
EGFR-Tyrosine Kinase Inhibitors Induced Activation of the Autocrine CXCL10/CXCR3 Pathway through Crosstalk between the Tumor and the Microenvironment in EGFR-Mutant Lung Cancer
 , ,
, ,
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
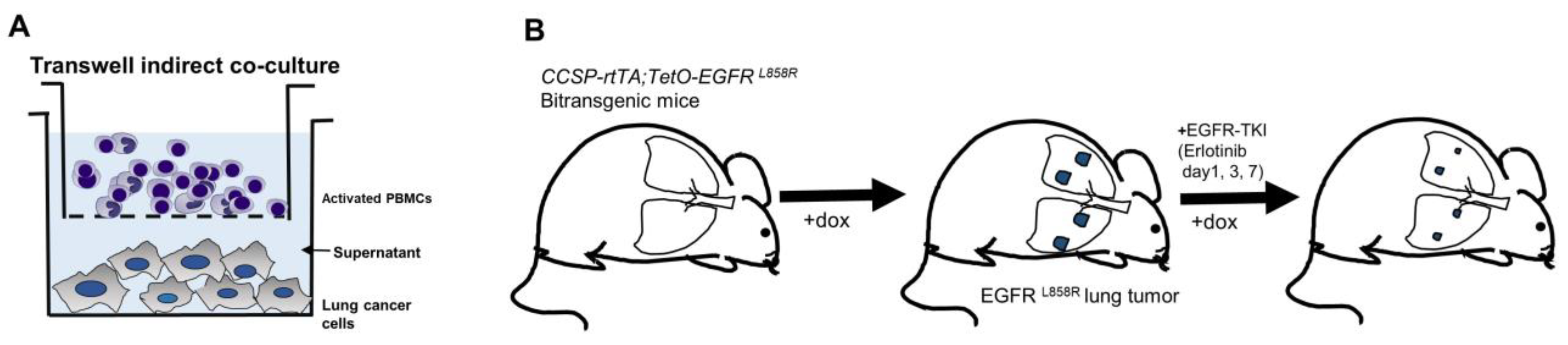
2.2. Preparation of Human PBMC Cocultures
2.3. Tyrosine Kinase Inhibitors and Cell Treatments
2.4. siRNA Treatment and CXCL10 Knockdown Validation
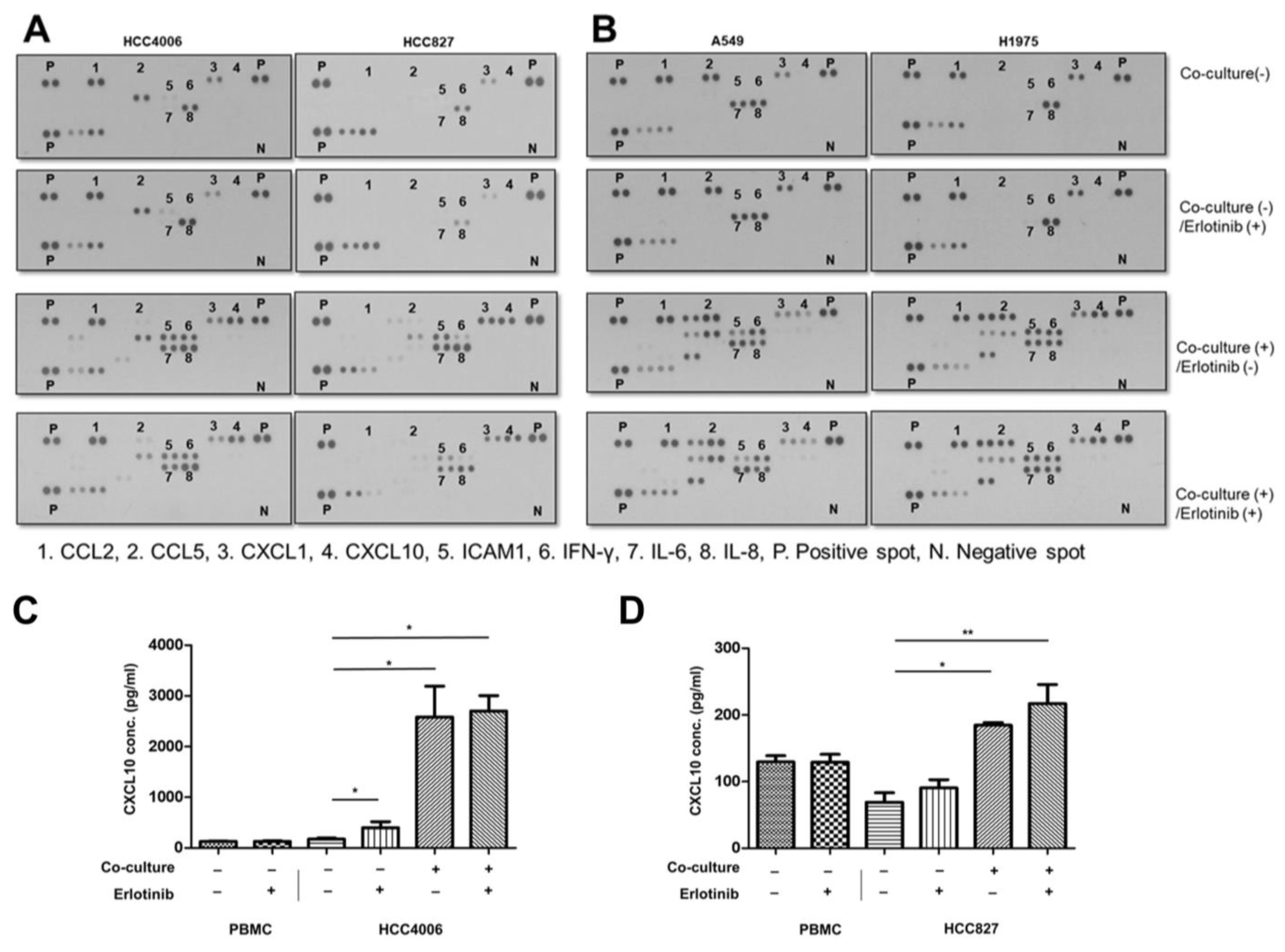
2.5. Cytokine Array Analysis
2.6. ELISA Analysis
2.7. Western Blotting
2.8. Immunofluorescence Staining
2.9. Real-Time RT-PCR
2.10. RT-PCR
2.11. Mice and Immunohistochemistry
2.12. Statistical Analysis
3. Results
3.1. Chemokine Screening in EGFR-Mutant Lung Cancer on EGFR-TKI Treatment
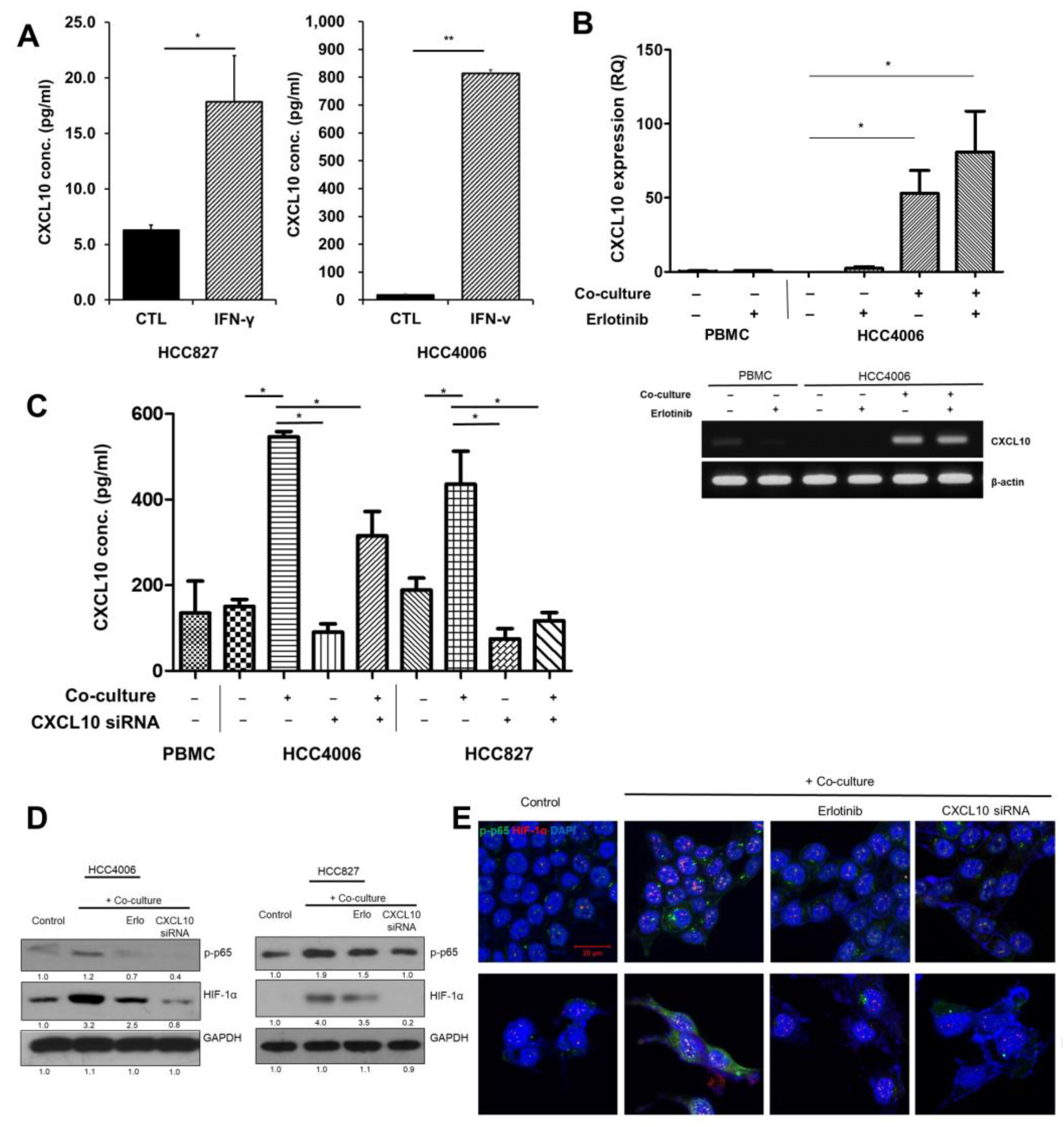
3.2. Effects of Oncogenic Src Phosphorylation by CXCL10 in EGFR-Mutant Lung Cancer
3.3. Autocrine Signaling by CXCL10 Affects Oncogenic Signaling
3.4. Effects of CXCL10 on the Oncogenic Pathway in a Transgenic Mouse Model of EGFR-Mutant Lung Cancer during EGFR-TKI Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, L.; Wang, C.; Li, S.; Bai, H.; Wang, J. Tumor immune microenvironment in epidermal growth factor receptor-mutated non-small cell lung cancer before and after epidermal growth factor receptor tyrosine kinase inhibitor treatment: A narrative review. Transl. Lung Cancer Res. 2021, 10, 3823–3839. [Google Scholar] [CrossRef] [PubMed]
- Busch, S.E.; Hanke, M.L.; Kargl, J.; Metz, H.E.; MacPherson, D.; Houghton, A.M. Lung Cancer Subtypes Generate Unique Immune Responses. J. Immunol. 2016, 197, 4493–4503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isomoto, K.; Haratani, K.; Hayashi, H.; Shimizu, S.; Tomida, S.; Niwa, T.; Yokoyama, T.; Fukuda, Y.; Chiba, Y.; Kato, R.; et al. Impact of EGFR-TKI Treatment on the Tumor Immune Microenvironment in EGFR Mutation-Positive Non-Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 2037–2046. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, E.; Togashi, Y.; Takeuchi, Y.; Shinya, S.; Tada, Y.; Kataoka, K.; Tane, K.; Sato, E.; Ishii, G.; Goto, K.; et al. Blockade of EGFR improves responsiveness to PD-1 blockade in EGFR-mutated non-small cell lung cancer. Sci. Immunol. 2020, 5, eaav3937. [Google Scholar] [CrossRef]
