

Inhibition of Anaplastic Lymphoma Kinase (Alk) as Therapeutic Target to Improve Brain Function in Neurofibromatosis Type 1 (Nf1)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
Neurofibromin Function in Neurofibromatosis and Its Link to Anaplastic Lymphoma Kinase
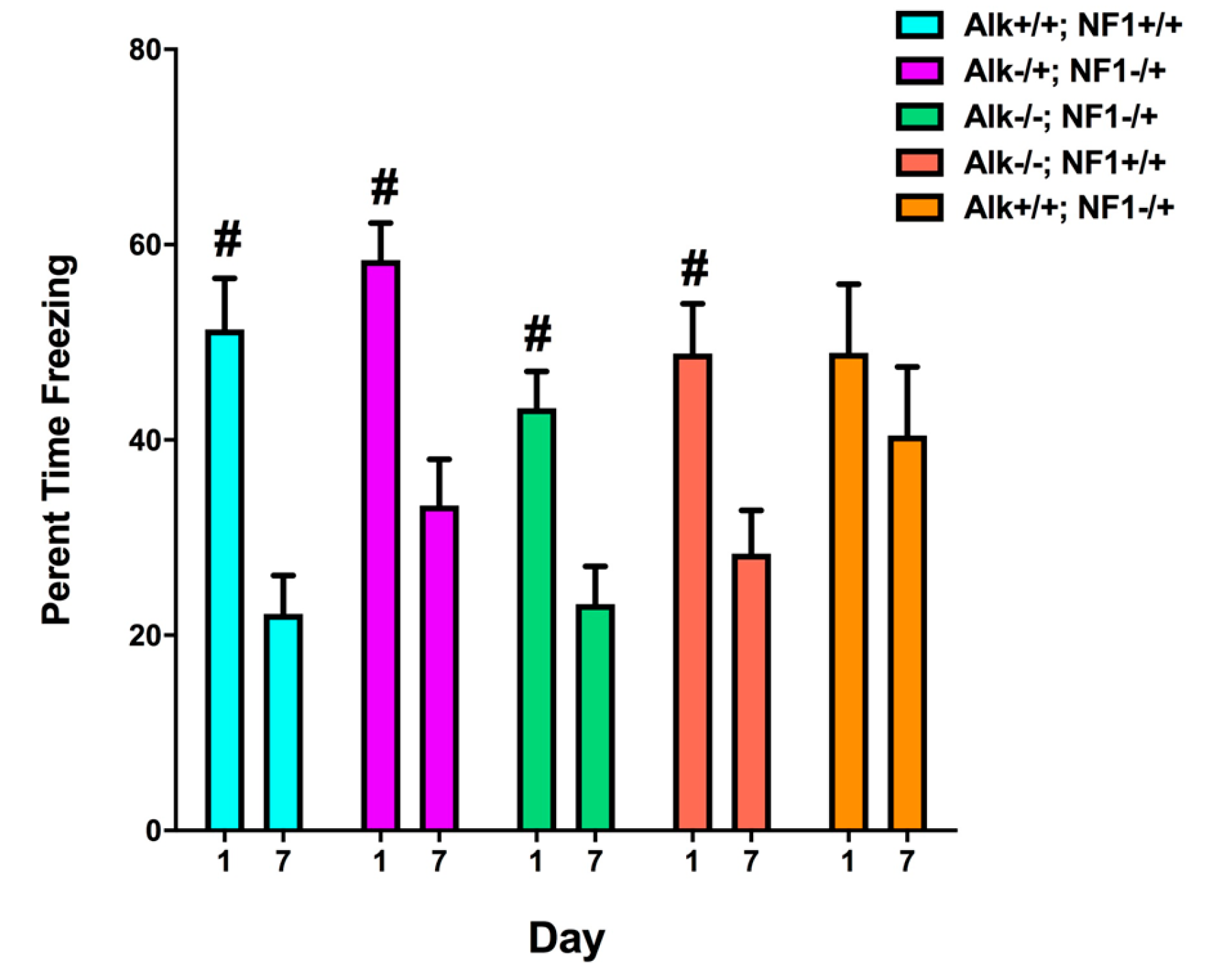
2. Genetic Inhibition of Alk Rescues Learning and Sleep Behaviors in Nf1 Heterozygous Mutant Mice
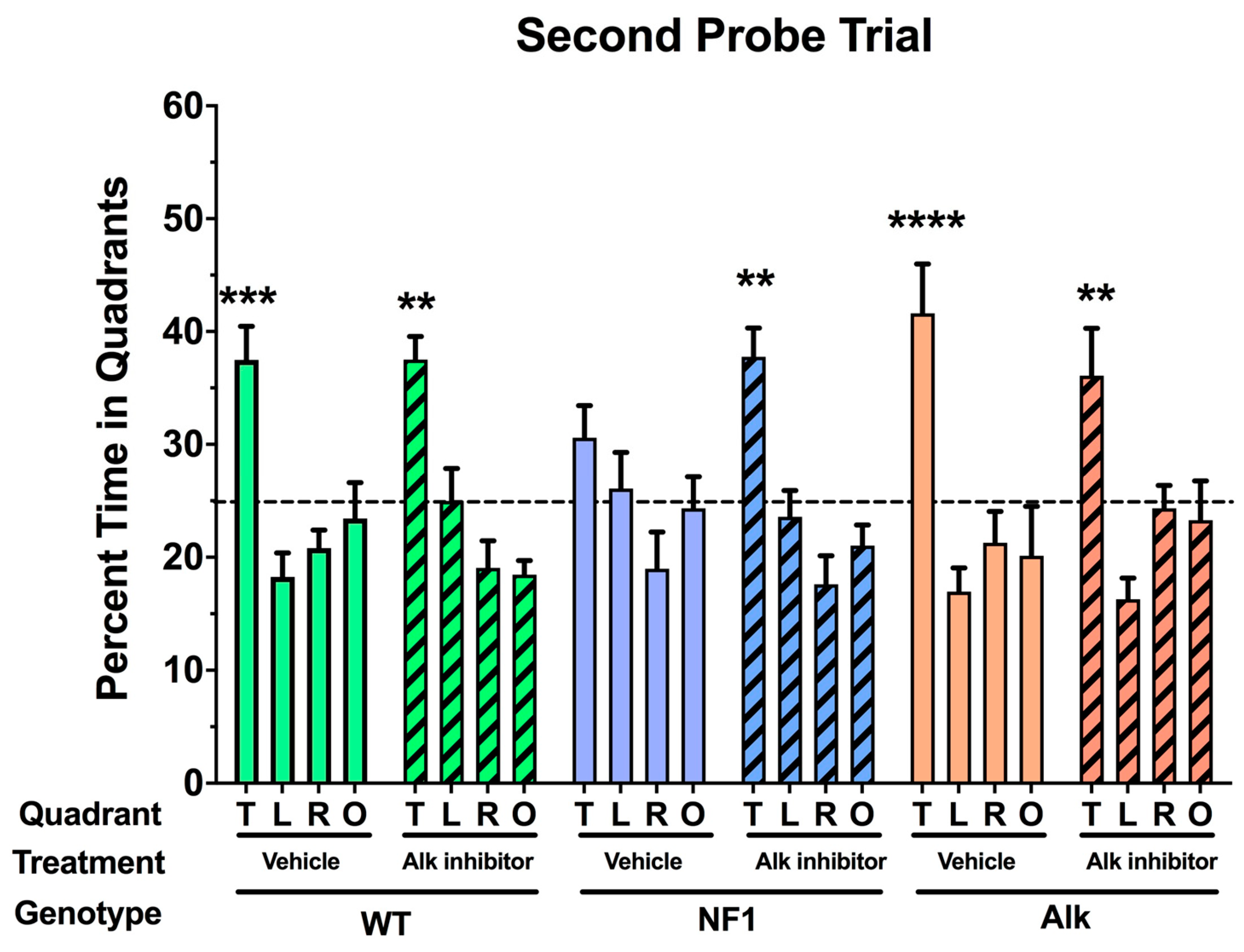
3. Pharmacologic Inhibition of Alk Rescues a Learning Defect in Nf1 Heterozygous Mice
4. Development of Alternative Therapeutic Strategies to Inhibit Alk
5. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Page, M.; McKenzie, J.; Bossuyt, P.; Hoffmann, T.; Mulrow, C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Wang, W.; Wei, C.J.; Cui, X.W.; Li, Y.H.; Gu, Y.H.; Gu, B.; Li, Q.F.; Wang, Z.C. Impacts of NF1 Gene Mutations and Genetic Modifiers in Neurofibromatosis Type 1. Front. Neurol. 2021, 12, 704639. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.-Y.; Moye, S.; McKaye, R.; Le, L. Neurofibromin and suppression of tumorigenesis: Beyond the GAP. Oncogene 2022, 41, 1235–1251. [Google Scholar] [CrossRef] [PubMed]
- Agaimi, A.; Semrau, S.; Heinz, M. Intra-abdominal ALK-positive anaplastic large cell lymphoma in a patient with neurofibromatosis type 1. Histopathology 2016, 68, 752–754. [Google Scholar] [CrossRef]
- Tao, J.; Sun, D.; Dong, L.-M.; Zhu, H.; Hou, H.H. Advancement in research and therapy of NF1 mutant malignant tumors. Cancer Cell Intern. 2020, 20, 492. [Google Scholar] [CrossRef]
- Tian, H.X.; Chen, Z.H.; Jie, G.L.; Wang, Z.; Yan, H.H.; Wu, S.P.; Zhang, S.L.; Lu, D.X.; Zhang, X.C.; Wu, Y.L. Prognostic features and comprehensive genomic analysis of NF1 mutations in EGFR mutant lung cancer patients. Cancer Med. 2023, 12, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Ratner, N.; Miller, S. A RASopathy gene commonly mutated in cancer: The neurofibromatosis type 1 tumour suppressor. Nat. Rev. Cancer 2016, 15, 290–301. [Google Scholar] [CrossRef]
- AlAnsari, G.; Bukhari, N.; Abdulkader, M.; Alotain, I.; Taha, M. Malignant anaplastic meningioma in neurofibromatosis type 1 patient: A rare case report. AME Case Rep. 2022, 6, 36. [Google Scholar] [CrossRef]
- Scheer, M.; Leisz, S.; Sorge, E.; Storozhuk, O.; Prell, J.; Ho, I.; Harder, A. Neurofibromatosis Type 1 Gene Alterations Define Specific Features of a Subset of Glioblastomas. Int. J. Mol. Sci. 2022, 23, 352. [Google Scholar] [CrossRef]
