
Alternative Lengthening of Telomeres in Pediatric High-Grade Glioma and Therapeutic Implications

{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Tumor Tissue Samples, Cell Culture and Reagents
2.2. Proliferation Assay
2.3. Western Blotting
2.4. Rolling C-Circle Amplification
2.5. TRAP Assay
2.6. hTERT Expression
2.7. IF-FISH Assay (Immunofluorescence—Fluorescence In Situ Hybridization)
2.8. Immunofluorescence
2.9. H3-3A Gene Sequencing
2.10. Whole Genome Sequencing
2.11. Orthotopic Implantation
2.12. Statistical Analyses
3. Results
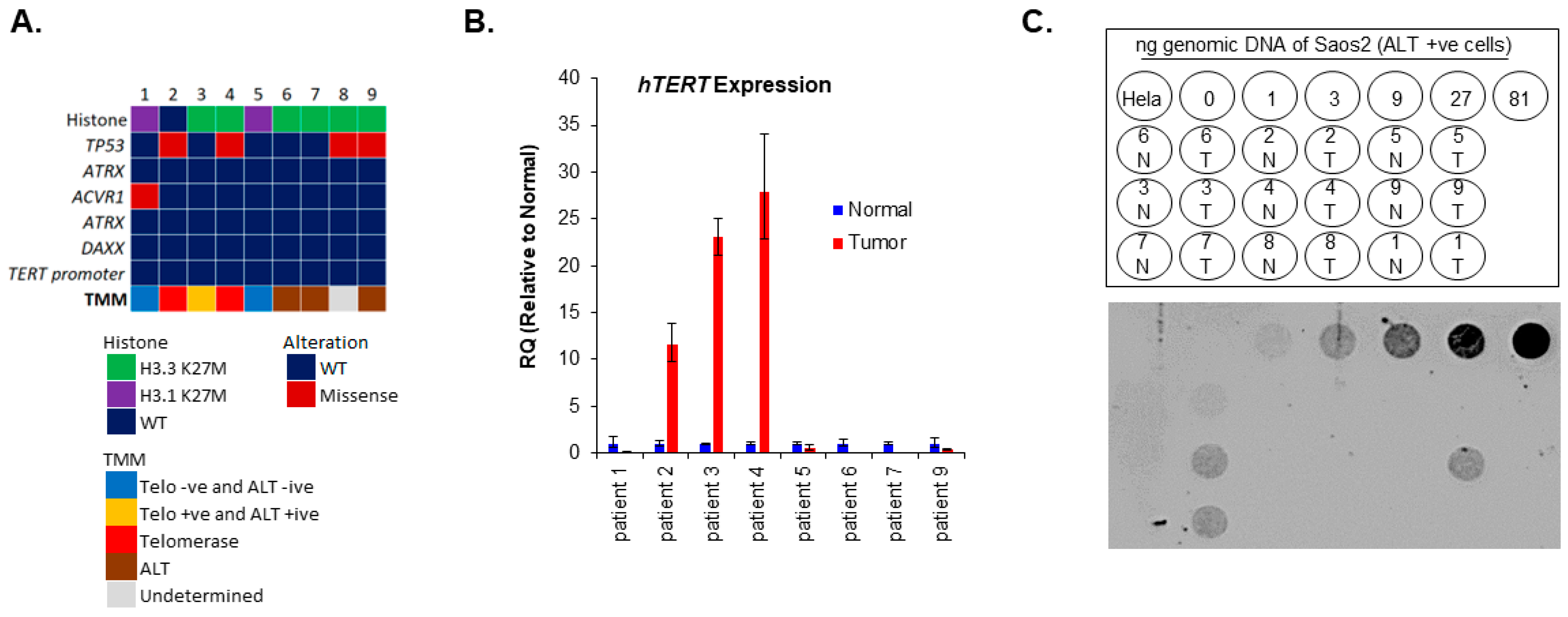
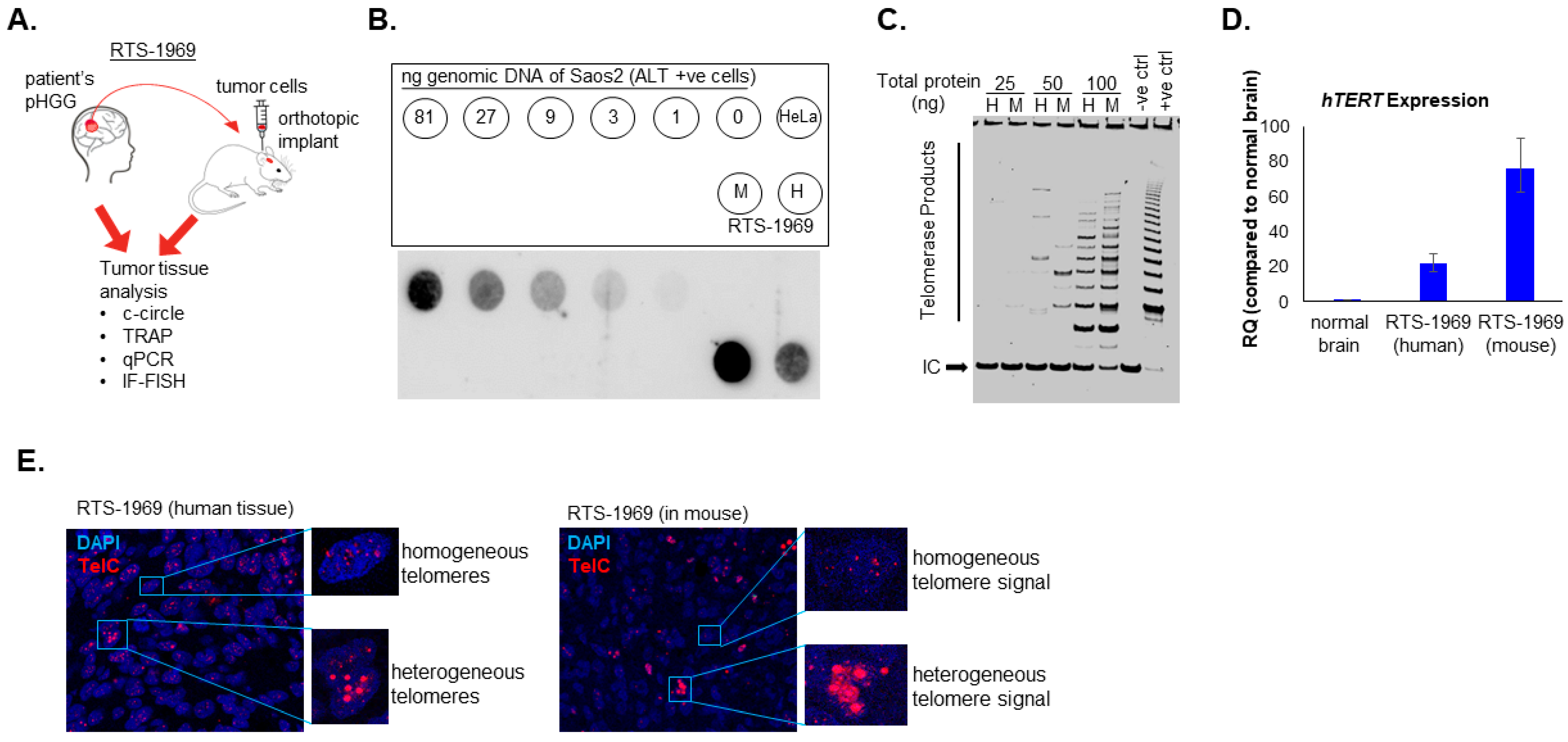
3.1. Presence of Both ALT and Telomerase in pHGG Patients
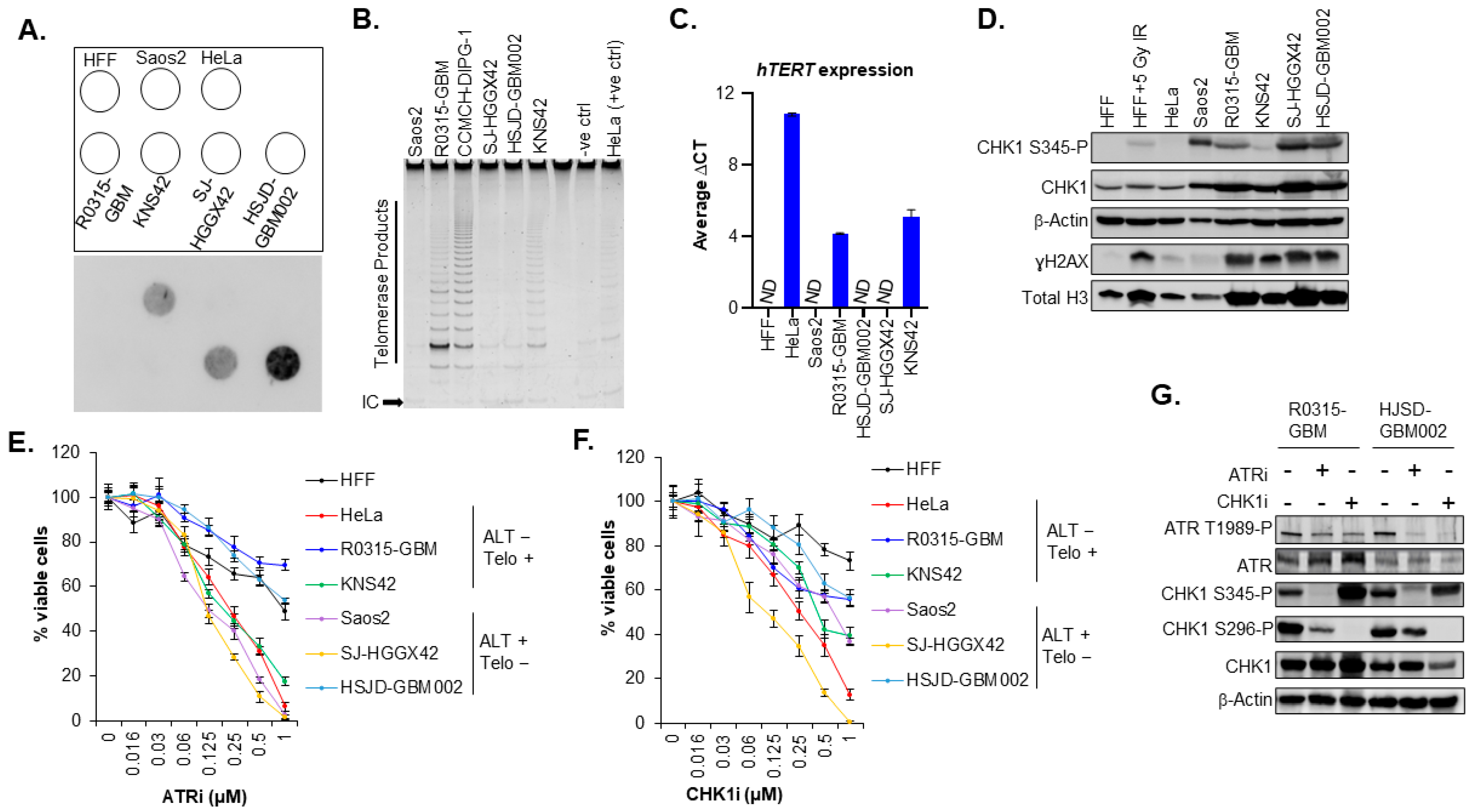
3.2. The Sensitivity of pHGG Cells to ATR and CHK1 Inhibitors Is Not Specific to the Presence of ALT Activity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cohen, K.J.; Pollack, I.F.; Zhou, T.; Buxton, A.; Holmes, E.J.; Burger, P.C.; Brat, D.J.; Rosenblum, M.K.; Hamilton, R.L.; Lavey, R.S.; et al. Temozolomide in the treatment of high-grade gliomas in children: A report from the Children’s Oncology Group. Neuro-Oncology 2011, 13, 317–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeNunzio, N.J.; Yock, T.I. Modern Radiotherapy for Pediatric Brain Tumors. Cancers 2020, 12, 1533. [Google Scholar] [CrossRef] [PubMed]
- Fangusaro, J. Pediatric high grade glioma: A review and update on tumor clinical characteristics and biology. Front. Oncol. 2012, 2, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warren, K.E. Diffuse intrinsic pontine glioma: Poised for progress. Front. Oncol. 2012, 2, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, C.; Karajannis, M.A.; Jones, D.T.W.; Kieran, M.W.; Monje, M.; Baker, S.J.; Becher, O.J.; Cho, Y.J.; Gupta, N.; Hawkins, C.; et al. Pediatric high-grade glioma: Biologically and clinically in need of new thinking. Neuro-Oncology 2017, 19, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef]
- Shay, J.W.; Bacchetti, S. A survey of telomerase activity in human cancer. Eur. J. Cancer 1997, 33, 787–791. [Google Scholar] [CrossRef]