- Ayeni, D.; Miller, B.; Kuhlmann, A.; Ho, P.C.; Robles-Oteiza, C.; Gaefele, M.; Levy, S.; de Miguel, F.J.; Perry, C.; Guan, T.; et al. Tumor regression mediated by oncogene withdrawal or erlotinib stimulates infiltration of inflammatory immune cells in EGFR mutant lung tumors. J. Immunother. Cancer 2019, 7, 172. [Google Scholar] [CrossRef] [Green Version]
- Gurule, N.J.; McCoach, C.E.; Hinz, T.K.; Merrick, D.T.; Van Bokhoven, A.; Kim, J.; Patil, T.; Calhoun, J.; Nemenoff, R.A.; Tan, A.C.; et al. A tyrosine kinase inhibitor-induced interferon response positively associates with clinical response in EGFR-mutant lung cancer. NPJ Precis. Oncol. 2021, 5, 41. [Google Scholar] [CrossRef]
- Hsu, W.H.; Yang, J.C.; Mok, T.S.; Loong, H.H. Overview of current systemic management of EGFR-mutant NSCLC. Ann. Oncol. 2018, 29, i3–i9. [Google Scholar] [CrossRef]
- Oxnard, G.R. The cellular origins of drug resistance in cancer. Nat. Med. 2016, 22, 232–234. [Google Scholar] [CrossRef]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.E.; Krishnamurthy Radhakrishna, V.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y. Tumor microenvironment and cancer therapy resistance. Cancer Lett. 2016, 380, 205–215. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, J.; Bai, J.; Ren, J. Reverse of non-small cell lung cancer drug resistance induced by cancer-associated fibroblasts via a paracrine pathway. Cancer Sci. 2018, 109, 944–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malfitano, A.M.; Pisanti, S.; Napolitano, F.; Di Somma, S.; Martinelli, R.; Portella, G. Tumor-Associated Macrophage Status in Cancer Treatment. Cancers 2020, 12, 1987. