Clinical Characteristics and Outcomes of Patients with Primary and Secondary Myelofibrosis According to the Genomic Classification Using Targeted Next-Generation Sequencing

 , , , , ,
, , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. General Characteristics
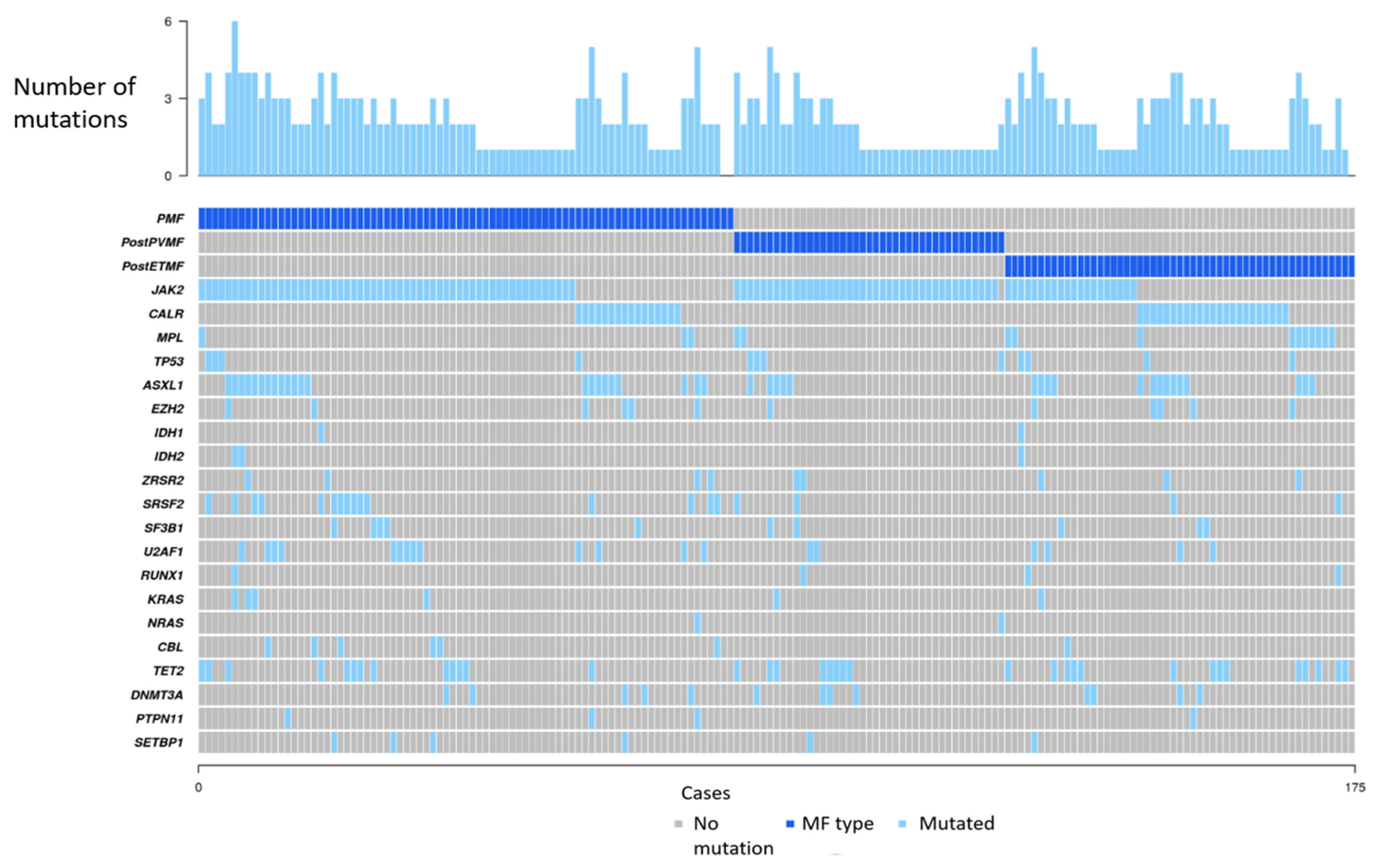
3.2. NGS Studies
3.3. Genomic Classification
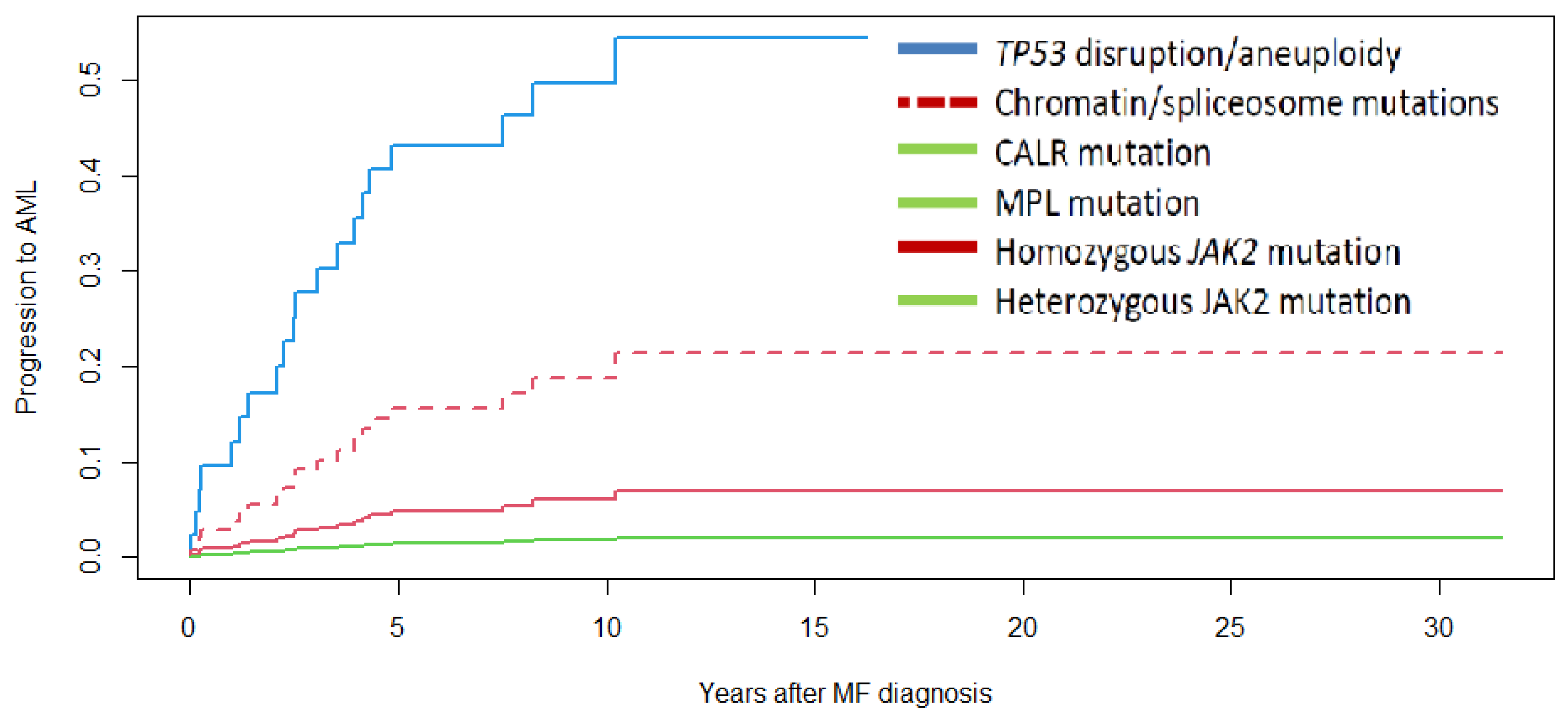
3.4. Survival and Progression to Acute Myeloid Leukemia
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cervantes, F. How I treat myelofibrosis. Blood 2014, 124, 2635–2642. [Google Scholar] [CrossRef]
- Pastor-Galán, I.; Hernández-Boluda, J.C.; Correa, J.G.; Alvarez-Larrán, A.; Ferrer-Marín, F.; Raya, J.M.; Ayala, R.; Velez, P.; Pérez-Encinas, M.; Estrada, N.; et al. Clinico-biological characteristics of patients with myelofibrosis: An analysis of 1000 cases from the Spanish Registry of Myelofibrosis. Med. Clin. 2020, 155, 152–158. [Google Scholar] [CrossRef]
- Barbui, T.; Tefferi, A.; Vannucchi, A.M.; Passamonti, F.; Silver, R.T.; Hoffman, R.; Verstovsek, S.; Mesa, R.; Kiladjian, J.J.; Hehlmann, R.; et al. Philadelphia chromosome-negative classical myeloproliferative neoplasms: Revised management recommendations from European LeukemiaNet. Leukemia 2018, 32, 1057–1069. [Google Scholar] [CrossRef] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.; Calvo, K.R.; Kvasnicka, H.M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef] [PubMed]
- Rumi, E.; Pietra, D.; Pascutto, C.; Guglielmelli, P.; Martínez-Trillos, A.; Casetti, I.; Colomer, D.; Pieri, L.; Pratcorona, M.; Rotunno, G.; et al. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood 2014, 124, 1062–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passamonti, F.; Mora, B.; Giorgino, T.; Guglielmelli, P.; Cazzola, M.; Maffioli, M.; Rambaldi, A.; Caramella, M.; Komrokji, R.; Gotlib, J.; et al. Driver mutations’ effect in secondary myelofibrosis: An international multicenter study based on 781 patients. Leukemia 2017, 31, 970–973. [Google Scholar] [CrossRef]