- Arita, H.; Narita, Y.; Fukushima, S.; Tateishi, K.; Matsushita, Y.; Yoshida, A.; Miyakita, Y.; Ohno, M.; Collins, V.P.; Kawahara, N.; et al. Upregulating mutations in the TERT promoter commonly occur in adult malignant gliomas and are strongly associated with total 1p19q loss. Acta Neuropathol. 2013, 126, 267–276. [Google Scholar] [CrossRef]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A., Jr.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef] [Green Version]
- Koelsche, C.; Sahm, F.; Capper, D.; Reuss, D.; Sturm, D.; Jones, D.T.; Kool, M.; Northcott, P.A.; Wiestler, B.; Bohmer, K.; et al. Distribution of TERT promoter mutations in pediatric and adult tumors of the nervous system. Acta Neuropathol. 2013, 126, 907–915. [Google Scholar] [CrossRef] [Green Version]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Boldrini, L.; Pistolesi, S.; Gisfredi, S.; Ursino, S.; Ali, G.; Pieracci, N.; Basolo, F.; Parenti, G.; Fontanini, G. Telomerase activity and hTERT mRNA expression in glial tumors. Int. J. Oncol. 2006, 28, 1555–1560. [Google Scholar] [CrossRef]
- Langford, L.A.; Piatyszek, M.A.; Xu, R.; Schold, S.C., Jr.; Shay, J.W. Telomerase activity in human brain tumours. Lancet 1995, 346, 1267–1268. [Google Scholar] [CrossRef]
- Maes, L.; Van Neste, L.; Van Damme, K.; Kalala, J.P.; De Ridder, L.; Bekaert, S.; Cornelissen, M. Relation between telomerase activity, hTERT and telomere length for intracranial tumours. Oncol. Rep. 2007, 18, 1571–1576. [Google Scholar] [CrossRef] [Green Version]
- Heaphy, C.M.; Subhawong, A.P.; Hong, S.M.; Goggins, M.G.; Montgomery, E.A.; Gabrielson, E.; Netto, G.J.; Epstein, J.I.; Lotan, T.L.; Westra, W.H.; et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am. J. Pathol. 2011, 179, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Henson, J.D.; Reddel, R.R. Assaying and investigating Alternative Lengthening of Telomeres activity in human cells and cancers. FEBS Lett. 2010, 584, 3800–3811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeager, T.R.; Neumann, A.A.; Englezou, A.; Huschtscha, L.I.; Noble, J.R.; Reddel, R.R. Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 1999, 59, 4175–4179. [Google Scholar] [PubMed]