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Guo, S.; Hibbert, J.M.; Jain, V.; Singh, N.; Wilson, N.O.; Stiles, J.K. CXCL10/IP-10 in infectious diseases pathogenesis and potential therapeutic implications. Cytokine Growth Factor Rev. 2011, 22, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Reschke, R.; Yu, J.; Flood, B.; Higgs, E.F.; Hatogai, K.; Gajewski, T.F. Immune cell and tumor cell-derived CXCL10 is indicative of immunotherapy response in metastatic melanoma. J. Immunother. Cancer 2021, 9, e003521. [Google Scholar] [CrossRef] [PubMed]
- Kawada, K.; Hosogi, H.; Sonoshita, M.; Sakashita, H.; Manabe, T.; Shimahara, Y.; Sakai, Y.; Takabayashi, A.; Oshima, M.; Taketo, M.M. Chemokine receptor CXCR3 promotes colon cancer metastasis to lymph nodes. Oncogene 2007, 26, 4679–4688. [Google Scholar] [CrossRef] [PubMed]
- Walser, T.C.; Rifat, S.; Ma, X.; Kundu, N.; Ward, C.; Goloubeva, O.; Johnson, M.G.; Medina, J.C.; Collins, T.L.; Fulton, A.M. Antagonism of CXCR3 inhibits lung metastasis in a murine model of metastatic breast cancer. Cancer Res. 2006, 66, 7701–7707. [Google Scholar] [CrossRef] [Green Version]
- Wightman, S.C.; Uppal, A.; Pitroda, S.P.; Ganai, S.; Burnette, B.; Stack, M.; Oshima, G.; Khan, S.; Huang, X.; Posner, M.C.; et al. Oncogenic CXCL10 signalling drives metastasis development and poor clinical outcome. Br. J. Cancer 2015, 113, 327–335. [Google Scholar] [CrossRef]
- Politi, K.; Zakowski, M.F.; Fan, P.D.; Schonfeld, E.A.; Pao, W.; Varmus, H.E. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev. 2006, 20, 1496–1510. [Google Scholar] [CrossRef] [Green Version]
- Bonacchi, A.; Romagnani, P.; Romanelli, R.G.; Efsen, E.; Annunziato, F.; Lasagni, L.; Francalanci, M.; Serio, M.; Laffi, G.; Pinzani, M.; et al. Signal transduction by the chemokine receptor CXCR3: Activation of Ras/ERK, Src, and phosphatidylinositol 3-kinase/Akt controls cell migration and proliferation in human vascular pericytes. J. Biol. Chem. 2001, 276, 9945–9954. [Google Scholar] [CrossRef] [Green Version]
- Passaro, A.; Janne, P.A.; Mok, T.; Peters, S. Overcoming therapy resistance in EGFR-mutant lung cancer. Nat. Cancer 2021, 2, 377–391. [Google Scholar] [CrossRef]