- Chuang, X.; Ch’an, D.; Oon, M.; Wang, S.; Chia, C. Inflammatory myofibroblastic tumour causing intestinal obstruction in a patient with neurofibromatosis type 1. Ann. R. Coll. Surg. 2021, 103, e53–e55. [Google Scholar] [CrossRef]
- North, K.; Joy, P.; Yuille, D.; Cocks, N.; Hutchins, P. Cognitive function and academic performance in children with neurofibromatosis type 1. Dev. Med. Child Neurol. 1995, 37, 427–436. [Google Scholar] [CrossRef]
- Acosta, M.T.; Gioia, G.A.; Silva, A.J. Neurofibromatosis type 1: New insights into neurocognitive issues. Curr. Neurol. Neurosci. Rep. 2006, 6, 136–143. [Google Scholar] [CrossRef]
- Rosser, T.L.; Packer, R.J. Neurocognitive dysfunction in children with neurofibromatosis type 1. Curr. Neurol. Neurosci. Rep. 2003, 3, 129–136. [Google Scholar] [CrossRef]
- Hofman, K.J.; Harris, E.L.; Bryan, R.N.; Denckla, M.B. Neurofibromatosis type 1: The cognitive phenotype. J. Pediatr. 1994, 124, S1–S8. [Google Scholar] [CrossRef]
- Hyman, S.L.; Shores, A.; North, K.N. The nature and frequency of cognitive deficits in children with neurofibromatosis type 1. Neurology 2005, 65, 1037–1044. [Google Scholar] [CrossRef]
- Zoller, M.E.; Rembeck, B.; Backman, L. Neuropsychological deficits in adults with neurofibromatosis type 1. Acta Neurol. Scand. 1997, 95, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Cawthon, R.M.; Weiss, R.; Xu, G.F.; Viskochil, D.; Culver, M.; Stevens, J.; Robertson, M.; Dunn, D.; Gesteland, R.; O’Connell, P.; et al. A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell 1990, 62, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.F.; O’Connell, P.; Viskochil, D.; Cawthon, R.; Robertson, M.; Culver, M.; Dunn, D.; Stevens, J.; Gesteland, R.; White, R.; et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell 1990, 62, 599–608. [Google Scholar] [CrossRef]
- Ballester, R.; Marchuk, D.; Boguski, M.; Saulino, A.; Letcher, R.; Wigler, M.; Collins, F. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell 1990, 63, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.A.; Viskochil, D.; Bollag, G.; McCabe, P.C.; Crosier, W.J.; Haubruck, H.; Conroy, L.; Clark, R.; O’Connell, P.; Cawthon, R.M.; et al. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell 1990, 63, 843–849. [Google Scholar] [CrossRef]
- Xu, G.F.; Lin, B.; Tanaka, K.; Dunn, D.; Wood, D.; Gesteland, R.; White, R.; Weiss, R.; Tamanoi, F. The catalytic domain of the neurofibromatosis type 1 gene product stimulates ras GTPase and complements ira mutants of S. cerevisiae. Cell 1990, 63, 835–841. [Google Scholar] [CrossRef]