- Finazzi, M.C.; Carobbio, A.; Cervantes, F.; Isola, I.M.; Vannucchi, A.M.; Guglielmelli, P.; Rambaldi, A.; Finazzi, G.; Barosi, G.; Barbui, T. CALR mutation, MPL mutation and triple negativity identify patients with the lowest vascular risk in primary myelofibrosis. Leukemia 2015, 29, 1209–1210. [Google Scholar] [CrossRef]
- Chowdhury, O.; O’Sullivan, J.; Barkas, N.; Wang, G.; Buck, G.; Hamblin, A.; Tefferi, A.; Al-Ali, H.K.; Barosi, G.; Devos, T.; et al. Spliceosome mutations are common in persons with myeloproliferative neoplasm-associated myelofibrosis with RBC-transfusion-dependence and correlate with response to pomalidomide. Leukemia 2021, 35, 1197–1202. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Lasho, T.L.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C.; et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 27, 1861–1869. [Google Scholar] [CrossRef] [Green Version]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef] [PubMed]
- Courtier, F.; Garnier, S.; Carbuccia, N.; Guille, A.; Adélaide, J.; Chaffanet, M.; Hirsch, P.; Paz, D.L.; Slama, B.; Vey, N.; et al. Targeted molecular characterization shows differences between primary and secondary myelofibrosis. Genes Chromosomes Cancer 2020, 59, 30–39. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Cervantes, F.; Dupriez, B.; Pereira, A.; Passamonti, F.; Reilly, J.T.; Morra, E.; Vannucchi, A.M.; Mesa, R.A.; Demory, J.L.; Barosi, G.; et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 2009, 113, 2895–2901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passamonti, F.; Giorgino, T.; Mora, B.; Guglielmelli, P.; Rumi, E.; Maffioli, M.; Rambaldi, A.; Caramella, M.; Komrokji, R.; Gotlib, J.; et al. A clinical-molecular prognostic model to predict survival in patients with post polycythemia vera and post essential thrombocythemia myelofibrosis. Leukemia 2017, 31, 2726–2731. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Li, F.; Ma, M.M.; Zhang, S.; Liu, Y.; Yan, Z.L.; Chen, W.; Cao, J.; Zeng, L.Y.; Wang, X.Y.; et al. Roles of T875N somatic mutation in the activity, structural stability of JAK2 and the transformation of OCI-AML3 cells. Int. J. Biol. Macromol. 2019, 137, 1030–1040. [Google Scholar] [CrossRef]
