
Leveraging Gene Redundancy to Find New Histone Drivers in Cancer
 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mutation Data Collection and Processing
2.2. Comparisons of the Observed Mutation Rates at the Protein Level
2.3. Mutation Rate Comparison between Histones and Known Cancer Drivers at the Protein Level
2.4. Mutation Rate Variation across Histone Genes Encoding Common Proteins
2.5. Identification of New Driver Histone Genes
2.6. Mutational Profiles and Mutational Signature Detection
3. Results
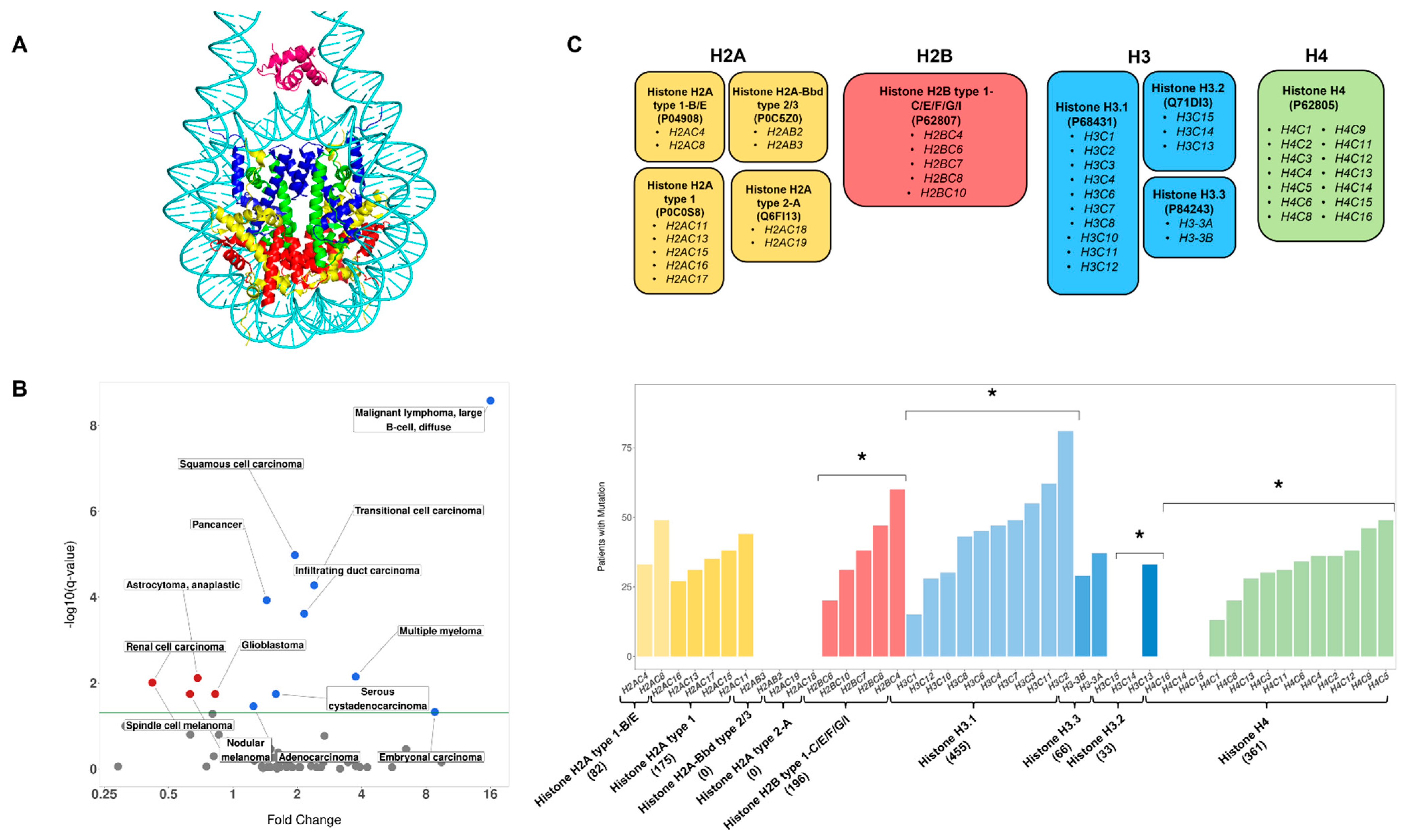
3.1. Histone Mutation Rates Vary across Cancer Types
3.2. Comparison of Histone Mutation Rates to List of Cancer Drivers
3.3. Observed Mutation Rates Do Not Vary across Histone Families
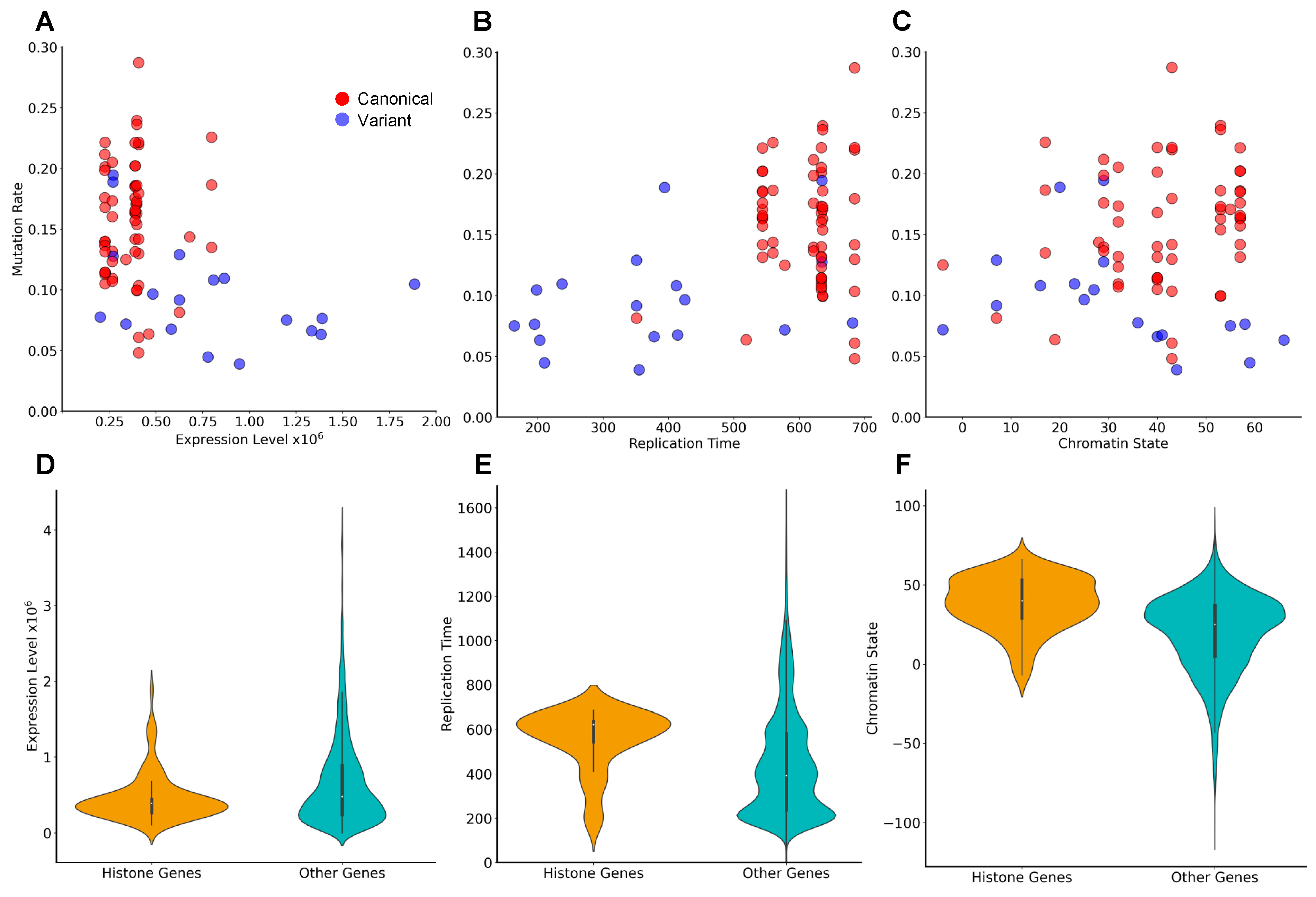
3.4. Genes Encoding the Same Histone Protein Differ in Mutation Frequencies
3.5. Identification of Novel Histone Gene Drivers across Various Cancers
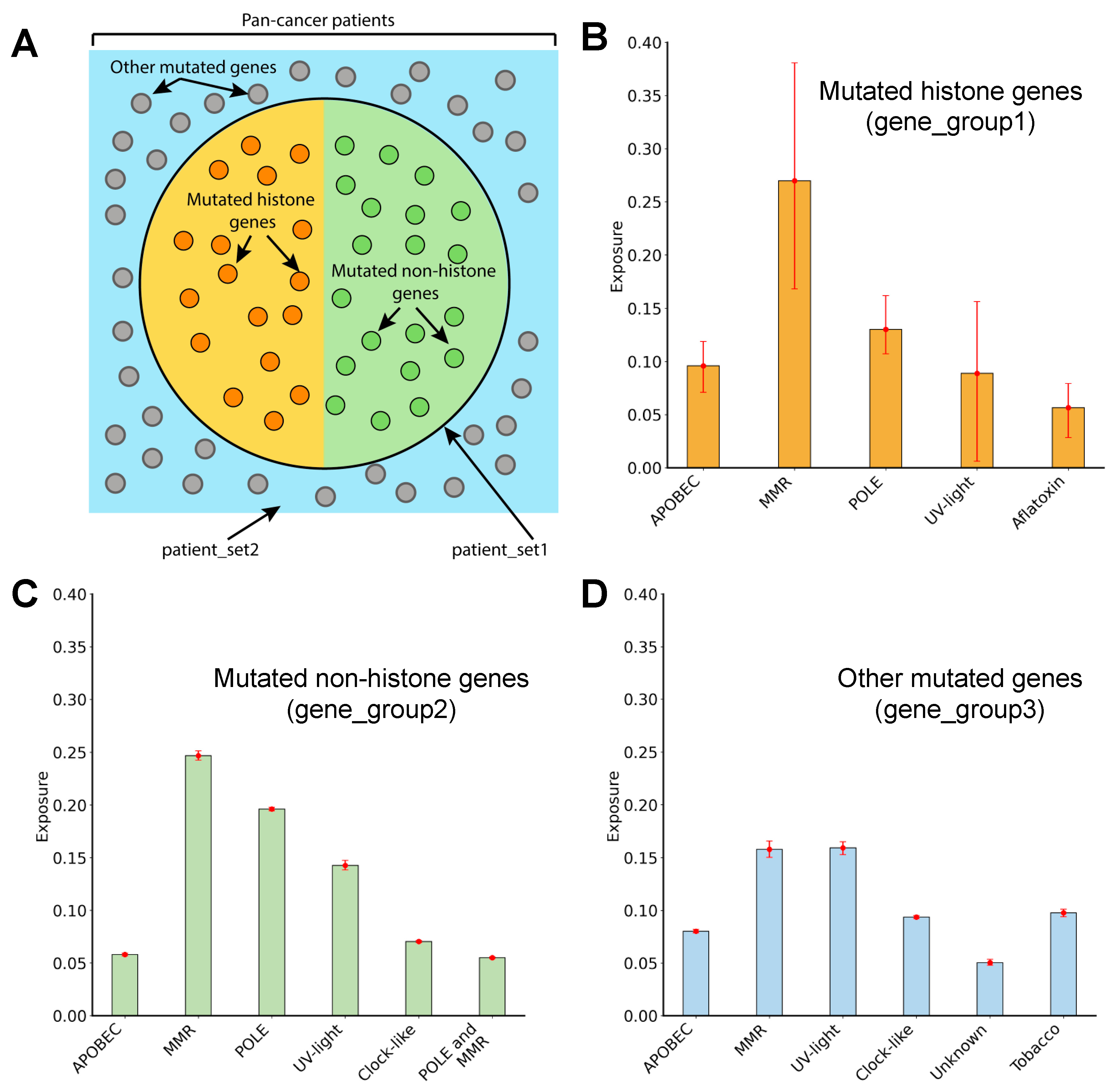
3.6. Mutagenic Processes Shaping the Mutational Landscape of Histone Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, B.R.; Feng, H.; Kale, S.; Fox, T.; Khant, H.; de Val, N.; Ghirlando, R.; Panchenko, A.R.; Bai, Y. Distinct Structures and Dynamics of Chromatosomes with Different Human Linker Histone Isoforms. Mol. Cell 2021, 81, 166–182.e6. [Google Scholar] [CrossRef]
- Nacev, B.A.; Feng, L.; Bagert, J.D.; Lemiesz, A.E.; Gao, J.; Soshnev, A.A.; Kundra, R.; Schultz, N.; Muir, T.W.; Allis, C.D. The expanding landscape of ‘oncohistone’ mutations in human cancers. Nature 2019, 567, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.L.; Bele, A.; Small, E.C.; Will, C.M.; Nabet, B.; Oyer, J.A.; Huang, X.; Ghosh, R.P.; Grzybowski, A.T.; Yu, T.; et al. A Mutation in Histone H2B Represents a New Class of Oncogenic Driver. Cancer Discov. 2019, 9, 1438–1451. [Google Scholar] [CrossRef]