- Cabanos, H.F.; Hata, A.N. Emerging Insights into Targeted Therapy-Tolerant Persister Cells in Cancer. Cancers 2021, 13, 2666. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Liu, J. Immunological effect of tyrosine kinase inhibitors on the tumor immune environment in non-small cell lung cancer. Oncol. Lett. 2022, 23, 165. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Kwon, O.J.; Hong, Y.K.; Kim, J.H.; Solca, F.; Ha, S.J.; Soo, R.A.; Christensen, J.G.; Lee, J.H.; Cho, B.C. Activation of IL-6R/JAK1/STAT3 signaling induces de novo resistance to irreversible EGFR inhibitors in non-small cell lung cancer with T790M resistance mutation. Mol. Cancer Ther. 2012, 11, 2254–2264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernando, R.I.; Hamilton, D.H.; Dominguez, C.; David, J.M.; McCampbell, K.K.; Palena, C. IL-8 signaling is involved in resistance of lung carcinoma cells to erlotinib. Oncotarget 2016, 7, 42031–42044. [Google Scholar] [CrossRef] [Green Version]
- Becker, J.H.; Gao, Y.; Soucheray, M.; Pulido, I.; Kikuchi, E.; Rodriguez, M.L.; Gandhi, R.; Lafuente-Sanchis, A.; Aupi, M.; Alcacer Fernandez-Coronado, J.; et al. CXCR7 Reactivates ERK Signaling to Promote Resistance to EGFR Kinase Inhibitors in NSCLC. Cancer Res. 2019, 79, 4439–4452. [Google Scholar] [CrossRef]
- Jin, W.J.; Kim, B.; Kim, D.; Park Choo, H.Y.; Kim, H.H.; Ha, H.; Lee, Z.H. NF-kappaB signaling regulates cell-autonomous regulation of CXCL10 in breast cancer 4T1 cells. Exp. Mol. Med. 2017, 49, e295. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Ao, X.; Shen, Z.; Ao, L.; Wu, X.; Pu, C.; Guo, W.; Xing, W.; He, M.; Yuan, H.; et al. TNF-alpha augments CXCL10/CXCR3 axis activity to induce Epithelial-Mesenchymal Transition in colon cancer cell. Int. J. Biol. Sci. 2021, 17, 2683–2702. [Google Scholar] [CrossRef]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Sato, A.; Aoto, M.; Fukami, Y. c-Src phosphorylates epidermal growth factor receptor on tyrosine 845. Biochem. Biophys. Res. Commun. 1995, 215, 1078–1087. [Google Scholar] [CrossRef]