- Parada, L.F. Neurofibromatosis type 1. Biochim. Biophys. Acta 2000, 1471, M13–M19. [Google Scholar] [CrossRef]
- Gouzi, J.Y.; Moressis, A.; Walker, J.A.; Apostolopoulo, A.A.; Palmer, R.H.; Bernards, A.; Soulakis, E.M. The receptorkinase alk control neurofibromin functions in Drosophila growth and learning. PLoS Genet. 2011, 9, e1002281. [Google Scholar] [CrossRef]
- Walker, J.A.; Gouzi, J.Y.; Long, J.B.; Huang, S.; Maher, R.C.; Zxia, H.; Khalil, K.; Ray, A.; Van Vacor, D.; Bernards, R.; et al. Genetic and functional studies implicate synaptic overgrowth and ring gland cAMP/PKA signaling defects in the Drosophila melanogaster Neurofibromatosis-1 growth deficiency. PLoS Genet. 2013, 9, e1003958. [Google Scholar] [CrossRef] [PubMed]
- Bollag, G.; Clapp, D.W.; Shih, S.; Adler, F.; Zhang, Y.Y.; Thompson, P.; Lange, B.J.; Freedman, M.H.; McCormick, F.; Jacks, T.; et al. Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat. Genet. 1996, 12, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Gianino, S.M.; Gutmann, D.H. Neurofibromatosis-1 regulation of neural stem cell proliferation and multilineage differentiation operates through distinct RAS effector pathways. Genes Dev. 2015, 29, 1677–1682. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, B.; Dasgupta, B.; Shin, J.E.; Emnett, R.J.; Hart-Mahon, E.K.; Elghazi, L.; Bernal-Mizrachi, E.; Gutmann, D.H. Neurofibromatosis-1 regulates neuronal and glial cell differentiation from neuroglial progenitors in vivo by both cAMP- and Ras-dependent mechanisms. Cell Stem Cell 2007, 1, 443–457. [Google Scholar] [CrossRef]
- Lau, N.; Feldkamp, M.M.; Roncari, L.; Loehr, A.H.; Shannon, P.; Gutmann, D.H.; Guha, A. Loss of neurofibromin is associated with activation of RAS/MAPK and PI3-K/AKT signaling in a neurofibromatosis 1 astrocytoma. J. Neuropathol. Exp. Neurol. 2000, 59, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Wu, X.; Rhodes, S.D.; Chen, S.; He, Y.; Yuan, J.; Li, J.; Yang, X.; Li, X.; Jiang, L.; et al. Hyperactive Ras/MAPK signaling is critical for tibial nonunion fracture in neurofibromin-deficient mice. Hum. Mol. Genet. 2013, 22, 4818–4828. [Google Scholar] [CrossRef]