- Cesare, A.J.; Griffith, J.D. Telomeric DNA in ALT cells is characterized by free telomeric circles and heterogeneous t-loops. Mol. Cell. Biol. 2004, 24, 9948–9957. [Google Scholar] [CrossRef] [Green Version]
- Henson, J.D.; Cao, Y.; Huschtscha, L.I.; Chang, A.C.; Au, A.Y.; Pickett, H.A.; Reddel, R.R. DNA C-circles are specific and quantifiable markers of alternative-lengthening-of-telomeres activity. Nat. Biotechnol. 2009, 27, 1181–1185. [Google Scholar] [CrossRef]
- Sobinoff, A.P.; Pickett, H.A. Alternative Lengthening of Telomeres: DNA Repair Pathways Converge. Trends Genet. 2017, 33, 921–932. [Google Scholar] [CrossRef]
- Dorris, K.; Sobo, M.; Onar-Thomas, A.; Panditharatna, E.; Stevenson, C.B.; Gardner, S.L.; Dewire, M.D.; Pierson, C.R.; Olshefski, R.; Rempel, S.A.; et al. Prognostic significance of telomere maintenance mechanisms in pediatric high-grade gliomas. J. Neurooncol. 2014, 117, 67–76. [Google Scholar] [CrossRef]
- MacKenzie, D., Jr.; Watters, A.K.; To, J.T.; Young, M.W.; Muratori, J.; Wilkoff, M.H.; Abraham, R.G.; Plummer, M.M.; Zhang, D. ALT Positivity in Human Cancers: Prevalence and Clinical Insights. Cancers 2021, 13, 2384. [Google Scholar] [CrossRef]
- McDonald, K.L.; McDonnell, J.; Muntoni, A.; Henson, J.D.; Hegi, M.E.; von Deimling, A.; Wheeler, H.R.; Cook, R.J.; Biggs, M.T.; Little, N.S.; et al. Presence of alternative lengthening of telomeres mechanism in patients with glioblastoma identifies a less aggressive tumor type with longer survival. J. Neuropathol. Exp. Neurol. 2010, 69, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Henson, J.D.; Hannay, J.A.; McCarthy, S.W.; Royds, J.A.; Yeager, T.R.; Robinson, R.A.; Wharton, S.B.; Jellinek, D.A.; Arbuckle, S.M.; Yoo, J.; et al. A robust assay for alternative lengthening of telomeres in tumors shows the significance of alternative lengthening of telomeres in sarcomas and astrocytomas. Clin. Cancer Res. 2005, 11, 217–225. [Google Scholar] [CrossRef]