- Yoshimitsu, M.; Hachiman, M.; Uchida, Y.; Arima, N.; Arai, A.; Kamada, Y.; Shide, K.; Ito, M.; Shimoda, K.; Ishitsuka, K. Essential thrombocytosis attributed to JAK2-T875N germline mutation. Int. J. Hematol. 2019, 110, 584–590. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Coltro, G.; Mannelli, F.; Rotunno, G.; Loscocco, G.G.; Mannarelli, C.; Maccari, C.; Paoli, C.; Romagnoli, S.; Bartalucci, N.; et al. ASXL1 mutations are prognostically significant in PMF, but not MF following essential thrombocythemia or polycythemia vera. Blood Adv. 2022, 6, 2927–2931. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Mudireddy, M.; Mannarelli, C.; Nicolosi, M.; Pacilli, A.; Pardanani, A.; Rumi, E.; Rosti, V.; et al. MIPSS70: Mutation-Enhanced International Prognostic Score System for Transplantation-Age Patients with Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 310–318. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Gangat, N.; Ketterling, R.P.; Pardanani, A.; Vannucchi, A.M. MIPSS70+ Version 2.0: Mutation and Karyotype-Enhanced International Prognostic Scoring System for Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 1769–1770. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Nicolosi, M.; Mannelli, F.; Mudireddy, M.; Bartalucci, N.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; et al. GIPSS: Genetically inspired prognostic scoring system for primary myelofibrosis. Leukemia 2018, 32, 1631–1642. [Google Scholar] [CrossRef] [Green Version]
- Luque Paz, D.; Riou, J.; Verger, E.; Cassinat, B.; Chauveau, A.; Ianotto, J.C.; Dupriez, B.; Boyer, F.; Renard, M.; Mansier, O.; et al. Genomic analysis of primary and secondary myelofibrosis redefines the prognostic impact of ASXL1 mutations: A FIM study. Blood Adv. 2021, 5, 1442–1451. [Google Scholar] [CrossRef] [PubMed]
- Coltro, G.; Rotunno, G.; Mannelli, L.; Mannarelli, C.; Fiaccabrino, S.; Romagnoli, S.; Bartalucci, N.; Ravenda, E.; Gelli, E.; Sant’Antonio, E.; et al. RAS/CBL mutations predict resistance to JAK inhibitors in myelofibrosis and are associated with poor prognostic features. Blood Adv. 2020, 4, 3677–3687. [Google Scholar] [CrossRef] [PubMed]