- Pan, Z.; Wang, K.; Wang, X.; Jia, Z.; Yang, Y.; Duan, Y.; Huang, L.; Wu, Z.X.; Zhang, J.Y.; Ding, X. Cholesterol promotes EGFR-TKIs resistance in NSCLC by inducing EGFR/Src/Erk/SP1 signaling-mediated ERRalpha re-expression. Mol. Cancer 2022, 21, 77. [Google Scholar] [CrossRef]
- Drube, S.; Stirnweiss, J.; Valkova, C.; Liebmann, C. Ligand-independent and EGF receptor-supported transactivation: Lessons from beta2-adrenergic receptor signalling. Cell. Signal. 2006, 18, 1633–1646. [Google Scholar] [CrossRef] [PubMed]
- Prenzel, N.; Zwick, E.; Leserer, M.; Ullrich, A. Tyrosine kinase signalling in breast cancer. Epidermal growth factor receptor: Convergence point for signal integration and diversification. Breast Cancer Res. 2000, 2, 184–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracht, J.W.P.; Karachaliou, N.; Berenguer, J.; Pedraz-Valdunciel, C.; Filipska, M.; Codony-Servat, C.; Codony-Servat, J.; Rosell, R. Osimertinib and pterostilbene in EGFR-mutation-positive non-small cell lung cancer (NSCLC). Int. J. Biol. Sci. 2019, 15, 2607–2614. [Google Scholar] [CrossRef] [Green Version]
- Ohmori, Y.; Hamilton, T.A. Cooperative interaction between interferon (IFN) stimulus response element and kappa B sequence motifs controls IFN gamma- and lipopolysaccharide-stimulated transcription from the murine IP-10 promoter. J. Biol. Chem. 1993, 268, 6677–6688. [Google Scholar] [CrossRef] [PubMed]
- Yeruva, S.; Ramadori, G.; Raddatz, D. NF-kappaB-dependent synergistic regulation of CXCL10 gene expression by IL-1beta and IFN-gamma in human intestinal epithelial cell lines. Int. J. Color. Dis. 2008, 23, 305–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bivona, T.G.; Hieronymus, H.; Parker, J.; Chang, K.; Taron, M.; Rosell, R.; Moonsamy, P.; Dahlman, K.; Miller, V.A.; Costa, C.; et al. FAS and NF-kappaB signalling modulate dependence of lung cancers on mutant EGFR. Nature 2011, 471, 523–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blakely, C.M.; Pazarentzos, E.; Olivas, V.; Asthana, S.; Yan, J.J.; Tan, I.; Hrustanovic, G.; Chan, E.; Lin, L.; Neel, D.S.; et al. NF-kappaB-activating complex engaged in response to EGFR oncogene inhibition drives tumor cell survival and residual disease in lung cancer. Cell Rep. 2015, 11, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef] [Green Version]