- Hakin-Smith, V.; Jellinek, D.A.; Levy, D.; Carroll, T.; Teo, M.; Timperley, W.R.; McKay, M.J.; Reddel, R.R.; Royds, J.A. Alternative lengthening of telomeres and survival in patients with glioblastoma multiforme. Lancet 2003, 361, 836–838. [Google Scholar] [CrossRef]
- Stundon, J.L.; Ijaz, H.; Gaonkar, K.S.; Kaufman, R.S.; Jin, R.; Karras, A.; Vaksman, Z.; Kim, J.; Corbett, R.J.; Lueder, M.R.; et al. ALT in Pediatric High-Grade Gliomas Can Occur without ATRX Mutation and is Enriched in Patients with Pathogenic Germline MMR Variants. Neuro-Oncology 2022, noac278. [Google Scholar] [CrossRef] [PubMed]
- Heaphy, C.M.; de Wilde, R.F.; Jiao, Y.; Klein, A.P.; Edil, B.H.; Shi, C.; Bettegowda, C.; Rodriguez, F.J.; Eberhart, C.G.; Hebbar, S.; et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science 2011, 333, 425. [Google Scholar] [CrossRef] [Green Version]
- Brosnan-Cashman, J.A.; Yuan, M.; Graham, M.K.; Rizzo, A.J.; Myers, K.M.; Davis, C.; Zhang, R.; Esopi, D.M.; Raabe, E.H.; Eberhart, C.G.; et al. ATRX loss induces multiple hallmarks of the alternative lengthening of telomeres (ALT) phenotype in human glioma cell lines in a cell line-specific manner. PLoS ONE 2018, 13, e0204159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udugama, M.; Hii, L.; Garvie, A.; Cervini, M.; Vinod, B.; Chan, F.L.; Das, P.P.; Mann, J.R.; Collas, P.; Voon, H.P.J.; et al. Mutations inhibiting KDM4B drive ALT activation in ATRX-mutated glioblastomas. Nat. Commun. 2021, 12, 2584. [Google Scholar] [CrossRef]
- de Nonneville, A.; Reddel, R.R. Alternative lengthening of telomeres is not synonymous with mutations in ATRX/DAXX. Nat. Commun. 2021, 12, 1552. [Google Scholar] [CrossRef]