- Kim, B.S.; Lee, J.; Bang, M.; Am Seo, B.; Khalid, A.; Jung, M.W.; Jeon, D. Differential regulation of observational fear and neural oscillations by serotonin and dopamine in the mouse anterior cingulate cortex. Psychopharmacology 2014, 231, 4371–4381. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, L.J.; Guha, G.; Li, S.; Kyrylkova, K.; Kioussi, C.; Leid, M.; Ganguli-Indra, G.; Indra, A.K. Selective ablation of Ctip2/Bcl11b in epidermal keratinocytes triggers atopic dermatitis-like skin inflammatory responses in adult mice. PLoS ONE 2012, 7, e51262. [Google Scholar] [CrossRef] [PubMed]
- Georganta, E.-M.; Moressis, A.; Skoulakis, E.M. Associative Learning Requires Neurofibromin to Modulate GABAergic Inputs to Drosophila Mushroom Bodies. J. Neurosci. 2021, 41, 5274–5286. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, P.; Cates-Gatto, C.; Varodoyan, F. Inflammatory myofibroblastic tumour causing intestinal obstruction in a patient with neurofibromatosis type 1. Neuropharmacology 2016, 107, 108. [Google Scholar]
- Violante, I.R.; Patricio, M.; Bernardino, I.; Rebola, J.; Abrunhosa, A.J.; Ferreira, N. and Castelo-Branco, M. GABA deficiency in NF1. Neurology 2016, 87, 897–904. [Google Scholar] [CrossRef]
- Ribeiro, M.; Violante, I.; Bernardino, I.; Edden, R.; Castelo-Branco, M. Abnormal relationship between GABA, neurophysiology and impulsive behavior in neurofibromatosis type 1. Cortex 2015, 64, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Licis, A.K.; Vallorani, A.; Gao, F.; Chen, C.; Lenox, J.; Yamada, K.A.; Duntley, S.P.; Gutmann, D.H. Prevalence of Sleep Disturbances in Children With Neurofibromatosis Type 1. J. Child Neurol. 2013, 28, 1400–1405. [Google Scholar] [CrossRef]
- Leschziner, G.D.; Golding, J.F.; Ferner, R.E. Sleep disturbance as part of the neurofibromatosis type 1 phenotype in adults. Am. J. Med. Genet. A 2013, 161, 1319–1322. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.; Wiggs, L.; Stores, G.; Huson, S.M. Psychological disturbance and sleep disorders in children with neurofibromatosis type 1. Dev. Med. Child Neurol. 2005, 47, 237–242. [Google Scholar] [CrossRef]
- Bai, L.; Sehgal, A. Anaplastic Lymphoma Kinase Acts in the Drosophila Mushroom Body to Negatively Regulate Sleep. PLoS Genet. 2015, 11, e1005611. [Google Scholar] [CrossRef]
- Anastasaki, C.; Rensing, N.; Johnson, K.; Wong, M.; Gutman, D. Neurofibromatosis type 1 (Nf1)-mutant mice exhibit increased sleep fragmentation. J. Sleep. Res. 2019, 28, e12816. [Google Scholar] [CrossRef]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Takita, J.; Choi, Y.L.; Kato, M.; Ohira, M.; Sanada, M.; Wang, L.; Soda, M.; Kikuchi, A.; Igarashi, T.; et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008, 455, 971–974. [Google Scholar] [CrossRef] [PubMed]