- Jin, Q.; Huang, F.; Xu, X.; He, H.; Zhang, Y. High expression of hypoxia inducible factor 1alpha related with acquired resistant to EGFR tyrosine kinase inhibitors in NSCLC. Sci. Rep. 2021, 11, 1199. [Google Scholar] [CrossRef]
- Meng, S.; Wang, G.; Lu, Y.; Fan, Z. Functional cooperation between HIF-1alpha and c-Jun in mediating primary and acquired resistance to gefitinib in NSCLC cells with activating mutation of EGFR. Lung Cancer 2018, 121, 82–90. [Google Scholar] [CrossRef]
- Nilsson, M.B.; Robichaux, J.; Herynk, M.H.; Cascone, T.; Le, X.; Elamin, Y.; Patel, S.; Zhang, F.; Xu, L.; Hu, L.; et al. Altered Regulation of HIF-1alpha in Naive- and Drug-Resistant EGFR-Mutant NSCLC: Implications for a Vascular Endothelial Growth Factor-Dependent Phenotype. J. Thorac. Oncol. 2021, 16, 439–451. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, S.-h.; Kang, N.; Kim, O.; Hong, S.A.; Park, J.; Kim, J.; Lee, M.-A.; Kang, J. EGFR-Tyrosine Kinase Inhibitors Induced Activation of the Autocrine CXCL10/CXCR3 Pathway through Crosstalk between the Tumor and the Microenvironment in EGFR-Mutant Lung Cancer. Cancers 2023, 15, 124. https://doi.org/10.3390/cancers15010124
Hong S-h, Kang N, Kim O, Hong SA, Park J, Kim J, Lee M-A, Kang J. EGFR-Tyrosine Kinase Inhibitors Induced Activation of the Autocrine CXCL10/CXCR3 Pathway through Crosstalk between the Tumor and the Microenvironment in EGFR-Mutant Lung Cancer. Cancers. 2023; 15(1):124. https://doi.org/10.3390/cancers15010124
Chicago/Turabian StyleHong, Sook-hee, Nahyeon Kang, Okran Kim, Soon Auck Hong, Juyeon Park, Joori Kim, Myung-Ah Lee, and Jinhyoung Kang. 2023. "EGFR-Tyrosine Kinase Inhibitors Induced Activation of the Autocrine CXCL10/CXCR3 Pathway through Crosstalk between the Tumor and the Microenvironment in EGFR-Mutant Lung Cancer" Cancers 15, no. 1: 124. https://doi.org/10.3390/cancers15010124