- Pezzolo, A.; Pistorio, A.; Gambini, C.; Haupt, R.; Ferraro, M.; Erminio, G.; De Bernardi, B.; Garaventa, A.; Pistoia, V. Intratumoral diversity of telomere length in individual neuroblastoma tumors. Oncotarget 2015, 6, 7493–7503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gocha, A.R.; Nuovo, G.; Iwenofu, O.H.; Groden, J. Human sarcomas are mosaic for telomerase-dependent and telomerase-independent telomere maintenance mechanisms: Implications for telomere-based therapies. Am. J. Pathol. 2013, 182, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, B.; Peng, M.; Song, Q. The co-expression of telomerase and ALT pathway in human breast cancer tissues. Tumour Biol. 2014, 35, 4087–4093. [Google Scholar] [CrossRef]
- Hoffman, L.M.; DeWire, M.; Ryall, S.; Buczkowicz, P.; Leach, J.; Miles, L.; Ramani, A.; Brudno, M.; Kumar, S.S.; Drissi, R.; et al. Spatial genomic heterogeneity in diffuse intrinsic pontine and midline high-grade glioma: Implications for diagnostic biopsy and targeted therapeutics. Acta Neuropathol. Commun. 2016, 4, 1. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.S.; Sengupta, S.; Lee, K.; Hura, N.; Fuller, C.; DeWire, M.; Stevenson, C.B.; Fouladi, M.; Drissi, R. BMI-1 is a potential therapeutic target in diffuse intrinsic pontine glioma. Oncotarget 2017, 8, 62962–62975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monje, M.; Mitra, S.S.; Freret, M.E.; Raveh, T.B.; Kim, J.; Masek, M.; Attema, J.L.; Li, G.; Haddix, T.; Edwards, M.S.; et al. Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc. Natl. Acad. Sci. USA 2011, 108, 4453–4458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengupta, S.; Senthil Kumar, S.; Bondra, K.; Sobo, M.; Mo, X.; Drissi, R. Limitations of radiosensitization by direct telomerase inhibition to treat high-risk medulloblastoma. Front. Oncol. 2023, 13, 1104670. [Google Scholar] [CrossRef] [PubMed]
- Senthil Kumar, S.; Sengupta, S.; Zhu, X.; Mishra, D.K.; Phoenix, T.; Dyer, L.; Fuller, C.; Stevenson, C.B.; DeWire, M.; Fouladi, M.; et al. Diffuse Intrinsic Pontine Glioma Cells Are Vulnerable to Mitotic Abnormalities Associated with BMI-1 Modulation. Mol. Cancer Res. 2020, 18, 1711–1723. [Google Scholar] [CrossRef]
- Minasi, S.; Baldi, C.; Gianno, F.; Antonelli, M.; Buccoliero, A.M.; Pietsch, T.; Massimino, M.; Buttarelli, F.R. Alternative lengthening of telomeres in molecular subgroups of paediatric high-grade glioma. Childs Nerv. Syst. 2021, 37, 809–818. [Google Scholar] [CrossRef]