- George, R.E.; Sanda, T.; Hanna, M.; Frèohling, S.; Luther, W., II; Zhang, J.; Ahn, Y.; Zhou, W.; London, W.B.; McGrady, P.; et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008, 455, 975–978. [Google Scholar] [CrossRef]
- Mosse, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008, 455, 930–935. [Google Scholar] [CrossRef]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Sirvent, N.; Hawkins, A.L.; Moeglin, D.; Coindre, J.M.; Kurzenne, J.Y.; Michiels, J.F.; Barcelo, G.; Turc-Carel, C.; Griffin, C.A.; Pedeutour, F. ALK probe rearrangement in a t(2;11;2)(p23;p15;q31) translocation found in a prenatal myofibroblastic fibrous lesion: Toward a molecular definition of an inflammatory myofibroblastic tumor family? Genes Chromosomes Cancer 2001, 31, 85–90. [Google Scholar] [CrossRef]
- Palmer, R.H.; Vernersson, E.; Grabbe, C.; Hallberg, B. Anaplastic lymphoma kinase: Signalling in development and disease. Biochem. J. 2009, 420, 345–361. [Google Scholar] [CrossRef]
- Kwak, E.L.; Bang, Y.J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.H.; Dezube, B.J.; Janne, P.A.; Costa, D.B.; et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef]
- Butrynski, J.E.; D’Adamo, D.R.; Hornick, J.L.; Dal Cin, P.; Antonescu, C.R.; Jhanwar, S.C.; Ladanyi, M.; Capelletti, M.; Rodig, S.J.; Ramaiya, N.; et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N. Engl. J. Med. 2010, 363, 1727–1733. [Google Scholar] [CrossRef]
- Umapathy, G.; Mendoza0Garcia, P.; Hallberg, B.; Palmer, R. Targeting anaplastic lymphoma kinase in neuroblastoma. APMIS 2019, 127, 288–302. [Google Scholar] [CrossRef]
- Kodama, T.; Hasegawa, M.; Takanashi, K.; Sakurai, Y.; Kondoh, O.; Sakamoto, H. Antitumor activity of the selective ALK inhibitor alectinib in models of intracranial metastases. Cancer Chemother. Pharmacol. 2014, 74, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Metro, G.; Lunardi, G.; Bennati, C.; Chiarini, P.; Sperduti, I.; Ricciuti, B.; Marcomigni, L.; Costa, C.; Crino, L.; Floridi, P.; et al. Alectinib’s activity against CNS metastases from ALK-positive non-small cell lung cancer: A single institution case series. J. Neurooncol 2016, 129, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.; Halloran, M. Mechanistic insights from animal models of neurofibromatosis type 1 cognitive impairment. Dis. Model. Mech. 2022, 15, dmm049422. [Google Scholar] [CrossRef]
