
Functional Characterization of Transforming Growth Factor-β Signaling in Dasatinib Resistance and Pre-BCR+ Acute Lymphoblastic Leukemia
 , ,
, ,
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Human Precursor B-ALL Cell Lines
2.2. Gene Expression Analysis
2.3. Western Blot Analysis
2.4. Phospho-Specific Intracellular Flow Cytometry Assay
2.5. Cell Proliferation Assays
2.6. Cell Cycle and Apoptosis Analysis
2.7. Statistical Analysis
3. Results
3.1. Upregulation of the TGFβ Signaling Pathway in Acquired Dasatinib Resistance
3.2. Dasatinib Partially Inhibits TGF-β1-Induced SMAD2/3 Phosphorylation in Pre-BCR+/E2A-PBX1+ ALL Cells
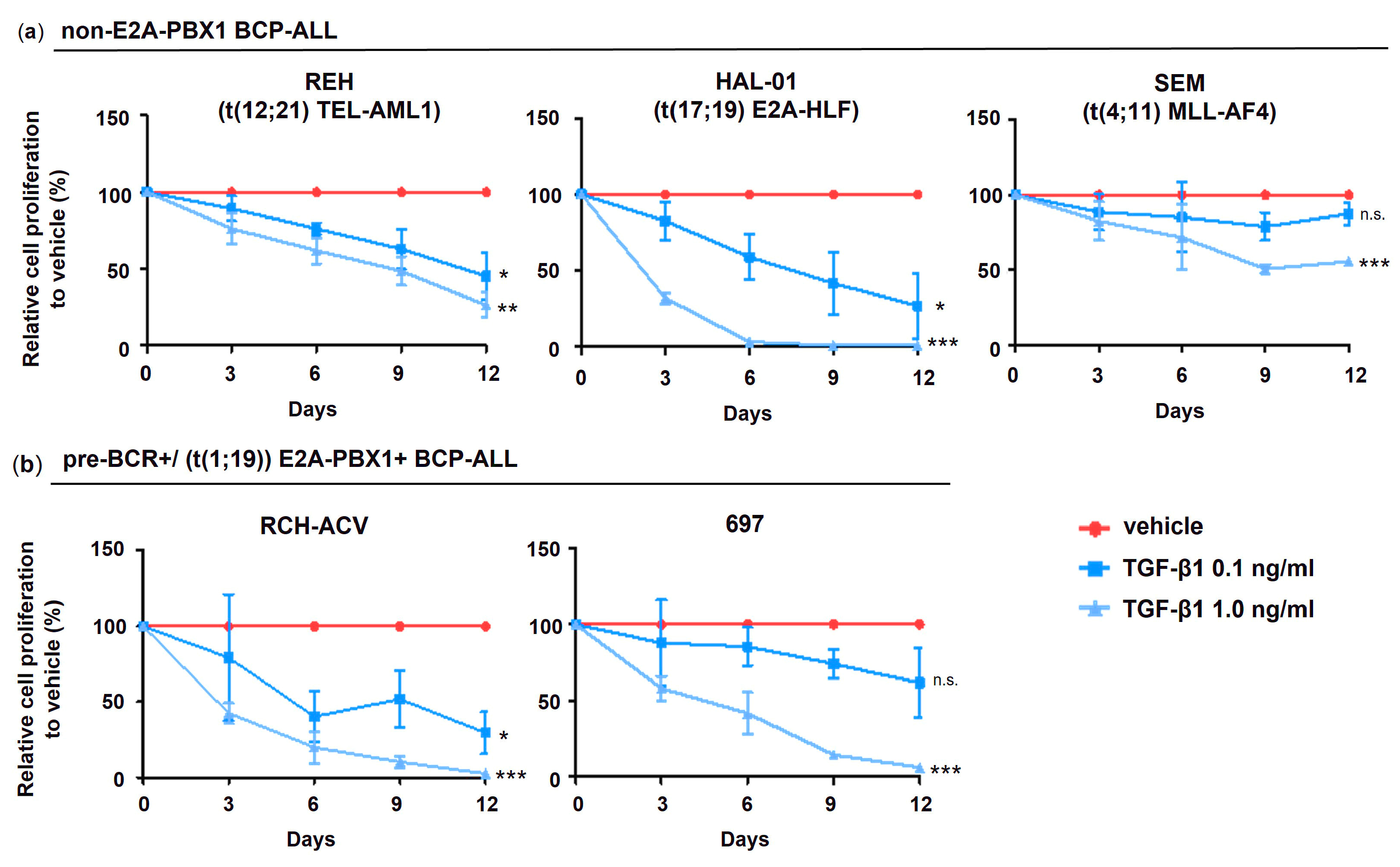
3.3. Heterogeneous TGFβ Signaling Activity and Effects in Human BCP-ALL Cell Lines
3.4. Cell Proliferation of Dasatinib-Resistant RCH-ACV Cells Inhibited by TGF-β1
4. Discussion
4.1. Heterogeneous TGFβ Signaling Activity and TGF-β1-Induced Growth Inhibition Imply Functionally Distinct BCP-ALL Subsets
4.2. Dasatinib Blocks TGF-β1-Induced SMAD2/3 Phosphorylation by Interacting with TGFβ Type I Receptors
4.3. Differential Mechanisms Underlying TGF-β1 Growth-Inhibitory Effects in Dasatinib-Sensitive and -Resistant RCH-ACV Cells
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Hunger, S.P.; Lu, X.; Devidas, M.; Camitta, B.M.; Gaynon, P.S.; Winick, N.J.; Reaman, G.H.; Carroll, W.L. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: A report from the children’s oncology group. J. Clin. Oncol. 2012, 30, 1663–1669. [Google Scholar] [CrossRef] [PubMed]
- Teachey, D.T.; Hunger, S.P. Predicting relapse risk in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 2013, 162, 606–620. [Google Scholar] [CrossRef]
- Bhojwani, D.; Pui, C.-H. Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol. 2013, 14, e205–e217. [Google Scholar] [CrossRef]
- Sun, W.; Malvar, J.; Sposto, R.; Verma, A.; Wilkes, J.J.; Dennis, R.; Heym, K.; Laetsch, T.W.; Widener, M.; Rheingold, S.R.; et al. Outcome of children with multiply relapsed B-cell acute lymphoblastic leukemia: A therapeutic advances in childhood leukemia & lymphoma study. Leukemia 2018, 32, 2316–2325. [Google Scholar] [PubMed]
- Hochhaus, A.; Baccarani, M.; Silver, R.T.; Schiffer, C.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Deininger, M.W.; Guilhot, F.; et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 2020, 34, 966–984. [Google Scholar]
- Cerchione, C.; Locatelli, F.; Martinelli, G. Dasatinib in the Management of Pediatric Patients with Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia. Front. Oncol. 2021, 11, 632231. [Google Scholar] [CrossRef] [PubMed]
- Geng, H.; Hurtz, C.; Lenz, K.B.; Chen, Z.; Baumjohann, D.; Thompson, S.; Goloviznina, N.A.; Chen, W.-Y.; Huan, J.; LaTocha, D.; et al. Self-enforcing feedback activation between BCL6 and pre-B cell receptor signaling defines a distinct subtype of acute lymphoblastic leukemia. Cancer Cell 2015, 27, 409–425. [Google Scholar] [CrossRef]