- Mackay, A.; Burford, A.; Molinari, V.; Jones, D.T.W.; Izquierdo, E.; Brouwer-Visser, J.; Giangaspero, F.; Haberler, C.; Pietsch, T.; Jacques, T.S.; et al. Molecular, Pathological, Radiological, and Immune Profiling of Non-brainstem Pediatric High-Grade Glioma from the HERBY Phase II Randomized Trial. Cancer Cell 2018, 33, 829–842.e825. [Google Scholar] [CrossRef] [Green Version]
- Flynn, R.L.; Cox, K.E.; Jeitany, M.; Wakimoto, H.; Bryll, A.R.; Ganem, N.J.; Bersani, F.; Pineda, J.R.; Suva, M.L.; Benes, C.H.; et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 2015, 347, 273–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeg, K.I.; Chung, I.; Bauer, C.; Rippe, K. Cancer Cells with Alternative Lengthening of Telomeres Do Not Display a General Hypersensitivity to ATR Inhibition. Front. Oncol. 2016, 6, 186. [Google Scholar] [CrossRef] [Green Version]
- Laroche-Clary, A.; Chaire, V.; Verbeke, S.; Algeo, M.P.; Malykh, A.; Le Loarer, F.; Italiano, A. ATR Inhibition Broadly Sensitizes Soft-Tissue Sarcoma Cells to Chemotherapy Independent of Alternative Lengthening Telomere (ALT) Status. Sci. Rep. 2020, 10, 7488. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.T.; Chan, F.L.; JD, R.M.; Udugama, M.; Mayne, L.; Collas, P.; Mann, J.R.; Wong, L.H. CHK1-driven histone H3.3 serine 31 phosphorylation is important for chromatin maintenance and cell survival in human ALT cancer cells. Nucleic Acids Res. 2015, 43, 2603–2614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, J.; Johannessen, T.C.; Ohba, S.; Chow, T.T.; Jones, L.; Pandita, A.; Pieper, R.O. Mutant IDH1 Cooperates with ATRX Loss to Drive the Alternative Lengthening of Telomere Phenotype in Glioma. Cancer Res. 2018, 78, 2966–2977. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, R.J.; Arnoult, N.; Lackner, D.H.; Oganesian, L.; Haggblom, C.; Corpet, A.; Almouzni, G.; Karlseder, J. Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat. Struct. Mol. Biol. 2014, 21, 167–174. [Google Scholar] [CrossRef]