- Bagert, J.D.; Mitchener, M.M.; Patriotis, A.L.; Dul, B.E.; Wojcik, F.; Nacev, B.A.; Feng, L.; Allis, C.D.; Muir, T.W. Oncohistone mutations enhance chromatin remodeling and alter cell fates. Nat. Chem. Biol. 2021, 17, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Espiritu, D.; Gribkova, A.K.; Gupta, S.; Shaytan, A.K.; Panchenko, A.R. Molecular Mechanisms of Oncogenesis through the Lens of Nucleosomes and Histones. J. Phys. Chem. B 2021, 125, 3963–3976. [Google Scholar] [CrossRef]
- Khazaei, S.; Chen, C.C.L.; Andrade, A.F.; Kabir, N.; Azarafshar, P.; Morcos, S.M.; Franca, J.A.; Lopes, M.; Lund, P.J.; Danieau, G.; et al. Single substitution in H3.3G34 alters DNMT3A recruitment to cause progressive neurodegeneration. Cell 2023, 186, 1162–1178.e20. [Google Scholar] [CrossRef]
- Amgalan, B.; Wojtowicz, D.; Kim, Y.A.; Przytycka, T.M. Influence network model uncovers relations between biological processes and mutational signatures. Genome Med. 2023, 15, 15. [Google Scholar] [CrossRef] [PubMed]
- Petljak, M.; Dananberg, A.; Chu, K.; Bergstrom, E.N.; Striepen, J.; von Morgen, P.; Chen, Y.; Shah, H.; Sale, J.E.; Alexandrov, L.B.; et al. Mechanisms of APOBEC3 mutagenesis in human cancer cells. Nature 2022, 607, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Bulock, C.R.; Xing, X.; Shcherbakova, P.V. DNA polymerase delta proofreads errors made by DNA polymerase epsilon. Proc. Natl. Acad. Sci. USA 2020, 117, 6035–6041. [Google Scholar] [CrossRef] [Green Version]
- Ostroverkhova, D.; Przytycka, T.M.; Panchenko, A.R. Cancer driver mutations: Predictions and reality. Trends Mol. Med. 2023, 29, 554–566. [Google Scholar] [CrossRef]
- Francioli, L.C.; Polak, P.P.; Koren, A.; Menelaou, A.; Chun, S.; Renkens, I.; Genome of the Netherlands Consortium; van Duijn, C.M.; Swertz, M.; Wijmenga, C.; et al. Genome-wide patterns and properties of de novo mutations in humans. Nat. Genet. 2015, 47, 822–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seal, R.L.; Denny, P.; Bruford, E.A.; Gribkova, A.K.; Landsman, D.; Marzluff, W.F.; McAndrews, M.; Panchenko, A.R.; Shaytan, A.K.; Talbert, P.B. A standardized nomenclature for mammalian histone genes. Epigenet. Chromatin 2022, 15, 34. [Google Scholar] [CrossRef] [PubMed]