- Weiss, J.; Weber, S.; Torres, E.; Marzulla, T.; Raber, J. Genetic inhibition of Anaplastic Lymphoma Kinase rescues cognitive impairments in Neurofibromatosis 1 mutant mice. Behav. Brain Res. 2017, 321, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Marana Perez, A.I.; Duat Rodriguez, A.; Soto Insuga, V.; Dominguez Carral, J.; Puertas Martin, V.; Gonzalez Gutierrez Solana, L. Prevalence of sleep disorders in patients with neurofibromatosis type 1. Neurologia 2015, 30, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Setabutr, D.; Perez, M.R.; Truong, M.T.; Senders, C.W.; Rubinstein, B.K. Neurofibromatosis of the larynx causing stridor and sleep apnea. Am. J. Otolaryngol. 2014, 35, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Sforza, E.; Colamaria, V.; Lugaresi, E. Neurofibromatosis associated with central alveolar hypoventilation syndrome during sleep. Acta Paediatr. 1994, 83, 794–796. [Google Scholar] [CrossRef]
- Stradling, J.R.; Huddart, S.; Arnold, A.G. Sleep apnoea syndrome caused by neurofibromatosis and superior vena caval obstruction. Thorax 1981, 36, 634–635. [Google Scholar] [CrossRef]
- Webb, T.R.; Slavish, J.; George, R.E.; Look, A.T.; Xue, L.; Jiang, Q.; Cui, X.; Rentrop, W.B.; Morris, S.W. Anaplastic lymphoma kinase: Role in cancer pathogenesis and small-molecule inhibitor development for therapy. Expert Rev. Anticancer Ther. 2009, 9, 331–356. [Google Scholar] [CrossRef]
- Hallberg, B.; Palmer, R. The role of the ALK receptor in cancer biology. Ann. Oncol. 2016, 27, iii4–iii5. [Google Scholar] [CrossRef]
- Chun, S.; Choe, K.; Ivengyar, P.; Yordy, J.; Timmerman, R. Isolated central nervous system progression on Crizotinib. Cancer Biol. Ther. 2012, 13, 1376–1383. [Google Scholar] [CrossRef]
- Zou, Z.; Xing, P.; Hao, X.; Wang, Y.; Song, X.; Shan, L.; Zhang, C.; Liu, Z.; Ma, K.; Dong, G. Intracranial efficacy of alectinib in ALK-positive NSCLC patients with CNS metastases—A multicenter retrospective study. BMC Med. 2022, 20, 12. [Google Scholar] [CrossRef]
- Weiss, J.B.; Xue, C.; Benice, T.; Xue, L.; Morris, S.W.; Raber, J. Anaplastic lymphoma kinase and leukocyte tyrosine kinase: Functions and genetic interactions in learning, memory and adult neurogenesis. Pharmacol. Biochem. Behav. 2012, 100, 566–574. [Google Scholar] [CrossRef]