- Onitake, Y.; Hiyama, E.; Kamei, N.; Yamaoka, H.; Sueda, T.; Hiyama, K. Telomere biology in neuroblastoma: Telomere binding proteins and alternative strengthening of telomeres. J. Pediatr. Surg. 2009, 44, 2258–2266. [Google Scholar] [CrossRef]
- Lawlor, R.T.; Veronese, N.; Pea, A.; Nottegar, A.; Smith, L.; Pilati, C.; Demurtas, J.; Fassan, M.; Cheng, L.; Luchini, C. Alternative lengthening of telomeres (ALT) influences survival in soft tissue sarcomas: A systematic review with meta-analysis. BMC Cancer 2019, 19, 232. [Google Scholar] [CrossRef] [Green Version]
- Mangerel, J.; Price, A.; Castelo-Branco, P.; Brzezinski, J.; Buczkowicz, P.; Rakopoulos, P.; Merino, D.; Baskin, B.; Wasserman, J.; Mistry, M.; et al. Alternative lengthening of telomeres is enriched in, and impacts survival of TP53 mutant pediatric malignant brain tumors. Acta Neuropathol. 2014, 128, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Liu, I.; Jiang, L.; Samuelsson, E.R.; Marco Salas, S.; Beck, A.; Hack, O.A.; Jeong, D.; Shaw, M.L.; Englinger, B.; LaBelle, J.; et al. The landscape of tumor cell states and spatial organization in H3-K27M mutant diffuse midline glioma across age and location. Nat. Genet. 2022, 54, 1881–1894. [Google Scholar] [CrossRef]
- Vallero, S.G.; Bertero, L.; Morana, G.; Sciortino, P.; Bertin, D.; Mussano, A.; Ricci, F.S.; Peretta, P.; Fagioli, F. Pediatric diffuse midline glioma H3K27- altered: A complex clinical and biological landscape behind a neatly defined tumor type. Front. Oncol. 2022, 12, 1082062. [Google Scholar] [CrossRef] [PubMed]
- Dagg, R.A.; Pickett, H.A.; Neumann, A.A.; Napier, C.E.; Henson, J.D.; Teber, E.T.; Arthur, J.W.; Reynolds, C.P.; Murray, J.; Haber, M.; et al. Extensive Proliferation of Human Cancer Cells with Ever-Shorter Telomeres. Cell Rep. 2017, 19, 2544–2556. [Google Scholar] [CrossRef] [Green Version]