- Dominski, Z.; Marzluff, W.F. Formation of the 3′ end of histone mRNA: Getting closer to the end. Gene 2007, 396, 373–390. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.K.; Kabbaj, M.H.; Paik, J.; Gunjan, A. Histone levels are regulated by phosphorylation and ubiquitylation-dependent proteolysis. Nat. Cell Biol. 2009, 11, 925–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rattray, A.M.; Muller, B. The control of histone gene expression. Biochem. Soc. Trans. 2012, 40, 880–885. [Google Scholar] [CrossRef] [Green Version]
- Duronio, R.J.; Marzluff, W.F. Coordinating cell cycle-regulated histone gene expression through assembly and function of the Histone Locus Body. RNA Biol. 2017, 14, 726–738. [Google Scholar] [CrossRef] [Green Version]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef]
- Tweedie, S.; Braschi, B.; Gray, K.; Jones, T.E.M.; Seal, R.L.; Yates, B.; Bruford, E.A. Genenames.org: The HGNC and VGNC resources in 2021. Nucleic Acids Res. 2021, 49, D939–D946. [Google Scholar] [CrossRef]
- Draizen, E.J.; Shaytan, A.K.; Marino-Ramirez, L.; Talbert, P.B.; Landsman, D.; Panchenko, A.R. HistoneDB 2.0: A histone database with variants—An integrated resource to explore histones and their variants. Database 2016, 2016, baw014. [Google Scholar] [CrossRef] [Green Version]
- Pruitt, K.D.; Harrow, J.; Harte, R.A.; Wallin, C.; Diekhans, M.; Maglott, D.R.; Searle, S.; Farrell, C.M.; Loveland, J.E.; Ruef, B.J.; et al. The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genome Res. 2009, 19, 1316–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef]
- UniProt, C. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Adery, C.A.H. A Simplified Monte Carlo Significance Test Procedure. J. R. Stat. Soc. Ser. B 1968, 30, 582–598. [Google Scholar]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.L.; Rappailles, A.; Duquenne, L.; Huvet, M.; Guilbaud, G.; Farinelli, L.; Audit, B.; d’Aubenton-Carafa, Y.; Arneodo, A.; Hyrien, O.; et al. Impact of replication timing on non-CpG and CpG substitution rates in mammalian genomes. Genome Res. 2010, 20, 447–457. [Google Scholar] [CrossRef] [Green Version]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Malladi, V.S.; Strattan, J.S.; Hitz, B.C.; Gabdank, I.; Narayanan, A.K.; Ho, M.; Lee, B.T.; et al. ENCODE data at the ENCODE portal. Nucleic Acids Res. 2016, 44, D726–D732. [Google Scholar] [CrossRef] [Green Version]