- Weiss, J.; Weber, S.; Marzulla, T.; Raber, J. Pharmacological inhibition of Anaplastic Lymphoma Kinase rescues spatial memory impairments in Neurofibromatosis 1 mutant mice. Behav. Brain Res. 2017, 332, 337–342. [Google Scholar] [CrossRef]
- Diggs-Andrews, K.; Brown, J.; Giamino, S.; Rubin, J.; Wozniak, D.; Gutman, D. Sex is a major determinant of neuronal dysfunction in Neurofibromatosis Type 1. Ann. Neurol. 2014, 75, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Elmadany, N.; Logiacco, F.; Buonfiglioli, A.; Haage, V.C.; Wright-Jin, E.C.; Schattenberg, A.; Papawassiliou, R.M.; Kettenmann, H.; Semtner, M.; Gutmann, D.H. Mutant microglia exhibit sexually-dimorphic cyclic AMP-dependent purinergic defects. Neurobiol. Dis. 2020, 144, 105030. [Google Scholar] [CrossRef] [PubMed]
- Warrington, N.M.; Sun, T.; Luo, J.; McKinstry, R.C.; Parkin, P.C.; Ganzhorn, S.; Spoljaric, D.; Albers, A.C.; Merkelson, A.; Stewart, D.R. The Cyclic AMP Pathway Is a Sex-Specific Modifier of Glioma Risk in Type I Neurofibromatosis Patients. Cancer Res. 2015, 75, 16–21. [Google Scholar] [CrossRef]
- Krenik, D.; Weiss, J.; Raber, J. Role of the parental NF1 carrier in effects of pharmacological inhibition of anaplastic lymphoma kinase in Neurofibromatosis 1 mutant mice. Brain Res. 2021, 1769, 147594. [Google Scholar] [CrossRef]
- Friedman, J.; Arbiser, J.; Epstein, J.; Gutmann, D.; Huot, S.; Lin, A.; Mcmanus, B.; Korf, B. Cardiovascular disease in neurofibromatosis 1: Report of the NF1 cardiovascular task force. Genet. Med. 2002, 4, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, A.; Lee, J.; Ismat, F.; Lu, M.; Lawson, N.; Kanki, J.; Look, A.; Epstein, J. Cardiac and vascular functions of the zebrafish orthologues of the type I neurofibromatosis gene NFI. Proc. Natl. Acad. Sci. USA 2009, 106, 22305–22310. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiss, J.B.; Raber, J. Inhibition of Anaplastic Lymphoma Kinase (Alk) as Therapeutic Target to Improve Brain Function in Neurofibromatosis Type 1 (Nf1). Cancers 2023, 15, 4579. https://doi.org/10.3390/cancers15184579
Weiss JB, Raber J. Inhibition of Anaplastic Lymphoma Kinase (Alk) as Therapeutic Target to Improve Brain Function in Neurofibromatosis Type 1 (Nf1). Cancers. 2023; 15(18):4579. https://doi.org/10.3390/cancers15184579
Chicago/Turabian StyleWeiss, Joseph B., and Jacob Raber. 2023. "Inhibition of Anaplastic Lymphoma Kinase (Alk) as Therapeutic Target to Improve Brain Function in Neurofibromatosis Type 1 (Nf1)" Cancers 15, no. 18: 4579. https://doi.org/10.3390/cancers15184579