- Viceconte, N.; Dheur, M.S.; Majerova, E.; Pierreux, C.E.; Baurain, J.F.; van Baren, N.; Decottignies, A. Highly Aggressive Metastatic Melanoma Cells Unable to Maintain Telomere Length. Cell Rep. 2017, 19, 2529–2543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Royds, J.A.; Al Nadaf, S.; Wiles, A.K.; Chen, Y.J.; Ahn, A.; Shaw, A.; Bowie, S.; Lam, F.; Baguley, B.C.; Braithwaite, A.W.; et al. The CDKN2A G500 allele is more frequent in GBM patients with no defined telomere maintenance mechanism tumors and is associated with poorer survival. PLoS ONE 2011, 6, e26737. [Google Scholar] [CrossRef]
- Thompson, P.A.; Drissi, R.; Muscal, J.A.; Panditharatna, E.; Fouladi, M.; Ingle, A.M.; Ahern, C.H.; Reid, J.M.; Lin, T.; Weigel, B.J.; et al. A phase I trial of imetelstat in children with refractory or recurrent solid tumors: A Children’s Oncology Group Phase I Consortium Study (ADVL1112). Clin. Cancer Res. 2013, 19, 6578–6584. [Google Scholar] [CrossRef] [Green Version]
- Asai, A.; Oshima, Y.; Yamamoto, Y.; Uochi, T.A.; Kusaka, H.; Akinaga, S.; Yamashita, Y.; Pongracz, K.; Pruzan, R.; Wunder, E.; et al. A novel telomerase template antagonist (GRN163) as a potential anticancer agent. Cancer Res. 2003, 63, 3931–3939. [Google Scholar] [PubMed]
- Silvestre, D.C.; Pineda, J.R.; Hoffschir, F.; Studler, J.M.; Mouthon, M.A.; Pflumio, F.; Junier, M.P.; Chneiweiss, H.; Boussin, F.D. Alternative lengthening of telomeres in human glioma stem cells. Stem Cells 2011, 29, 440–451. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Umaru, B.; Sengupta, S.; Senthil Kumar, S.; Drissi, R. Alternative Lengthening of Telomeres in Pediatric High-Grade Glioma and Therapeutic Implications. Cancers 2023, 15, 3070. https://doi.org/10.3390/cancers15123070
Umaru B, Sengupta S, Senthil Kumar S, Drissi R. Alternative Lengthening of Telomeres in Pediatric High-Grade Glioma and Therapeutic Implications. Cancers. 2023; 15(12):3070. https://doi.org/10.3390/cancers15123070
Chicago/Turabian StyleUmaru, Banlanjo, Satarupa Sengupta, Shiva Senthil Kumar, and Rachid Drissi. 2023. "Alternative Lengthening of Telomeres in Pediatric High-Grade Glioma and Therapeutic Implications" Cancers 15, no. 12: 3070. https://doi.org/10.3390/cancers15123070