- Bicocca, V.T.; Chang, B.H.; Masouleh, B.K.; Muschen, M.; Loriaux, M.M.; Druker, B.J.; Tyner, J.W. Crosstalk between ROR1 and the Pre-B cell receptor promotes survival of t(1;19) acute lymphoblastic leukemia. Cancer Cell 2012, 22, 656–667. [Google Scholar] [CrossRef]
- Duque-Afonso, J.; Feng, J.; Scherer, F.; Lin, C.-H.; Wong, S.H.K.; Wang, Z.; Iwasaki, M.; Cleary, M.L. Comparative genomics reveals multistep pathogenesis of E2A-PBX1 acute lymphoblastic leukemia. J. Clin. Investig. 2015, 125, 3667–3680. [Google Scholar] [CrossRef]
- Duque-Afonso, J.; Lin, C.-H.; Han, K.; Wei, M.C.; Feng, J.; Kurzer, J.H.; Schneidawind, C.; Wong, S.H.-K.; Bassik, M.C.; Cleary, M.L. E2A-PBX1 Remodels Oncogenic Signaling Networks in B-cell Precursor Acute Lymphoid Leukemia. Cancer Res. 2016, 76, 6937–6949. [Google Scholar] [CrossRef] [PubMed]
- Duque-Afonso, J.; Lin, C.-H.; Han, K.; Morgens, D.W.; Jeng, E.E.; Weng, Z.; Jeong, J.; Wong, S.H.K.; Zhu, L.; Wei, M.C.; et al. CBP Modulates Sensitivity to Dasatinib in Pre-BCR+ Acute Lymphoblastic Leukemia. Cancer Res. 2018, 78, 6497–6508. [Google Scholar] [CrossRef] [PubMed]
- Fischer, U.; Forster, M.; Rinaldi, A.; Risch, T.; Sungalee, S.; Warnatz, H.-J.; Bornhauser, B.; Gombert, M.; Kratsch, C.; Stütz, A.M.; et al. Genomics and drug profiling of fatal TCF3-HLF−positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat. Genet. 2015, 47, 1020–1029. [Google Scholar] [PubMed]
- Eldfors, S.; Kuusanmäki, H.; Kontro, M.; Majumder, M.M.; Parsons, A.; Edgren, H.; Pemovska, T.; Kallioniemi, O.; Wennerberg, K.; Gökbuget, N.; et al. Idelalisib sensitivity and mechanisms of disease progression in relapsed TCF3-PBX1 acute lymphoblastic leukemia. Leukemia 2017, 31, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Buchner, M.; Swaminathan, S.; Chen, Z.; Müschen, M. Mechanisms of pre-B-cell receptor checkpoint control and its oncogenic subversion in acute lymphoblastic leukemia. Immunol. Rev. 2015, 263, 192–209. [Google Scholar]
- Wood, K.C. Mapping the Pathways of Resistance to Targeted Therapies. Cancer Res. 2015, 75, 4247–4251. [Google Scholar] [CrossRef]
- Braun, T.P.; Eide, C.A.; Druker, B.J. Response and Resistance to BCR-ABL1-Targeted Therapies. Cancer Cell 2020, 37, 530–542. [Google Scholar] [CrossRef]
- Ricordel, C.; Friboulet, L.; Facchinetti, F.; Soria, J.-C. Molecular mechanisms of acquired resistance to third-generation EGFR-TKIs in EGFR T790M-mutant lung cancer. Ann. Oncol. 2018, 29 (Suppl. S1), i28–i37. [Google Scholar] [CrossRef]
- Krutzik, P.O.; Nolan, G.P. Intracellular phospho-protein staining techniques for flow cytometry: Monitoring single cell signaling events. Cytometry 2003, 55, 61–70. [Google Scholar] [CrossRef]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. Available online: https://cshperspectives.cshlp.org/content/8/5/a021873.short (accessed on 10 May 2021). [CrossRef]