- Amatori, S.; Tavolaro, S.; Gambardella, S.; Fanelli, M. The dark side of histones: Genomic organization and role of oncohistones in cancer. Clin. Epigenet. 2021, 13, 71. [Google Scholar] [CrossRef]
- Goncearenco, A.; Rager, S.L.; Li, M.; Sang, Q.X.; Rogozin, I.B.; Panchenko, A.R. Exploring background mutational processes to decipher cancer genetic heterogeneity. Nucleic Acids Res. 2017, 45, W514–W522. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Beare, D.; Gunasekaran, P.; Leung, K.; Bindal, N.; Boutselakis, H.; Ding, M.; Bamford, S.; Cole, C.; Ward, S.; et al. COSMIC: Exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015, 43, D805–D811. [Google Scholar] [CrossRef] [Green Version]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusufova, N.; Kloetgen, A.; Teater, M.; Osunsade, A.; Camarillo, J.M.; Chin, C.R.; Doane, A.S.; Venters, B.J.; Portillo-Ledesma, S.; Conway, J.; et al. Histone H1 loss drives lymphoma by disrupting 3D chromatin architecture. Nature 2021, 589, 299–305. [Google Scholar] [CrossRef]
- Okosun, J.; Bodor, C.; Wang, J.; Araf, S.; Yang, C.Y.; Pan, C.; Boller, S.; Cittaro, D.; Bozek, M.; Iqbal, S.; et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat. Genet. 2014, 46, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Behjati, S.; Tarpey, P.S.; Presneau, N.; Scheipl, S.; Pillay, N.; Van Loo, P.; Wedge, D.C.; Cooke, S.L.; Gundem, G.; Davies, H.; et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet. 2013, 45, 1479–1482. [Google Scholar] [CrossRef]
- Brown, A.L.; Li, M.; Goncearenco, A.; Panchenko, A.R. Finding driver mutations in cancer: Elucidating the role of background mutational processes. PLoS Comput. Biol. 2019, 15, e1006981. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Bellone, S.; Lopez, S.; Thakral, D.; Schwab, C.; English, D.P.; Black, J.; Cocco, E.; Choi, J.; Zammataro, L.; et al. Mutational landscape of uterine and ovarian carcinosarcomas implicates histone genes in epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2016, 113, 12238–12243. [Google Scholar] [CrossRef] [Green Version]
