar] [CrossRef]
- Park, K.-S.; Hong, Y.S.; Choi, J.; Yoon, S.; Kang, J.; Kim, D.; Lee, K.-P.; Im, H.-S.; Lee, C.H.; Seo, S.; et al. HSP90 inhibitor, AUY922, debilitates intrinsic and acquired lapatinib-resistant HER2-positive gastric cancer cells. BMB Rep. 2018, 51, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Skuli, S.J.; Alomari, S.; Gaitsch, H.; Bakayoko, A.; Skuli, N.; Tyler, B.M. Metformin and Cancer, an Ambiguanidous Relationship. Pharmaceuticals 2022, 15, 626. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Klempner, S.J.; Chao, J. Progress and challenges in HER2-positive gastroesophageal adenocarcinoma. J. Hematol. Oncol. 2019, 12, 50. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Wang, X.; Guo, J.; Yang, B.; Li, Y. Challenges and future of HER2-positive gastric cancer therapy. Front. Oncol. 2023, 13, 1080990. [Google Scholar] [CrossRef] [PubMed]
- Cunha Junior, A.D.; Bragagnoli, A.C.; Costa, F.O.; Carvalheira, J.B.C. Repurposing metformin for the treatment of gastrointestinal cancer. World J. Gastroenterol. 2021, 27, 1883–1904. [Google Scholar] [CrossRef]
- Wang, J.; Li, G.; Wang, Y.; Tang, S.; Sun, X.; Feng, X.; Li, Y.; Bao, G.; Li, P.; Mao, X.; et al. Suppression of tumor angiogenesis by metformin treatment via a mechanism linked to targeting of HER2/HIF-1alpha/VEGF secretion axis. Oncotarget 2015, 6, 44579–44592. [Google Scholar] [CrossRef]
- Kim, J.; Lee, J.; Kim, C.; Choi, J.; Kim, A. Anti-cancer effect of metformin by suppressing signaling pathway of HER2 and HER3 in tamoxifen-resistant breast cancer cells. Tumour Biol. 2016, 37, 5811–5819. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; del Barco, S.; Martin-Castillo, B.; Menendez, J.A. The antidiabetic drug metformin: A pharmaceutical AMPK activator to overcome breast cancer resistance to HER2 inhibitors while decreasing risk of cardiomyopathy. Ann. Oncol. 2009, 20, 592–595. [Google Scholar] [CrossRef]
- Jhaveri, T.Z.; Woo, J.; Shang, X.; Park, B.H.; Gabrielson, E. AMP-activated kinase (AMPK) regulates activity of HER2 and EGFR in breast cancer. Oncotarget 2015, 6, 14754–14765. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Viollet, B. Metformin: Update on mechanisms of action and repurposing potential. Nat. Rev. Endocrinol. 2023, 19, 460–476. [Google Scholar] [CrossRef]
- Algire, C.; Amrein, L.; Bazile, M.; David, S.; Zakikhani, M.; Pollak, M. Diet and tumor LKB1 expression interact to determine sensitivity to anti-neoplastic effects of metformin in vivo. Oncogene 2011, 30, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Zhao, T.; Liu, J.; Zou, Z.; Xu, Q.; Gong, P.; Guo, H. Decreased expression of LKB1 is associated with epithelial-mesenchymal transition and led to an unfavorable prognosis in gastric cancer. Hum. Pathol. 2019, 83, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Morgillo, F.; Sasso, F.C.; Della Corte, C.M.; Vitagliano, D.; D'Aiuto, E.; Troiani, T.; Martinelli, E.; De Vita, F.; Orditura, M.; De Palma, R.; et al. Synergistic effects of metformin treatment in combination with gefitinib, a selective EGFR tyrosine kinase inhibitor, in LKB1 wild-type NSCLC cell lines. Clin. Cancer Res. 2013, 19, 3508–3519. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | Trastuzumab | Metformin | Combination | Index § | ||||
|---|---|---|---|---|---|---|---|---|
| MGI * | p † | MGI | p † | Expected | Observed | p † | ||
| N87 | 0.575 | <0.001 | 0.458 | <0.001 | 0.264 | 0.120 | <0.001 | 2.189 |
| OE19 | 0.690 | <0.001 | 0.423 | <0.001 | 0.292 | 0.019 | <0.001 | 15.621 |
| YCC-19 | 0.465 | <0.001 | 0.702 | 0.004 | 0.326 | 0.049 | <0.001 | 6.666 |
| YCC-38 | 0.536 | <0.001 | 0.603 | <0.001 | 0.324 | 0.267 | <0.001 | 1.211 |
| AGS | 0.808 | 0.106 | 0.747 | 0.017 | 0.604 | 0.687 | 0.003 | 0.879 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-S.; Kim, M.Y.; Hong, S. Synergistic Effects of Metformin and Trastuzumab on HER2 Positive Gastroesophageal Adenocarcinoma Cells In Vitro and In Vivo. Cancers 2023, 15, 4768. https://doi.org/10.3390/cancers15194768
Kim J-S, Kim MY, Hong S. Synergistic Effects of Metformin and Trastuzumab on HER2 Positive Gastroesophageal Adenocarcinoma Cells In Vitro and In Vivo. Cancers. 2023; 15(19):4768. https://doi.org/10.3390/cancers15194768
Chicago/Turabian StyleKim, Jin-Soo, Mi Young Kim, and Sungyoul Hong. 2023. "Synergistic Effects of Metformin and Trastuzumab on HER2 Positive Gastroesophageal Adenocarcinoma Cells In Vitro and In Vivo" Cancers 15, no. 19: 4768. https://doi.org/10.3390/cancers15194768