- Loscocco, G.G.; Guglielmelli, P.; Mannelli, F.; Mora, B.; Mannarelli, C.; Rotunno, G.; Pancani, F.; Maccari, C.; Bartalucci, N.; Romagnoli, S.; et al. SF3B1 mutations in primary and secondary myelofibrosis: Clinical, molecular and prognostic correlates. Am. J. Hematol. 2022, 97, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Nicolosi, M.; Mudireddy, M.; Szuber, N.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Gangat, N.; et al. Driver mutations and prognosis in primary myelofibrosis: Mayo-Careggi MPN alliance study of 1,095 patients. Am. J. Hematol. 2018, 93, 348–355. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Lasho, T.L.; Tischer, A.; Wassie, E.A.; Finke, C.M.; Belachew, A.A.; Ketterling, R.P.; Hanson, C.A.; Pardanani, A.D. The prognostic advantage of calreticulin mutations in myelofibrosis might be confined to type 1 or type 1-like CALR variants. Blood 2014, 124, 2465–2466. [Google Scholar] [CrossRef] [Green Version]
- Guglielmelli, P.; Maccari, C.; Sordi, B.; Balliu, M.; Atanasio, A.; Mannarelli, C.; Capecchi, G.; Sestini, I.; Coltro, G.; Loscocco, G.G.; et al. Phenotypic correlations of CALR mutation variant allele frequency in patients with myelofibrosis. Blood Cancer J. 2023, 13, 21. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Global n = 175 | TP53 Disruption n = 16 | Chromatin/Splicing Mutation n = 72 | CALR Mutation n = 19 | MPL Mutation n = 4 | JAK2 Homozygous Mutation n = 37 | JAK2 Heterozygous Mutation n = 22 | Other Mutation n = 2 | No Mutation n = 3 | p Value ¶ | |
|---|---|---|---|---|---|---|---|---|---|---|
| Age * | 66 (24–93) | 69 (42–88) | 66 (31–89) | 57 (24–86) | 52 (39–62) | 70 (35–88) | 66 (25–93) | 43 (43–44) | 62 (48–78) | 0.009 |
| Male sex ** | 85 (49) | 10 (62.5) | 45 (62.5) | 6 (32) | 2 (50) | 14 (38) | 6 (27) | 1 (50) | 1 (33) | 0.04 |
Type of MF **
| 81 (46) 53 (30) 41 (24) | 5 (31) 5 (31) 6 (37.5) | 43 (60) 19 (26) 10 (14) | 7 (37) 12 (63) - | - 4 (100) - | 10 (27) 5 (13.5) 22 (59.5) | 13 (59) 6 (27) 3 (14) | 1 (50) 1 (50) - | 2 (66) 1 (33) - | <0.0001 |
| Symptomatic splenomegaly ** | 33 (19) | 1 (6) | 15 (21) | 2 (10.5) | - | 8 (22) | 6 (27) | 1 (50) | - | 0.06 |
| Constitutional symptoms ** | 62 (35) | 6 (37.5) | 32 (44) | 2 (10.5) | - | 16 (43) | 4 (18) | 1 (50) | 1 (33) | 0.05 |