- Polak, P.; Karlic, R.; Koren, A.; Thurman, R.; Sandstrom, R.; Lawrence, M.; Reynolds, A.; Rynes, E.; Vlahovicek, K.; Stamatoyannopoulos, J.A.; et al. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature 2015, 518, 360–364. [Google Scholar] [CrossRef] [Green Version]
- Marino-Ramirez, L.; Jordan, I.K.; Landsman, D. Multiple independent evolutionary solutions to core histone gene regulation. Genome Biol. 2006, 7, R122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, L.J.; Jensen, T.S.; de Lichtenberg, U.; Brunak, S.; Bork, P. Co-evolution of transcriptional and post-translational cell-cycle regulation. Nature 2006, 443, 594–597. [Google Scholar] [CrossRef]
- Soshnev, A.A.; Allis, C.D.; Cesarman, E.; Melnick, A.M. Histone H1 Mutations in Lymphoma: A Link(er) between Chromatin Organization, Developmental Reprogramming, and Cancer. Cancer Res. 2021, 81, 6061–6070. [Google Scholar] [CrossRef]
- Saha, A.; Dalal, Y. A glitch in the snitch: The role of linker histone H1 in shaping the epigenome in normal and diseased cells. Open Biol. 2021, 11, 210124. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Lowe, B.R.; Yadav, R.K.; Henry, R.A.; Schreiner, P.; Matsuda, A.; Fernandez, A.G.; Finkelstein, D.; Campbell, M.; Kallappagoudar, S.; Jablonowski, C.M.; et al. Surprising phenotypic diversity of cancer-associated mutations of Gly 34 in the histone H3 tail. Elife 2021, 10, e65369. [Google Scholar] [CrossRef]
- Arimura, Y.; Ikura, M.; Fujita, R.; Noda, M.; Kobayashi, W.; Horikoshi, N.; Sun, J.; Shi, L.; Kusakabe, M.; Harata, M.; et al. Cancer-associated mutations of histones H2B, H3.1 and H2A.Z.1 affect the structure and stability of the nucleosome. Nucleic Acids Res. 2018, 46, 10007–10018. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Li, S.; Onufriev, A.; Landsman, D.; Panchenko, A.R. Binding of regulatory proteins to nucleosomes is modulated by dynamic histone tails. Nat. Commun. 2021, 12, 5280. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.A.; Boerlijst, M.C.; Cooke, J.; Smith, J.M. Evolution of genetic redundancy. Nature 1997, 388, 167–171. [Google Scholar] [CrossRef]