- Mullen, A.C.; Wrana, J.L. TGF-β Family Signaling in Embryonic and Somatic Stem-Cell Renewal and Differentiation. Cold Spring Harb. Perspect. Biol. 2017, 9, a022186. [Google Scholar]
- Seoane, J.; Gomis, R.R. TGF-β Family Signaling in Tumor Suppression and Cancer Progression. Cold Spring Harb. Perspect. Biol. 2017, 9, a022277. Available online: https://cshperspectives.cshlp.org/content/9/12/a022277.short (accessed on 10 May 2021). [CrossRef] [PubMed]
- Derynck, R.; Budi, E.H. Specificity, versatility and control of TGF-β family signaling. Sci. Signal 2019, 12, eaav5183. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.S. Transcriptional Control by the SMADs. Cold Spring Harb. Perspect. Biol. 2016, 8, a022079. [Google Scholar] [CrossRef]
- Zhang, Y.; Alexander, P.B.; Wang, X.-F. TGF-β Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harb. Perspect. Biol. 2017, 9, a022145. [Google Scholar] [CrossRef]
- Buske, C.; Becker, D.; Feuring-Buske, M.; Hannig, H.; Wulf, G.; Schäfer, C.; Hiddemann, W.; Wörmann, B. TGF-β inhibits growth and induces apoptosis in leukemic B cell precursors. Leukemia 1997, 11, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Ford, A.M.; Palmi, C.; Bueno, C.; Hong, D.; Cardus, P.; Knight, D.; Cazzaniga, G.; Enver, T.; Greaves, M. The TEL-AML1 leukemia fusion gene dysregulates the TGF-beta pathway in early B lineage progenitor cells. J. Clin. Investig. 2009, 119, 826–836. [Google Scholar] [PubMed]
- Lombardo, L.J.; Lee, F.Y.; Chen, P.; Norris, D.; Barrish, J.C.; Behnia, K.; Castaneda, S.; Cornelius, L.A.M.; Das, J.; Doweyko, A.M.; et al. Discovery of N-(2-Chloro-6-methyl-phenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (BMS-354825), a Dual Src/Abl Kinase Inhibitor with Potent Antitumor Activity in Preclinical Assays. J. Med. Chem. 2004, 47, 6658–6661. [Google Scholar] [CrossRef]
- Lee, F.Y.; Lombardo, L.; Camuso, A.; Castaneda, S.; Fager, K.; Flefleh, C.; Inigo, I.; Johnson, K.; Kan, D.; Luo, R.; et al. BMS-354825 potently inhibits multiple selected oncogenic tyrosine kinases and possesses broad-spectrum antitumor activities in vitro and in vivo. Cancer Res. 2005, 65 (Suppl. S9), 159. [Google Scholar]
- Li, J.; Rix, U.; Fang, B.; Bai, Y.; Edwards, A.; Colinge, J.; Bennett, K.L.; Gao, J.; Song, L.; Eschrich, S.; et al. A chemical and phosphoproteomic characterization of dasatinib action in lung cancer. Nat. Chem. Biol. 2010, 6, 291–299. [Google Scholar] [CrossRef]
- Gordian, E.; Li, J.; Pevzner, Y.; Mediavilla-Varela, M.; Luddy, K.; Ohaegbulam, K.; Daniel, K.G.; Haura, E.B.; Muñoz-Antonia, T. Transforming growth factor β signaling overcomes dasatinib resistance in lung cancer. PLoS ONE 2014, 9, e114131. [Google Scholar] [CrossRef]