| Hb, g/L * | 106 (63–174) | 102 (77–174) | 104 (63–150) | 102 (84–140) | 105 (93–116) | 116 (80–159) | 108 (87–148) | 120 (107–134) | 90 (78–92) | 0.02 |
| WBC, ×109/L * | 10.3 (2–148) | 8.6 (2–41) | 10 (2.2–133) | 6.4 (2.5–26) | 8.8 (5.3–15.4) | 15.8 (2–148) | 8 (4–23) | 7.2 (5.2–9.2) | 4.5 (4.1–4.6) | ns |
| Platelets, ×109/L * | 307 (18–1640) | 157 (18–1640) | 269 (35–1086) | 467 (78–1359) | 254 (112–684) | 316 (41–849) | 402 (64–1200) | 248 (81–416) | 255 (27–309) | 0.04 |
| Transfusion dependence ** | 66 (38) | 6 (37.5) | 42 (58) | 5 (26) | 3 (75) | 7 (20) | 2 (9) | 1 (50) | 0 (0) | <0.0001 |
| JAKi ** | 109 (62) | 9 (56) | 46 (64) | 16 (84) | 2 (50) | 22 (59.5) | 11 (50) | - | 3 (100) | ns |
| SRV at 5 years | 63% | 45% | 59% | 89% | 100% | 55% | 67% | 100% | 100% | <0.0001 |
| AML at 5 years | 14% | 49% | 16% | 0% | 0% | 13% | 0% | 0% | 0% | 0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garrote, M.; López-Guerra, M.; Arellano-Rodrigo, E.; Rozman, M.; Carbonell, S.; Guijarro, F.; Santaliestra, M.; Triguero, A.; Colomer, D.; Cervantes, F.; et al. Clinical Characteristics and Outcomes of Patients with Primary and Secondary Myelofibrosis According to the Genomic Classification Using Targeted Next-Generation Sequencing. Cancers 2023, 15, 3904. https://doi.org/10.3390/cancers15153904
Garrote M, López-Guerra M, Arellano-Rodrigo E, Rozman M, Carbonell S, Guijarro F, Santaliestra M, Triguero A, Colomer D, Cervantes F, et al. Clinical Characteristics and Outcomes of Patients with Primary and Secondary Myelofibrosis According to the Genomic Classification Using Targeted Next-Generation Sequencing. Cancers. 2023; 15(15):3904. https://doi.org/10.3390/cancers15153904
Chicago/Turabian StyleGarrote, Marta, Mónica López-Guerra, Eduardo Arellano-Rodrigo, María Rozman, Sara Carbonell, Francesca Guijarro, Marta Santaliestra, Ana Triguero, Dolors Colomer, Francisco Cervantes, and et al. 2023. "Clinical Characteristics and Outcomes of Patients with Primary and Secondary Myelofibrosis According to the Genomic Classification Using Targeted Next-Generation Sequencing" Cancers 15, no. 15: 3904. https://doi.org/10.3390/cancers15153904