- Rose Li, Y.; Halliwill, K.D.; Adams, C.J.; Iyer, V.; Riva, L.; Mamunur, R.; Jen, K.Y.; Del Rosario, R.; Fredlund, E.; Hirst, G.; et al. Mutational signatures in tumours induced by high and low energy radiation in Trp53 deficient mice. Nat. Commun. 2020, 11, 394. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Protein | Type | Chromosome Location | q-Value | Cancer Type |
|---|---|---|---|---|---|
| H1-4 | H1.4 linker histone, cluster member | Canonical | Chr6 | 0.002 | Multiple myeloma |
| H2AC16 | H2A clustered histone 16 | Canonical | Chr7 | 0.004 | Squamous cell carcinoma |
| H1-8 | H1.8 linker histone | Variant | Chr3 | 0.006 | Squamous cell carcinoma |
| H1-5 | H1.5 linker histone, cluster member | Canonical | Chr6 | 0.009 | Squamous cell carcinoma |
| H2AC16 | H2A clustered histone 16 | Canonical | Chr7 | 0.049 | Adenocarcinoma |
| H4C4 | H4 clustered histone 4 | Canonical | Chr6 | 0.062 | Infiltrating duct carcinoma |
| H3C3 | H3 clustered histone 3 | Canonical | Chr6 | 0.072 | Infiltrating duct carcinoma |
| H1-2 | H1.2 linker histone, cluster member | Canonical | Chr6 | 0.099 | Hepatocellular carcinoma |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ostroverkhova, D.; Espiritu, D.; Aristizabal, M.J.; Panchenko, A.R. Leveraging Gene Redundancy to Find New Histone Drivers in Cancer. Cancers 2023, 15, 3437. https://doi.org/10.3390/cancers15133437
Ostroverkhova D, Espiritu D, Aristizabal MJ, Panchenko AR. Leveraging Gene Redundancy to Find New Histone Drivers in Cancer. Cancers. 2023; 15(13):3437. https://doi.org/10.3390/cancers15133437
Chicago/Turabian StyleOstroverkhova, Daria, Daniel Espiritu, Maria J. Aristizabal, and Anna R. Panchenko. 2023. "Leveraging Gene Redundancy to Find New Histone Drivers in Cancer" Cancers 15, no. 13: 3437. https://doi.org/10.3390/cancers15133437