- Bartscht, T.; Rosien, B.; Rades, D.; Kaufmann, R.; Biersack, H.; Lehnert, H.; Gieseler, F.; Ungefroren, H. Dasatinib blocks transcriptional and promigratory responses to transforming growth factor-beta in pancreatic adenocarcinoma cells through inhibition of Smad signalling: Implications for in vivo mode of action. Mol. Cancer 2015, 14, 199. [Google Scholar] [CrossRef]
- Siegel, P.M.; Massagué, J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat. Rev. Cancer 2003, 3, 807–821. [Google Scholar] [CrossRef]
- Massagué, J.; Blain, S.W.; Lo, R.S. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell 2000, 103, 295–309. [Google Scholar] [CrossRef]
- Grüninger, P.K.; Uhl, F.; Herzog, H.; Gentile, G.; Andrade-Martinez, M.; Schmidt, T.; Han, K.; Morgens, D.W.; Bassik, M.C.; Cleary, M.L.; et al. Functional characterization of the PI3K/AKT/MTOR signaling pathway for targeted therapy in B-precursor acute lymphoblastic leukemia. Cancer Gene Ther. 2022, 29, 1751–1760. [Google Scholar] [CrossRef] [PubMed]
- Köhrer, S.; Havranek, O.; Seyfried, F.; Hurtz, C.; Coffey, G.P.; Kim, E.; Ten Hacken, E.; Jäger, U.; Vanura, K.; O’Brien, S.; et al. Pre-BCR signaling in precursor B-cell acute lymphoblastic leukemia regulates PI3K/AKT, FOXO1 and MYC, and can be targeted by SYK inhibition. Leukemia 2016, 30, 1246–1254. [Google Scholar] [CrossRef] [PubMed]
- Seoane, J.; Le, H.-V.; Shen, L.; Anderson, S.A.; Massagué, J. Integration of Smad and Forkhead Pathways in the Control of Neuroepithelial and Glioblastoma Cell Proliferation. Cell 2004, 117, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Gomis, R.R.; Alarcón, C.; He, W.; Wang, Q.; Seoane, J.; Lash, A.; Massagué, J. A FoxO-Smad synexpression group in human keratinocytes. Proc. Natl. Acad. Sci. USA 2006, 103, 12747–12752. [Google Scholar] [CrossRef]
- Tominaga, K.; Suzuki, H.I. TGF-β Signaling in Cellular Senescence and Aging-Related Pathology. Int. J. Mol. Sci. 2019, 20, 5002. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mostufi-Zadeh-Haghighi, G.; Veratti, P.; Zodel, K.; Greve, G.; Waterhouse, M.; Zeiser, R.; Cleary, M.L.; Lübbert, M.; Duque-Afonso, J. Functional Characterization of Transforming Growth Factor-β Signaling in Dasatinib Resistance and Pre-BCR+ Acute Lymphoblastic Leukemia. Cancers 2023, 15, 4328. https://doi.org/10.3390/cancers15174328
Mostufi-Zadeh-Haghighi G, Veratti P, Zodel K, Greve G, Waterhouse M, Zeiser R, Cleary ML, Lübbert M, Duque-Afonso J. Functional Characterization of Transforming Growth Factor-β Signaling in Dasatinib Resistance and Pre-BCR+ Acute Lymphoblastic Leukemia. Cancers. 2023; 15(17):4328. https://doi.org/10.3390/cancers15174328
Chicago/Turabian StyleMostufi-Zadeh-Haghighi, Gila, Pia Veratti, Kyra Zodel, Gabriele Greve, Miguel Waterhouse, Robert Zeiser, Michael L. Cleary, Michael Lübbert, and Jesús Duque-Afonso. 2023. "Functional Characterization of Transforming Growth Factor-β Signaling in Dasatinib Resistance and Pre-BCR+ Acute Lymphoblastic Leukemia" Cancers 15, no. 17: 4328. https://doi.org/10.3390/cancers15174328